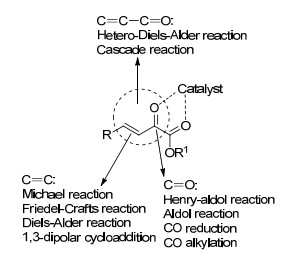
图 1.
β, γ-不饱和α-酮酸酯不同反应位点
Figure 1.
Different reaction sites of β, γ-unsaturated α-ketoester

不对称催化是当前化学的热门研究领域之一, 为众多光活性产品提供绿色、经济的合成手段, 为医药、农药、液晶材料和精细化工品等提供理论和实验基础[1].选择合适的底物是实现不对称催化反应的重要研究内容. β, γ-不饱和α-酮酸酯是一种多官能团合成子, 作为反应底物被广泛用于不对称催化反应中.其1, 2-双羰基结构可以与手性催化剂通过1, 4-配位的方式形成五元环中间体, 在活化了底物的同时诱导反应的立体选择性.此外, 它包含了不饱和α-酰基共轭体系、碳碳双键、羰基和酯基等多个活性反应位点(图 1), 在近20多年里被应用到不对称Friedel-Crafts反应[2, 3]、不对称Aldol反应[4]、不对称Diels-Alder反应[5]等多类反应, 合成了大量光活性化合物, 构筑了多种天然产物的手性骨架. 2015年, Eftekhari-Sis和Zirak[6]综述了α-酰基酮酸酯在合成杂环化合物中的应用, 其中描述了β, γ-不饱和α-酮酸酯2015年以前的进展.但是近年来, 随着各种新的催化体系的开发, β, γ-不饱和α-酮酸酯有了许多新的发展与应用.本文根据β, γ-不饱和α-酮酸酯中的不饱和酰基共轭体系、碳碳双键及羰基三种不同的反应位点分类, 对近几年来β, γ-不饱和α-酮酸酯中在不对称催化中的应用进行了综述.
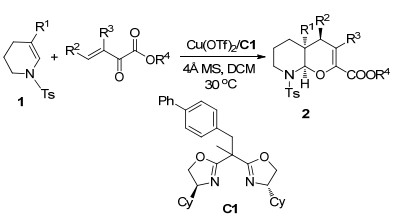
Diels-Alder (DA)反应是最常见的[4+2]环化反应之一, 通过协同过程可以生成不饱和六元环状化合物. β, γ-不饱和α-酮酸酯不仅可以通过碳碳双键作为亲双烯体与共轭双烯体进行DA反应, 且可以通过其杂共轭双烯体系与其他亲双烯体进行Hetero-Diels-Alder (HDA)反应, 生成二氢吡喃衍生物. 2016年, 唐勇课题组[7]报道了Cy-SaBox C1与Cu(OTf)2配合物催化的环状烯胺1与β, γ-不饱和α-酮酸酯的不对称HDA反应(Scheme 1), 以最高99%的产率和最高99% ee值获得双环N, O-缩醛2.当使用五元环状或七元环状烯胺时, 反应依然获得了85%~99%产率和95%~99% ee值.将反应扩大至克级规模, 可以在仅1 mol%的催化剂用量下, 以74%的产率和92% ee值得到对映产物, 表现出潜在的应用价值.
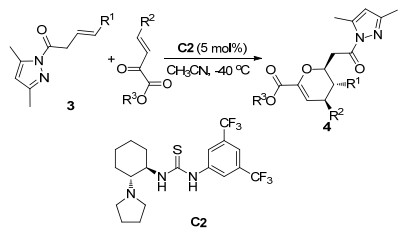
2019年, 黄慧才课题组[8]使用手性硫脲催化剂C2实现了β, γ-不饱和酰胺3与β, γ-不饱和α-酮酸酯的不对称HDA反应(Scheme 2).在最优条件下, 反应以中等以上的产率(61%~99%)和高对映选择性(99%~>99% ee)得到具有三个手性中心的二氢吡喃衍生物4.将底物3的R1扩展到噻吩环、呋喃环和萘环时, 反应依然以99%产率和99% ee值得到目标产物, 但是当3, 5-二甲基吡唑环扩展到其他氮杂环时, 反应的产率和对映选择性都显著降低, 底物3结构中不同杂环的位阻作用对反应有很大的影响.
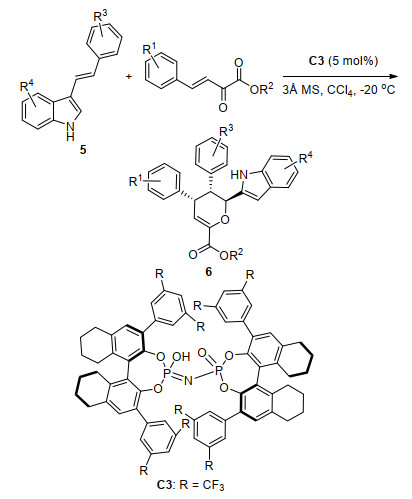
同年, 张锁秦小组[9]使用手性亚氨基二磷酸C3催化了3-乙烯基吲哚5与β, γ-不饱和α-酮酸酯的不对称HDA反应(Scheme 3), 合成了具有三个连续手性中心的一系列光学活性二氢吡喃衍生物6.在最优条件下, 获得了高收率(70%~99%)与非对映选择性(>20:1 dr), 以及中等到高的对映选择性(73%~99% ee值).他们推测该反应的机理是C3通过氢键与β, γ-不饱和酮酸酯的两个羰基相互作用.与此同时, 酮酸酯的Si面被C3的三氟甲基所阻碍, 5从酮酸酯的Re面通过协同途径进攻从而得到主要构型的产物6.
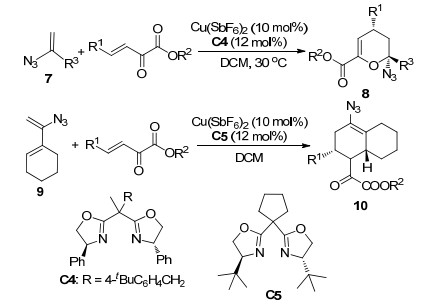
叠氮化合物是一类重要化合物, 可以作为各种药物合成的中间体. Thirupathi等[10]使用手性双噁唑啉配体C4与Cu(SbF6)2配合物实现了乙烯基叠氮化物7和β, γ-不饱和α-酮酸酯的不对称HDA反应(Scheme 4).该反应以中等到高的收率(58%~94%)和中等到高的对映选择性(72%~99% ee)得到了多种手性环状叠氮化物8.而当使用α, β-不饱和叠氮化物9与酮酸酯反应时, 酮酸酯提供碳碳双键作为亲烯体与提供共轭双烯体的9进行DA反应得到加成产物10 (Scheme 4), 进一步优化条件后, 对于不同取代基的底物, 该反应获得了61%~88%的产率和82%~91%的ee值.
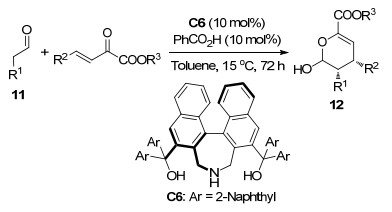
Maruoka小组[11]使用氨基二醇催化剂C6催化了β, γ-不饱和α-酮酸酯与醛11的不对称[4+2]环化反应, 以最高82%收率和99% ee值得到顺式光学纯二氢吡喃衍生物12 (Scheme 5).他们认为该反应可能的机理是C6的羟基与酮酸酯之间的氢键作用使得原本不稳定的顺式加成过渡态稳定化, 所以该反应得到了与常见的通过不对称HDA反应所得到的反式二氢吡喃衍生物不同的顺式加成产物12.但是在最优条件下使用脂肪醛作为底物时, 反应未显示出对映选择性.
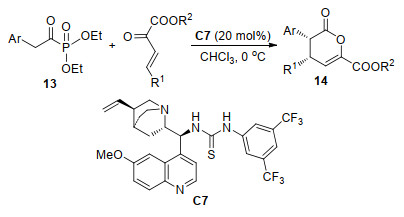
除了HDA反应之外, β, γ-不饱和α-酮酸酯还可以通过串联反应进行[4+2]环化从而构建出新的六元环骨架. 2016年, 袁伟成课题组[12]报道了手性硫脲C7催化的芳基乙酰基膦酸酯13与β, γ-不饱和α-酮酸酯的不对称Michael串联反应(Scheme 6).该反应利用芳基乙酰基膦酸酯作为亲核试剂进行不对称Michael反应, 然后发生酯化完成关环.在最优条件下, 对于不同取代基的底物以中等产率(54%~78%)和高对映选择性(92%~99% ee)得到了对映选择性产物二氢吡喃酮14.
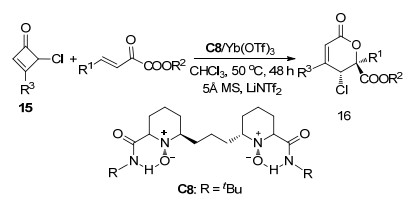
2018年, 冯小明课题组[13]报道了Yb(OTf)3和双手性氮氧配体C8配合物催化的β, γ-不饱和α-酮酸酯与环丁烯酮15的不对称[4+2]环加成反应, 以良好收率(38%~92%)、优异的对映选择性(64%~98% ee)和非对映选择性(8:1~19:1 dr)获得了二氢呋喃衍生物16 (Scheme 7).他们推断了该反应的机理, 首先配体C8与Yb3+形成四齿配位, 然后15的羰基与Yb3+配位并在LiNtf2与5Å分子筛的辅助下活化为烯酮中间体, 接下来酮酸酯的羰基与Yb3+配位并被活化, 最后15的Si面和酮酸酯的Re面之间发生对映选择性加成, 获得(5R, 6S)构型的加成产物16.
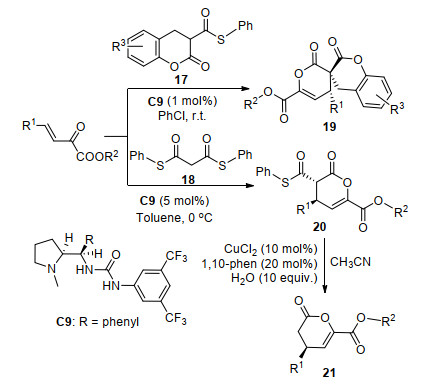
2018年, Ryu等[14]报道了手性脲C9催化的β, γ-不饱和α-酮酸酯与硫酯17和18的不对称串联反应(Scheme 8).在常温下, 以氯代苯为溶剂时, α-酮酸酯与硫酯17以54%~82%产率和97%~99% ee值生成对映产物19.而在0 ℃以甲苯为溶剂时, α-酮酸酯与硫酯18反应生成中间体20, 接着在氯化铜的作用下, 以乙腈为溶剂回流水解脱羧, 以54%~94%产率和84%~94% ee对映选择性得到产物21.该产物可用于抗高胆固醇药物的合成, 显示出良好的应用价值.
联烯是有机化学的研究热点之一, 但是不饱和共轭化合物与未活化的联烯进行不对称[4+2]环化反应一直难以实现. 2017年, 罗三中课题组[15]使用手性膦酸盐C9/InBr3在未活化的1, 1-双取代联烯22和β, γ-不饱和α-酮酸酯的[4+2]环化反应上取得突破(Scheme 9).在最优条件下, 该反应以最高90%的产率和99%的ee值得到二氢吡喃衍生物23.该反应具有良好的底物普适性, 当使用苯环以外的杂环或者烷基取代的联烯时, 依然能以中等收率(35%~60%)和高对映选择性(87%~96% ee)得到相应的加成产物.
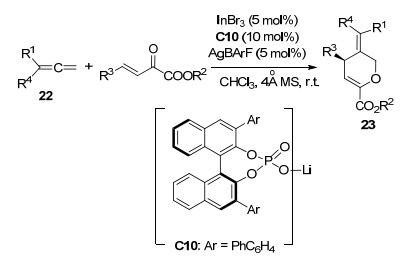
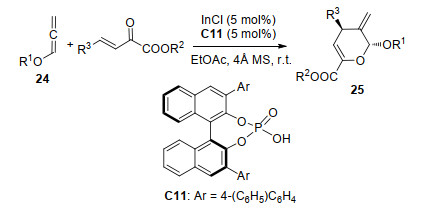
2018年, 该课题组[16]在此基础上进一步研究, 实现了InCl/手性磷酸C11络合物催化β, γ-不饱和α-酮酸酯与烷氧基联烯24的[4+2]环化反应(Scheme 10).在最优条件下, 以高产率(64%~99%)、高非对映选择性(>95:5 dr)和高对映选择性(>99% ee值)得到具有两个手性中心的产物25.他们认为, α-酮酸酯与InCl/手性磷酸C11络合物的单齿配位可能诱导了反应的立体选择性, 另外, 游离酸与烷氧基联烯24之间的氢键作用也选择性地诱导了烷氧基联烯24对于活化的α-酮酯的进攻.
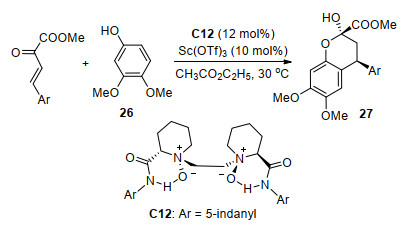
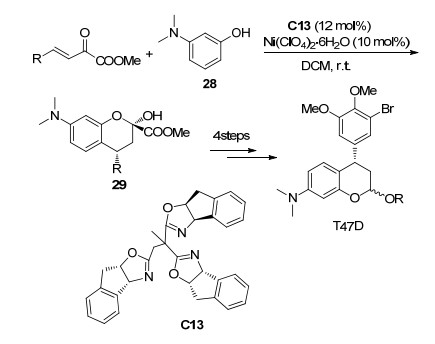
色烯骨架广泛存在于各种天然产物之中, 它们表现出广泛的生物活性, 如抗菌、抗肿瘤、抗氧化剂和抗衰老等[6].酚类或类似化合物与β, γ-不饱和α-酮酸酯的Friedel-Crafts/Aldol串联反应是合成苯并二氢吡喃(类色烯骨架)的重要方法之一. 2017年, 冯小明课题组[17]使用Sc(OTf)3与手性氮氧配体C12络合物催化了3, 4-二甲氧基苯酚26与β, γ-不饱和α-酮酸酯的不对称串联反应(Scheme 11), 以最高97%的产率和95% ee值以及中等的非对映选择性(6:1~14:1 dr)获得了一系列苯并二氢吡喃衍生物27.但是, 该反应体系对于其它取代酚的催化效果不佳, 特别是当使用3-氨基酚28作为底物时未获得对映选择性. 2018年, 唐勇课题组[18]用手性三噁唑啉C13/Ni(ClO4)2·6H2O配合物实现了3-氨基酚28和β, γ-不饱和α-酮酸酯的不对称串联反应(Scheme 12), 获得最高99%的产率和99% ee值, 并且加成产物经过4步反应可以得到潜在抗癌活性化合物T47D.
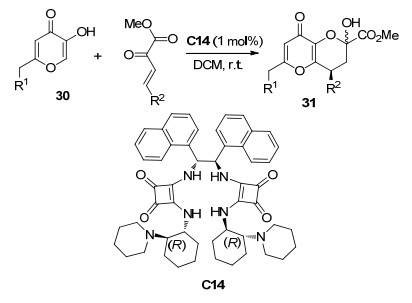
2018年, Zlotin课题组[19]报道了使用新颖的C2轴对称有机胺催化剂C14催化曲酸衍生物30与β, γ-不饱和α-酮酸酯的不对称Michael串联反应(Scheme 13).该反应以二氯甲烷作为溶剂在室温下反应3 h, 对于不同的底物能够以高产率(84%~99%)和高对映选择性(92%~99% ee)获得目标产物31.另外, 该反应催化剂负载量仅1 mol%, 且C14能够以萃取方式回收, 从而使催化剂能够循环利用. 2019年, 该课题组[20]报道了β, γ-不饱和α-酮酸酯与5-羟基苯并酚嗪(32)的不对称Michael串联反应(Scheme 14), 以85%~95%的产率和92%~98% ee的对映选择性获得目标产物33, 但是非对映选择性较差(1:1~1.5:1 dr).该催化反应的产物与抗癌药物sAJM589有着相同的关键单元结构.
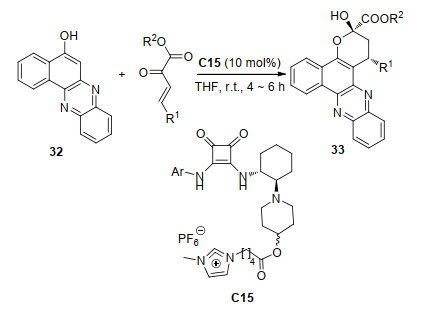
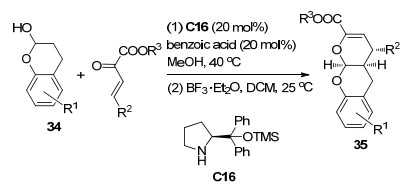
2019年, 刘延凯课题组[21]报道了手性胺C16催化的苯并二氢吡喃34与β, γ-不饱和α-酮酸酯的[3+3]环化反应(Scheme 15).他们认为, 该反应的途径是Michael加成反应之后, 酮酸酯的羰基与苯并二氢吡喃发生了半缩酮反应从而得到对映产物35.对于不同的底物, 在最优条件下该反应获得了53%~85%产率和93%~>99% ee值, 并且他们将催化剂大规模回收使用八次, 产率和立体选择性都没有受到显着影响.此外, 加成产物可以在保持光学纯度的同时转化为一系列结构复杂的分子.
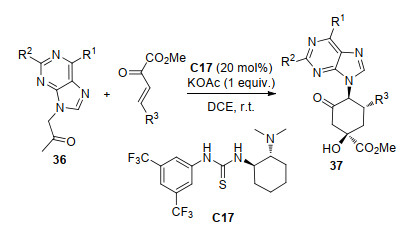
嘌呤核苷类似物在医药方面有着广泛的应用, 如Hudarabine和Cladribine可以用于治疗慢性淋巴白血病等血液癌症.迄今为止, 根据美国食品与药品管理局(FDA)统计, 上市的60多个抗肿瘤药物中有9个(15%)是核苷及其类似物, 29个抗病毒药物中有16个(55%)是核苷及其类似物[22]. 2018年, 郭海明课题组[23]报道了α-嘌呤取代丙酮36与β, γ-不饱和α-酮酸酯通过[3+3]环化反应得到具有三个手性立体中心的手性六元碳环嘌呤核苷类似物37的方法(Scheme 16).该反应使用手性硫脲C17为催化剂, 二氯乙烷作溶剂, 在常温下反应5 d以高收率(81%~87%)、高对映选择性(93%~98% ee)和中等到高的非对映选择性(75:25~90:10 dr)获得嘌呤核苷类似物37.但是, 当R3为非芳香基底物时, 在同样条件下反应不发生.
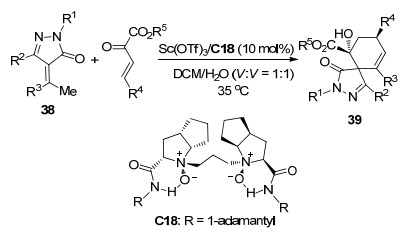
用水作溶剂一直是绿色化学的追求目标之一. 2019年, 冯小明课题组[24]报道了使用手性双氮氧配体C18与Sc(OTf)3络合物催化的α-亚芳基吡唑啉酮38与β, γ-不饱和α-酮酸酯的不对称Michael/Aldol串联反应(Scheme 17).该反应使用二氯甲烷与水作为混合溶剂(体积比1:1), 反应2 d, 对于不同取代基的底物以中等到高的产率(60%~99%)和中等到高的对映选择性(58%~94% ee)得到螺环化合物39.他们认为在该过程中水对于38 γ-氢的去质子化过程起着重要作用, 促进了双烯中间体的形成, 从而使Michael/Aldol串联反应进行.
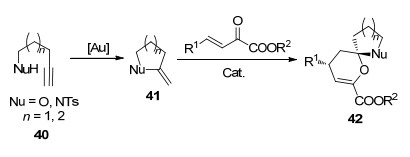
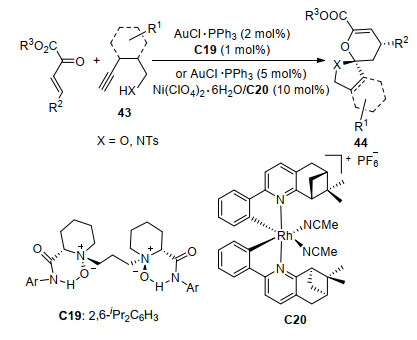
螺环是一种刚性环状结构, 在农药、医药和发光材料等多个方面有着重要的应用[25].炔基醇或炔基磺酰胺40可以在[Au]的作用下生成环状中间体41, 接着与β, γ-不饱和α-酮酸酯发生[4+2]环化反应得到螺环化合物42 (Scheme 18). 2016年, 冯小明课题组[26]使用Au(Ⅰ)和手性双氮氧配体C19/Ni(Ⅱ)配合物双金属催化剂体系实现了β, γ-不饱和α-酮酸酯与4, 5-炔基戊醇或4, 5-炔基戊磺酰胺43的不对称串联反应(Scheme 19).该反应以氯仿为溶剂反应24 h, 以最高99%产率和>99% ee合成了一系列对映的螺环产物44.他们[27]还用相似的体系催化了3, 4-炔基丁醇或3, 4-炔基丁酰胺45与β, γ-不饱和α-酮酸酯的不对称串联反应, 但是反应经过中间体46得到了稠环化合物47 (Scheme 20). 2018年, 康强等[28]用Au(Ⅰ)和手性Rh(Ⅲ)路易斯酸催化剂C20催化了该反应(Scheme 19), 同样取得了高收率和高对映选择性.与之前的研究相比, 由于C20由Rh(Ⅲ)中心离子与两个双齿配体不可逆地配位形成, 避免了使用双金属催化体系导致两种金属与配体竞争配位的情况.因此, 该反应的催化剂负载量较低[2 mol% Au(Ⅰ), 1 mol% Rh(Ⅲ)].
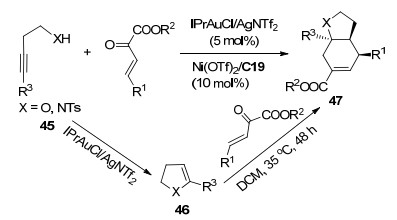
2019年, 冯小明课题组[29]使用Au(Ⅲ)和手性双氮氧配体C21/Mg(Ⅱ)配合物双金属催化剂体系成功实现了β-炔基酮48与β, γ-不饱和α-酮酸酯的不对称串联反应(Scheme 21).在最优条件下, 以39%~97%的收率以及中等至高的对映选择性(41%~93% ee)合成了6, 6-螺缩酮49.他们认为该反应的机理是: β-炔基酮48在Au(Ⅲ)的作用下烯醇化生成六元环中间体, 同时手性双氮氧配体C21/Mg(Ⅱ)配合物与酮酸酯的双羰基配位并活化, 然后发生[4+2]环加成反应得到对映选择性的目标化合物49.
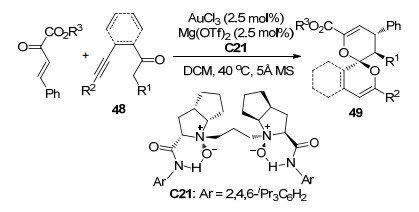
冯小明课题组[30]还报道了3-氯吲哚衍生物50与β, γ-不饱和α-酮酸酯之间不对称Michael/Alkylation串联反应(Scheme 22).当使用手性双氮氧配体C22/ Mg(OTf)2配合物作为催化剂时, 以52%~70%的产率以及84%~98%的ee值经过中间体51获得了热力学稳定的(1S, 2S, 3R)构型的加成产物52.当使用手性双氮氧配体C23/Sc(OTf)3配合物作为催化剂时, 以67%~99%的产率和72%~99%的ee值得到烷基化过程中通过SN2反应机理直接获得的(1R, 2S, 3R)构型的加成产物54.他们认为之所以得到不同构型的产物可能是因为不同的路易斯酸催化剂的特性不同.
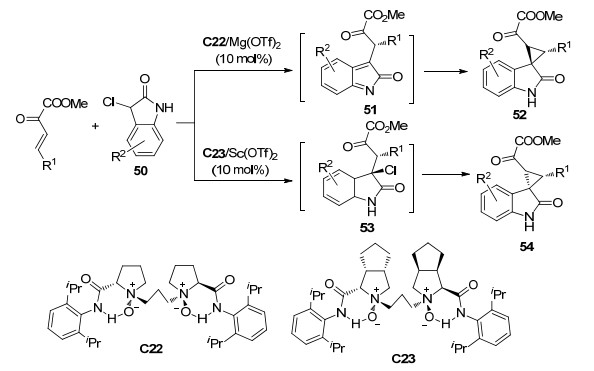
2019年, 袁伟成等[31]报道了使用手性硫脲C24催化的β, γ-不饱和α-酮酸酯与靛红衍生物55的不对称串联反应(Scheme 23), 以高产率(90%~98%)、中等到高的非对映选择性(73:27~>99:1 dr)和高对映选择性(91%~>99% ee值)得到了一系列具有两个连续手性中心的螺环化合物56.他们将所得产物对老鼠腹膜巨噬细胞进行了相关活性测试, 部分产物表现出优异的抗炎活性, 体现了该反应的良好的应用前景.
2019年, 冯小明课题组[32]使用手性双氮氧配体C25/Zn(OTf)2配合物实现了β, γ-不饱和α-酮酸酯与重氮酰亚胺57的[4+3]环化反应(Scheme 24).该反应提供了一种制备含有多个手性中心的含氧桥环的八元杂环化合物的方法, 在最优条件下, 当R2为芳香族时, 反应以67%~90%产率和93%~99% ee值获得对映产物; 但是当R2为环己基时反应并未发生.不同取代基的底物对于反应的影响较大.

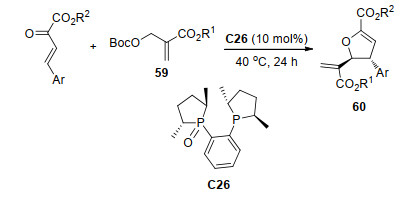
2017年, 李鹏飞课题组[33]使用有机磷催化剂C26催化了缺电子烯烃59与β, γ-不饱和α-酮酸酯的不对称[1+4]环化反应, 得到了一系列具有两个手性中心的二氢呋喃衍生物60 (Scheme 25).该反应有广泛的底物普适性, 在最优条件下, 对不同取代基的底物, 反应获得了中等到高的收率(22%~82%)以及高对映选择性(94%~99% ee), 并且将酮酸酯的酯基扩展为各类芳香基或者溴时, 依然可以达到53%~90%收率和94%~>99% ee值.将反应扩大至克级规模, 以62%的产率和96% ee值获得目标产物.对所得产物进行氢化, 在几乎不影响光学纯度的情况下得到旋光性的四氢呋喃衍生物.
2018年, 罗三中等[34]报道了InCl/手性磷酸C11双酸体系催化的简单烯烃61和β, γ-不饱和α-酮酸酯的不对称[4+2]环加成反应, 以高产率和高选择性(最高99:1 dr, 99% ee)生成相应的[4+2]环加成产物62 (Scheme 26).反应具有良好的底物普适性, 当使用桥环烯烃作为底物时, 依然能获得优异的非对映选择性(>95:5 dr)和对映选择性(最高99% ee值).他们认为InCl和C11的协同作用是反应成功的关键, 单独使用InCl或C11都不能催化该反应进行.

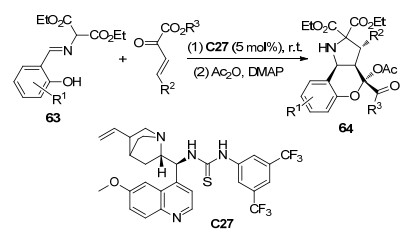
2019年, 许鹏飞课题组[35]实现了邻亚胺酚63与β, γ-不饱和α-酮酸酯的不对称串联反应, 得到具有四个连续手性中心(包括一个四级碳手性中心)的产物64 (Scheme 27).该反应使用硫脲C27作催化剂, 甲苯为溶剂, 常温下反应得到75%~98%的收率和高非对映选择性(>20:1)以及高对映选择性(93%~>99%).所得的产物容易衍生化, 具有生成生物活性分子的潜在应用.
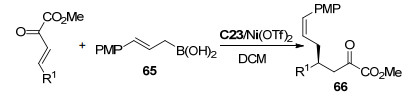
同年, 冯小明课题组[36]报道了烯丙基硼酸65与β, γ-不饱和α-酮酸酯烯丙基硼化/oxa-cope重排串联反应(Scheme 28).使用Ni(Ⅱ)/手性双氮氧配体C23络合物作为催化剂, 二氯甲烷为溶剂反应3 d, 以中等到高的收率(67%~92%)、高非对映选择性(96:4~>99:1 dr)和高对映选择性(92%~98% ee)得到重排产物66.该报道为如何使不饱和共轭化合物烯丙基化提供了另一种有效的合成策略.
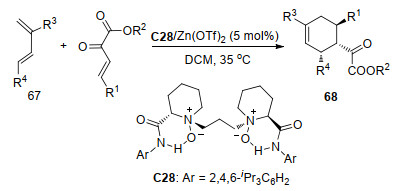
β, γ-不饱和α-酮酸酯可以通过碳碳双键作为亲双烯体与共轭双烯体进行D-A反应生成环己烯衍生物.冯小明课题组[37]使用Zn(OTf)2和手性双氮氧配体C28络合物催化了β, γ-不饱和α-酮酸酯与(E)-1-苯基二烯67的不对称D-A反应(Scheme 29), 取得最高99%的产率和99%的ee值.他们推测该反应的机理可能是手性氮氧配体C28和酮酸酯的两个氧配位于Zn(Ⅱ)中心, 同时降低了酮酸酯的最低未占分子轨道(LUMO)能量并促进D-A反应.由于酮酸酯的Si面被配体的邻近酰胺基阻挡, 因此, (E)-1-苯基二烯从酮酸酯的Re面通过协同途径进攻, 形成了(1R, 2S, 3R)构型的环己烯衍生物68.
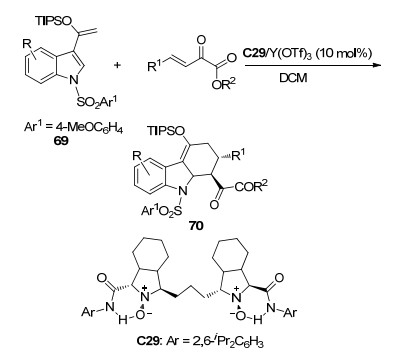
2016年, 冯小明课题组[38]使用Y(OTf)3与手性氮氧配体C29配合物催化了三异丙基硅烷氧基乙烯基吲哚69与β, γ-不饱和α-酮酸酯的不对称[4+2]环加成反应(Scheme 30).以47%~98%的收率和86%~99% ee值成功合成了具有三个连续手性中心的光活性咔唑骨架化合物70, 并且所得的产物可以很容易地转化成其它功能化合物.
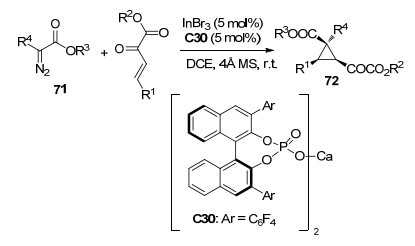
手性环丙烷结构广泛存在于各类活性天然产物中, 开发制备高度官能团化的手性环丙烷化合物一直是有机化学的研究热点之一. 2017年, 罗三中课题组[39]报道了InBr3与手性磷酸盐C30协同催化的β, γ-不饱和α-酮酸酯与重氮酯71的不对称环丙烷化反应, 得到了具有三个连续手性中心的环丙烷衍生物72 (Scheme 31).该反应的反应条件温和, 在室温下以二氯乙烷为溶剂, 对于不同取代的底物以中等收率(33%~77%)和高对映选择性(最高达99% ee)得到对映产物.该报道为获得高度能团化的手性环丙烷化合物提供了一种全新的便捷的方法.
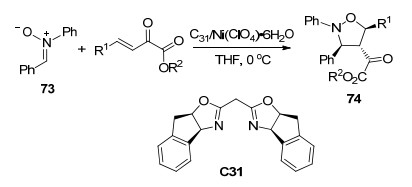
β, γ-不饱和α-酮酸酯中的碳碳双键还可以作为亲偶极体参与到1, 3-偶极环加成反应. 2017年, 傅滨等[40]报道了Ni(ClO4)2·6H2O与手性双噁唑啉C31络合物催化的β, γ-不饱和α-酮酸酯和硝酮73的不对称1, 3-偶极环加成反应(Scheme 32), 以中等程度以上的产率、高非对映选择性(>20:1 dr)和高对映选择性(88%~99% ee)得到异噁唑烷类化合物74.该反应可能的机理是酮酸酯与手性催化剂中心离子Ni(Ⅱ)以1, 4-配位方式络合, 由于C31的大位阻挡住了双键的Si面, 使得硝酮73更有利于从潜手性双键碳的Re面进攻, 发生1, 3-偶极环加成.
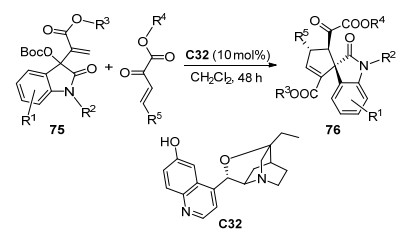
2019年, 陈永正等[41]报道了有机小分子C32催化的β, γ-不饱和α-酮酸酯与靛红衍生物75的不对称[3+2]环加成反应, 以94%的产率、>99:1 dr的非对映选择性和最高99% ee的对映选择性得到了手性螺环化合物76 (Scheme 33).在最优条件下, 将反应放大至克级, 依然可以得到优异的效果, 所得产物官能团易于转化, 具有潜在的应用价值.
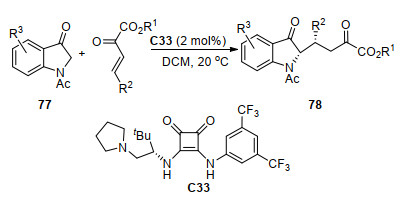
Michael加成反应是亲核物种对α, β-不饱和体系进行的1, 4-亲核加成, 是常见的构筑新手性中心的方法之一[42]. 2016年, 周正洪等[43]报道了有机小分子C33催化的1-乙酰基吲哚-3-酮77与β, γ-不饱和α-酮酸酯的Michael加成反应(Scheme 34), 构建了两个连续的叔碳手性中心, 获得最高99%的产率和99%的ee值.他们推测该反应机理是77烯醇化后与C33的叔胺基基团部分配位, 同时酮酸酯的双羰基通过氢键和C33相互作用形成过渡态, 然后77从Re面接近酮酸酯, 从而得到主要构型的产物78.
王敏灿课题组[44]使用新颖的AzePhenol C34与双核Zn络合物催化了4-羟基香豆素79和β, γ-不饱和α-酮酸酯的不对称Michae反应(Scheme 35).在最优条件下, 以最高99%的产率和94%的ee值得到一系列有药学意义的光活性香豆素和吡喃酮衍生物80.与之前的相关金属催化反应相比, 该催化体系在没有添加剂和更温和的反应条件下, 取得了更高的收率和对映选择性.

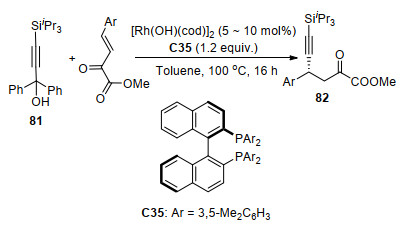
2017年, 窦晓巍课题组[45]报道了Rh/手性磷配体C35配合物催化的β, γ-不饱和α-酮酸酯炔基化反应(Scheme 36), 首次实现了使用sp杂化的碳原子作为亲核试剂与酮酸酯的不对称共轭加成, 获得了中等到高的催化活性(产率63%~88%)和高对映选择性(84%~93% ee).他们认为由于位于Si面的手性双膦配体芳环的空间位阻, 酮酸酯更易于与中心离子Rh配位, 从而使反应得到R构型的产物82.
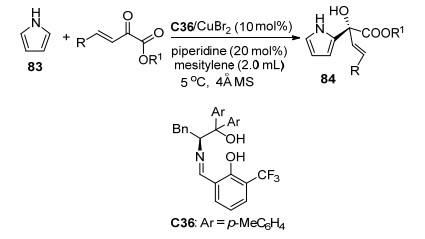
由于β, γ-不饱和α-酮酸酯中羰基参与的不对称反应在过去已经有了比较系统的报道[46~49], 所以近年来相关文献相对较少. 2017年, 汪志勇课题组[50]报道了CuBr2与手性小分子C36络合物催化的β, γ-不饱和α-酮酸酯中羰基与吡咯83的不对称Friedel-Crafts反应(Scheme 37), 获得了中等收率(33%~77%)和高对映选择性(75%~90% ee), 实现了吡咯与β, γ-不饱和α-酮酸酯中羰基的1, 2-加成, 但是在最优条件下, 当使用N-甲基保护的吡咯作为底物时反应不能发生.
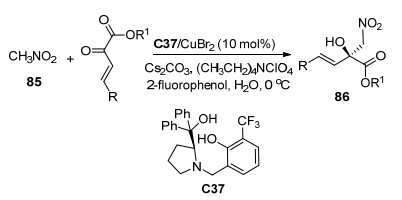
同年, 该课题组[51]报道了有机小分子C37与CuBr2络合物催化的β, γ-不饱和α-酮酸酯与硝基甲烷85的不对称Henry反应(Scheme 38), 在以水为溶剂的情况下, 以高收率(75%~91%)和优异的对映选择性(83%~94% ee)获得了一系列带有四取代碳立体中心的不饱和β-硝基-α-羟基酯86, 并且在最优条件下将反应放大至克级, 仍然能获得优异的结果.
近年来, β, γ-不饱和α-酮酸酯合成子在不对称催化中的应用得到了快速的发展, 特别是通过使用许多新骨架、新结构的有机小分子和路易斯酸催化剂催化, 进行了[4+2]、[3+3]、[3+2]和[1+4]环化等反应, 构建了一系列光活性的多手性中心复杂化合物, 其中对大部分反应机理进行了一定的探索, 为相关手性药物的合成提供了理论基础和学术依据, 有望使该底物进一步在新的反应中得到拓展应用.但是, 依然存在着反应底物局限性较大、催化剂循环利用较难、部分反应条件苛刻及所得产物实用性较低等诸多问题.因此, 寻找更加高效、高选择性、普适性好和可循环的催化体系是未来的发展趋势; 以药物、天然产物中间体为目标导向, 针对性地利用β, γ-不饱和α-酮酸酯合成子多官能团的优势, 设计开发条件温和、成本低廉和更具有实用价值的不对称催化反应也是未来的任务.
帕特里克J.沃尔什, 玛丽莎C.科兹洛夫斯基, 不对称催化基础, 赵金钵译, 化学工业出版社, 北京, 2018.Walsh, J. P.; Kozowski, M. C. Fundamentals of Asymmetric Catalysis, Translated by Zhao, Y. J., Chemical Industry Press, Beijing, 2018 (in Chinese).
Liu, L.; Ma, H.-L.; Xiao, Y.-M.; Du, F.-P.; Qin, Z.-H.; Li, N.; Fu, B. Chem. Commun. 2012, 48, 9281. doi: 10.1039/c2cc34803a
Liu, Y.; Shang, D. J.; Zhou, X.; Zhu, Y.; Lin, L.-L.; Liu, X.-H.; Feng, X.-M. Org. Lett. 2010, 12, 180. doi: 10.1021/ol902587t
Peng, L.; Wang, L.-L.; Bai, J.-F.; Jia, L.-N.; Guo, Y.-L; Luo, X.-Y.; Wang, F.-Y.; Yu, X. Y.; Wang, L. X. Org. Biomol. Chem. 2011, 9, 4774. doi: 10.1039/c1ob05607g
Xu, Z.-H.; Liu, L.; Wheeler, K.; Wang, H. Angew. Chem., Int. Ed. 2011, 50, 3484. doi: 10.1002/anie.201100160
Eftekhari-Sis, B.; Zirak, M. Chem. Rev. 2015, 115, 151. doi: 10.1021/cr5004216
Liu, Q.-J.; Wang, L.-J.; Kang, Q.-K.; Zhang, X.-P.; Tang, Y. Angew. Chem., Int. Ed. 2016, 55, 9220. doi: 10.1002/anie.201603911
Qin, J.-L.; Zhang, Y.-L.; Liu, G.-T.; Zhou, J.; Zhan, R.-T.; Chen, W.-W.; Huang, H. C. Org. Lett. 2019, 21, 7337. doi: 10.1021/acs.orglett.9b02629
Guo, X.-K.; Liu, G.-F.; An, D.; Zhang, H.; Zhang, S.-Q. Org. Lett. 2019, 21, 5438. doi: 10.1021/acs.orglett.9b01675
Thirupathi, N.; Wei, F.; Tung, C. H.; Xu, Z.-H. Nat. Commun. 2019, 10, 1. doi: 10.1038/s41467-018-07882-8
Kano, T.; Maruyama, H.; Homma, C.; Maruoka, K. Chem. Commun. 2018, 54, 3496. doi: 10.1039/C8CC01443D
Zhang, M.-J.; Wu, Z.-J.; Zhao, J.-Q.; Luo, Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W. C. Org. Lett. 2016, 18, 5110. doi: 10.1021/acs.orglett.6b02558
Yao, Q.; Yu, H.; Zhang, H.; Dong, S.-X.; Chang, F.-Z.; Lin, L.-L.; Liu, X.-H.; Feng, X.-M. Chem. Commun. 2018, 54, 3375. doi: 10.1039/C8CC01040D
Jin, H.; Lee, J.; Shi, H.; Li, J. Y.; Yoo, E. J.; Song, C. E.; Dyu, D. U. Org. Lett. 2018, 20, 1584. doi: 10.1021/acs.orglett.8b00331
Wang, L.; Lv, J.; Zhang, L.; Luo, S.-Z. Angew. Chem., Int. Ed. 2017, 56, 10867. doi: 10.1002/anie.201704020
Li, S.-J.; Lv, J.; Luo, S.-Z. Org. Chem. Front. 2018, 5, 1787. doi: 10.1039/C8QO00319J
Hao, X. Y.; Lin, L. L.; Tan, F.; Ge, S.-L.; Liu, X.-H.; Feng, X.-M. Org. Chem. Front. 2017, 4, 1647. doi: 10.1039/C7QO00323D
Ren, H.; Song, X.-Y.; Wang, S. R.; Wang, L.-J.; Tang, Y. Org. Lett. 2018, 20, 3858. doi: 10.1021/acs.orglett.8b01442
Kostenko, A. A.; Kucherenko, A. S.; Komogortsev, A. N.; Lichitsky, B. V.; Zlotin, S. G. Org. Biomol. Chem. 2018, 16, 9314. doi: 10.1039/C8OB02523A
Tukhvatshin, R. S.; Kucherenko, A. S.; Nelyubina, Y. V.; Zlotin, S. G. J. Org. Chem. 2019, 84, 13824. doi: 10.1021/acs.joc.9b02021
Lv, X.-J.; Zhao, W.-W.; Chen, Y.-H.; Wang, S.-B.; Liu, Y. K. Org. Chem. Front. 2019, 6, 1972. doi: 10.1039/C9QO00366E
唐杰, 董祥有, 欧阳文良, 朱云龙, 丁海新, 肖强, 有机化学, 2019, 39, 2609. doi: 10.6023/cjoc201901045Tang, J.; Dong, X.-Y.; Ouyang, W.-L.; Zhu, Y.-L.; Ding, H.-X.; Xiao, Q. Chin. J. Org. Chem. 2019, 39, 2609 (in Chinese). doi: 10.6023/cjoc201901045
Huang, K.-X.; Xie, M.-S.; Zhang, Q.-Y.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. Org. Lett. 2018, 20, 5398. doi: 10.1021/acs.orglett.8b02309
Xu, J.-X.; Hu, L.-F.; Hu, H.-P.; Ge, S.-L.; Liu, X.-H.; Feng, X.-M. Org. Lett. 2019, 21, 1632. doi: 10.1021/acs.orglett.9b00168
魏荣宝, 螺环化合物化学, 化学工业出版社, 北京, 2007.Wei, R.-B. Spiro Compound Chemistry, Chemical Industry Press, Beijing, 2007 (in Chinese).
Li, J.; Lin, L.-L.; Hu, B.; Lian, X.; Wang, G.; Liu, X.-H.; Feng, X.-M. Angew. Chem., Int. Ed. 2016, 55, 6075. doi: 10.1002/anie.201601701
Hu, B.; Li, J.; Gao, W.-D.; Lin, Q. C.; Yang, J.; Lin, L.-L.; Liu, X.-H.; Feng, X.-M. Adv. Synth. Catal. 2018, 360, 2831. doi: 10.1002/adsc.201800576
Gong, J.; Wan, Q.; Kang, Q. Adv. Synth. Catal. 2018, 360, 4031. doi: 10.1002/adsc.201800492
Ge, S.-L.; Cao, W.-D.; Kang, T.-F.; Hu, B.; Zhang, H.; Su, Z.-S.; Liu, X.-H.; Feng, X.-M. Angew. Chem., Int. Ed. 2019, 58, 4017. doi: 10.1002/anie.201812842
Kuang, Y.-L.; Shen, B.; Dai, L.; Yao, Q.; Liu, X.-H.; Lin, L.-L.; Feng, X.-M. Chem. Sci. 2018, 9, 688. doi: 10.1039/C7SC02757E
Bai, M.; Chen, Y.-Z.; Cui, B.-D.; Xu, X.-Y.; Yuan, W.-C. Tetrahedron 2019, 75, 2155. doi: 10.1016/j.tet.2019.02.036
Xu, C.-R.; Wang, K.-X.; Li, D. W.; Lin, L.-L.; Feng, X.-M. Angew. Chem., Int. Ed. 2019, 58, 1. doi: 10.1002/anie.201813481
Cheng, Y.-Y.; Han, Y.-Z.; Li, P.-F. Org. Lett. 2017, 19, 4774. doi: 10.1021/acs.orglett.7b02144
李速家, 吕健, 罗三中, 化学学报, 2018, 76, 869. doi: 10.7503/cjcu20170608Li, S.-J.; Lv, J.; Luo, S.-Z. Acta Chim. Sinica 2018, 76, 869 (in Chinese). doi: 10.7503/cjcu20170608
Cao, J.; Liu, Y.-J.; Zhang, Y.-M.; Wang, Z.-Y.; Xu, P.-F. Org. Chem. Front. 2019, 6, 674.
Tang, Q.; Fu, K.; Ruan, P.; Dong, S.-X.; Su, Z.-S.; Liu, X.-H.; Feng, X.-M. Angew. Chem., Int. Ed. 2019, 58, 11846. doi: 10.1002/anie.201905533
Xiong, Q.; Lin, L.-L.; Zhao, Y.-H.; Lang, J.-W.; Liu, X.-H.; Feng, X.-M. J. Org. Chem. 2018, 83, 12527. doi: 10.1021/acs.joc.8b01771
Zhao, X.-H.; Mei, H.-J.; Xiong, Q.; Fu, K.; Lin, L.-L.; Feng, X.-M. Chem. Commun. 2016, 52, 10692. doi: 10.1039/C6CC05328A
Zhong, X.-R.; Lv, J.; Luo, S.-Z. Org. Lett. 2017, 19, 3331. doi: 10.1021/acs.orglett.7b01577
Xie, L.; Yu, X.; Li, J.-Q.; Zhang, Z.-H.; Qin, Z.-H.; Fu, B. Eur. J. Org. Chem. 2017, 3, 657.
Chen, Y.; Cui. B. D.; Bai, M.; Han, W. Y.; Wan, N. W.; Chen, Y. Z. Tetrahedron 2019, 75, 2971. doi: 10.1016/j.tet.2019.04.040
白冰, 王龙, 杨静, 蔡莉莉, 刘前进, 席高磊, 赵志伟, 毛多斌, 陈芝飞, 有机化学, 2019, 39, 1053. doi: 10.6023/cjoc201809015Bai, B.; Wang, L.; Yang, J.; Cai, L.-L.; Liu, Q.-J.; Xi, G.-L.; Zhao, Z.-W.; Mao, D.-B.; Chen, Z.-F. Chin. J. Org. Chem. 2019, 39, 1053 (in Chinese). doi: 10.6023/cjoc201809015
Chen, S.-R.; Wang, Y.-M.; Zhou, Z. H. J. Org. Chem. 2016, 81, 11432. doi: 10.1021/acs.joc.6b02070
Liu, S.-S.; Xu, Z.-H.; Wang, X.; Zhu, H.-R.; Wang, M.-C. J. Org. Chem. 2019, 84, 13881. doi: 10.1021/acs.joc.9b02046
Zhi, Y.-L.; Huang, J.-H.; Liu, N.; Tao, L.; Dou, X.-W. Org. Lett. 2017, 19, 2378. doi: 10.1021/acs.orglett.7b00909
Li, P.-F.; Zhao, J.-L.; Li, F.-B.; Chan, A.-S. C.; Kwong, F. Y. Org. Lett. 2010, 12, 5616. doi: 10.1021/ol102254q
Wei, A.-J.; Nie, J.; Zheng, Y.; Ma, J.-A. J. Org. Chem. 2015, 80, 3766. doi: 10.1021/jo502741z
Li, P.-F.; Chan, S.-H.; Chan, A. S. C.; Yee, F. Adv. Synth. Catal. 2011, 353, 1179. doi: 10.1002/adsc.201000982
Luo, W.-W.; Zhao, J.-N.; Ji, J.; Lin, L.-L.; Liu, X.-H.; Mei, H.-J. Chem. Commun. 2015, 51, 10042. doi: 10.1039/C5CC02748A
Sun, J.-N.; Hu, Y.-B.; Li, Y.-N.; Zhang, S.; Zha, Z.-G.; Wang, Z.-Y. J. Org. Chem. 2017, 82, 5102. doi: 10.1021/acs.joc.7b00159
Li, Y.-N.; Huang, Y.-K.; Gui, Y.; Sun, J.-N.; Li, J.-D.; Zha, Z.-G.; Wang, Z.-Y. Org. Lett. 2017, 19, 6416. doi: 10.1021/acs.orglett.7b03299

图 1 β, γ-不饱和α-酮酸酯不同反应位点
Figure 1 Different reaction sites of β, γ-unsaturated α-ketoester

图式 1 环状烯胺与β, γ-不饱和α-酮酸酯的不对称Hetero-Diels-Alder (HDA)反应
Scheme 1 Catalytic asymmetric Hetero-Diels-Alder (HDA) reaction of cyclic enamine with β, γ-unsaturated α-ketoester

图式 2 β, γ-不饱和酰胺与β, γ-不饱和α-酮酸酯的不对称HDA反应
Scheme 2 Catalytic asymmetric HDA reaction of β, γ-unsaturated amide with β, γ-unsaturated α-ketoester

图式 3 3-乙烯基吲哚与β, γ-不饱和α-酮酸酯的不对称HDA反应
Scheme 3 Catalytic asymmetric HDA reaction of 3-vinylindole with β, γ-unsaturated α-ketoester

图式 4 叠氮化物与β, γ-不饱和α-酮酸酯的不对称DA反应
Scheme 4 Catalytic asymmetric DA reaction of azide with β, γ-unsaturated α-ketoester

图式 5 醛与β, γ-不饱和α-酮酸酯的不对称HDA反应
Scheme 5 Catalytic asymmetric HDA reaction of aldehyde with β, γ-unsaturated α-ketoester

图式 6 芳基乙酰基膦酸酯与β, γ-不饱和α-酮酸酯的不对称串联反应
Scheme 6 Catalytic asymmetric cascade reaction of arylacetyl phosphonate with β, γ-unsaturated α-ketoester

图式 7 环丁烯酮与β, γ-不饱和α-酮酸酯的不对称[4+2]环加成反应
Scheme 7 Catalytic asymmetric [4+2] cycloaddition reaction of cyclobutenones with β, γ-unsaturated α-ketoester

图式 8 硫酯与β, γ-不饱和α-酮酸酯的不对称串联反应
Scheme 8 Catalytic asymmetric cascade reaction of thioesters with β, γ-unsaturated α-ketoester

图式 9 未活化联烯与β, γ-不饱和α-酮酸酯的不对称[4+2]环化反应
Scheme 9 Catalytic asymmetric [4+2] annulation reaction of non-activated allenes with β, γ-unsaturated α-ketoester

图式 10 烷氧基联烯与β, γ-不饱和α-酮酸酯的不对称[4+2]环化反应
Scheme 10 Catalytic asymmetric [4+2] annulation reaction of alkoxyallenes with β, γ-unsaturated α-ketoester

图式 11 3, 4-二甲氧基苯酚与β, γ-不饱和α-酮酸酯的不对称Friedel-Crafts/aldol串联反应
Scheme 11 Catalytic asymmetric Friedel-Crafts/aldol reaction of 3, 4-dimethoxyphenol with β, γ-unsaturated α-ketoester

图式 12 3-氨基酚与β, γ-不饱和α-酮酸酯的不对称Friedel-Crafts/aldol串联反应
Scheme 12 Catalytic asymmetric Friedel-Crafts/aldol reaction of 3-aminophenol with β, γ-unsaturated α-ketoester

图式 13 曲酸衍生物与β, γ-不饱和α-酮酸酯的不对称Michael串联反应
Scheme 13 Catalytic asymmetric Michael cascade reaction of kojic acid derivatives with β, γ-unsaturated α-ketoester

图式 14 5-羟基-苯并酚嗪与β, γ-不饱和α-酮酸酯的不对称Michael串联反应
Scheme 14 Catalytic asymmetric Michael cascade reaction of Benzo[a]phenazin-5-ol with β, γ-unsaturated α-ketoester

图式 15 苯并二氢吡喃与β, γ-不饱和α-酮酸酯的不对称[3+3]环化反应
Scheme 15 Catalytic asymmetric [3+3] annulation reaction of benzodihydropyran with β, γ-unsaturated α-ketoester

图式 16 α-嘌呤取代丙酮与β, γ-不饱和α-酮酸酯的不对称[3+3]环化反应
Scheme 16 Catalytic asymmetric [3+3] annulation reaction of α-purine-substituted acetones with β, γ-unsaturated α-ketoester

图式 17 α-亚芳基吡唑啉酮与β, γ-不饱和α-酮酸酯的不对称Michael/Aldol串联反应
Scheme 17 Catalytic asymmetric Michael/aldol reaction of α-arylidene pyrazolinone acetones with β, γ-unsaturated α-ketoester

图式 18 炔基醇或炔基磺酰胺与β, γ-不饱和α-酮酸酯的不对称串联反应
Scheme 18 Catalytic asymmetric cascade reaction of alkynyl alcohols and amides with β, γ-unsaturated α-ketoester

图式 19 4, 5-炔基戊醇或4, 5-炔基戊磺酰胺与β, γ-不饱和α-酮酸酯的不对称串联反应
Scheme 19 Catalytic asymmetric cascade reaction of 4, 5-alkynyl pentanol and 4, 5-alkynyl pentylsulfonamide with β, γ-unsaturated α-ketoester

图式 20 3, 4-炔基丁醇或3, 4-炔基丁酰胺与β, γ-不饱和α-酮酸酯的不对称串联反应
Scheme 20 Catalytic asymmetric cascade reaction of 3, 4-alkynyl butanol and 3, 4-alkynyl butanamide with β, γ-unsaturated α-ketoester

图式 21 β-炔基酮与β, γ-不饱和α-酮酸酯的不对称串联反应
Scheme 21 Catalytic asymmetric cascade reaction of β-alkynyl ketone with β, γ-unsaturated α-ketoester

图式 22 3-氯吲哚衍生物与β, γ-不饱和α-酮酸酯的不对称Michael/alkylation串联反应
Scheme 22 Catalytic asymmetric Michael/alkylation cascade reaction reaction of 3-chloro-oxindoles with β, γ-unsaturated α-ketoester

图式 23 靛红衍生物与β, γ-不饱和α-酮酸酯的不对称串联反应
Scheme 23 Catalytic asymmetric cascade reaction of 3-isothiocyanato oxindoles with β, γ-unsaturated α-ketoester

图式 24 α-嘌呤取代丙酮与β, γ-不饱和α-酮酸酯的不对称[4+3]环化反应
Scheme 24 Catalytic asymmetric [4+3] annulation reaction of acyclic diazoimide with β, γ-unsaturated α-ketoester

图式 25 缺电子烯烃与β, γ-不饱和α-酮酸酯的不对称[1+4]环化反应
Scheme 25 Catalytic asymmetric [1+4] annulation reaction of electron-deficient olefins with β, γ-unsaturated α-ketoester

图式 26 简单烯烃与β, γ-不饱和α-酮酸酯的不对称[4+2]环加成反应
Scheme 26 Catalytic asymmetric [4+2] cycloaddition of simple olefin with β, γ-unsaturated α-ketoester

图式 27 邻亚胺酚与β, γ-不饱和α-酮酸酯的不对称串联反应
Scheme 27 Catalytic asymmetric cascade reaction of o-iminophenol with β, γ-unsaturated α-ketoester

图式 28 烯丙基硼酸与β, γ-不饱和α-酮酸酯的烯丙基硼化/ oxa-cope重排串联反应
Scheme 28 Catalytic asymmetric allylboration/oxy-cope rearrangement of allylboronic acids with β, γ-unsaturated α-ketoester

图式 29 (E)-1-苯基二烯与β, γ-不饱和α-酮酸酯的不对称DA反应
Scheme 29 Catalytic asymmetric DA reaction of (E)-1-phenyl dienes with β, γ-unsaturated α-ketoester

图式 30 三异丙基硅烷氧基乙烯基吲哚与β, γ-不饱和α-酮酸酯的不对称[4+2]环加成反应
Scheme 30 Catalytic asymmetric [4+2] cycloaddition of silyloxyvinylindoles with β, γ-unsaturated α-ketoester

图式 31 重氮酯与β, γ-不饱和α-酮酸酯的不对称环丙烷化反应
Scheme 31 Catalytic asymmetric [4+2] cycloaddition of diazoesters with β, γ-unsaturated α-ketoester

图式 32 硝酮与β, γ-不饱和α-酮酸酯的不对称1, 3-偶极环加成反应
Scheme 32 Catalytic asymmetric 1, 3-dipolar cycloaddition of nitrones with β, γ-unsaturated α-ketoester

图式 33 靛红衍生物与β, γ-不饱和α-酮酸酯的不对称[3+2]环加成反应
Scheme 33 Catalytic asymmetric [3+2] cycloaddition of isatin-derived MBH carbonates with β, γ-unsaturated α-ketoester

图式 34 1-乙酰基吲哚-3-酮与β, γ-不饱和α-酮酸酯的不对称Michael加成反应
Scheme 34 Catalytic asymmetric Michael addition of 1-acetylindolin-3ones with β, γ-unsaturated α-ketoester

图式 35 4-羟基香豆素与β, γ-不饱和α-酮酸酯的不对称Michael加成反应
Scheme 35 Catalytic asymmetric Michael addition of 4-hydroxycoumarins with β, γ-unsaturated α-ketoester

图式 36 β, γ-不饱和α-酮酸酯的不对称炔基化反应
Scheme 36 Catalytic asymmetric conjugate alkynylation of β, γ-unsaturated α-ketoester

图式 37 吡咯与β, γ-不饱和α-酮酸酯的不对称Friedel-Crafts反应
Scheme 37 Catalytic asymmetric Friedel-Crafts reaction of pyrrole with β, γ-unsaturated α-ketoester

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

 下载:
下载: