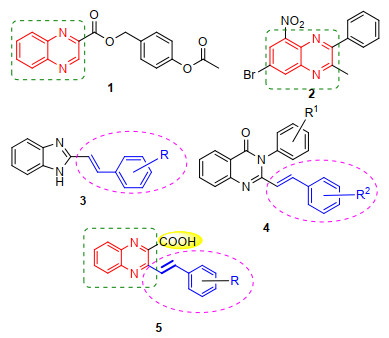
Figure 1.
Anti-tubercular quinoxaline, arylvinyl heterocyclic derivatives and the designed structure of targeted compounds
A Facile Synthesis and M. tuberculosis Leucyl-tRNA Synthetase Inhi-bitory Activity of Novel 3-Arylvinylquinoxaline-2-carboxylic Acids
Yang Li , Shiyu Dong , Hongwei Qin , Bingyue Tang , Wentao Gao , Yu Chen

Tuberculosis (TB) is a foremost respiratory disease mainly caused by Mycobacterium tuberculosis (Mtb). In 2019, World Health Organisation (WHO) declared about 9.0 million people developed tuberculosis worldwide, out of which India and China accounted for 24% and 11% of the total cases, respectively.[1] Especially, currently due to the widespread existence of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB), there is an increased concern that TB may once again become an incurable disease.[2] It is known that substitution of key amino acid residues in the enzyme active site would lead to the drug resistance.[3] Since Leucyl-tRNA synthetase (LeuRS) plays an essential role in charging leucine to its cognate tRNA correctly and don't mutate very easily, [4] these enzymes are thus less likely to become drug-resistant.[5] Thus, LeuRS has been paid much attention as a clinically validated and attractive therapeutic target.[6-7] Accordingly, to overcome multidrug resistance, Mycobacterium tuberculosis LeuRS (Mtb LeuRS) has been proposed recently as a promising target with great potential in the development of new and potent antitubercular drugs.[8-9] In this context, a review involving the development of leucyl-tRNA synthetase inhibitors as antimicrobial agents has been reported just recently by Zhang and coworker.[10]
It is well known that quinoxaline ring system is an exceptional class of nitrogen-containing heterocycles present in many pharmacologically relevant molecules, and has been acted as a valuable pharmacophore for drug design in the field of contemporary medicinal chemistry.[11-12] In this regard, Ajani[13] described the status of quinoxaline motifs as excellent pathfinders in therapeutic medicine. Recently, a recent advance on pharmacological activities of quinoxaline derivatives has been reviewed by Tariq et al.[14] Among diverse striking biological properties, numerous 2-substituted and 2, 3-disubstituted quinoxaline derivatives have been frequently reported to possess substantial anti-tubercular activity and have drawn considerable attention.[15-18] For example, Seitz et al.[19] ever reported the synthesis of a series of novel quinoxaline derivatives, among which 4'-acetoxybenzyl 2-quinoxalinecarboxylate (1, Figure 1) showed potent activity against M. tuberculosis (Mtb). In this context, Magnet et al.[20] found that quinoxaline-based derivatives such as 2-methyl-3-phenyl- 5-nitro-7-bromoquinoxaline (VI-9376, 2, Figure 1) displayed significant potential as good candidates for the development of new pharmaceutical products against M. tuberculosis through screening more than 12000 heterocyclic compounds with the aim of identifying new scaffolds active against M. tuberculosis. However, to the best of our knowledge, the synthesis of structurally new quinoxaline derivatives as Mtb LeuRS inhibitors for anti-tuberculosis remains unexplored.
On the other hand, arylvinyl moiety is an important structural motif existing in numerous bioactive natural products and medicinal agents and playing an essential role for biological activity.[21-22] Therefore, the efficient incorporation of an arylvinyl group into organic molecules has been a topic of great synthetic interest with the aim of enhancing the potency of this class of compounds.[23-24] For example, Shingalapur et al.[25] reported an easy access to a new series of novel (E)-2-arylvinyl-1H-benzo[d]imida- zoles (3, Figure 1), which were found to show promising anti-tubercular activity. In this regard, Jadhavar et al.[26] described the synthesis of various 2-styrylquinazolones (4, Figure 1) with potent anti-Mtb activity. For another, many potential anti-TB heteroaromatic compounds have carboxylic acid functionality as the essential structural feature, such as cinnamic acid, [27] quinoline-3-carboxylic acid[28] and thiophene-3-carboxylic acid derivatives.[29] Especially, Maresca et al.[30] have revealed that Mycobacterium tuberculosis is highly sensitive to weak aromatic/heterocy- clic acids, and many carboxylic acids incorporating various aromatic/heterocyclic scaffold strongly inhibited the growth of Mycobacterium tuberculosis, showing significant potential for developing antimycobacterial agents with a diverse mechanism of action compared to the clinically used drugs.
In light of the above findings, it would be an attractive template by combination of the structural features of quinoxaline, arylvinyl moiety and carboxyl functional group in a single molecule framework, because it might lead to a new dimension of structural diversity as potential candidates for anti-tubercular evaluation. Thus, in the context of our continuing interest in the construction of interesting types of heterocyclic compounds, we would like to report the synthesis of a series of structurally intriguing quinoxaline scaffolds 5 for anti-tubercular screening by integrating the arylvinyl moiety and carboxyl functional group at the position of two and three of the quinoxaline ring as represented in Figure 1. To our knowledge, except for our studies, the synthesis and anti-tubercular activity evaluation of such compounds has not previously been reported.
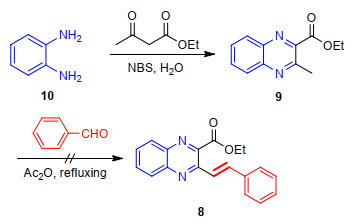
Arylvinyl-substituted compounds are commonly prepared through the condensation reaction of α-methyl nitrogen heterocyclic compounds with aromatic aldehydes due to the methyl group could be activated by the presence of the nitrogen atom.[31] Accordingly, our initial investigation towards the synthesis of ethyl 2-styrylquinoxaline-3- carboxylate (8) was conducted employing ethyl 2-methyl- quinoline-3-carboxylate (9) and benzaldehyde as substrates as shown in Scheme 1. The substrate 9 was readily synthesized in good yield based on the method described by Kumar et al.[32] involving the condensation reaction of phenylene diamine (10) with N-bromosuccinimide and ethyl acetoacetate. However, when 9 was subjected to the condensation reaction with benzaldehyde under the reaction conditions as described in literature[31] using boiling acetic anhydride as the media followed by hydrolysis in pyridine water mixture, this was not the case in our hands and the reaction proceed very poorly, giving an intractable complex mixture, from which the desired 8 could not be separated in any appreciable yield. Attempts to use other reaction conditions, such as NBS/TBHP (N-bromosuccinimide/ t-butyl hydroperoxide), [33] NaOAc in H2O/AcOH (V:V=1:1), [34] and Bmim[BF4], [35] were also unfruitful. In addition, we also attempted to use Aliquat 336 as the catalyst, [36] which was reported to yield good results in this type of condensation reaction, the corresponding 2-styryl- quinoxaline. Unfortunately, once again we obtained a highly impure mixture of products with no identifiable products being obtained.
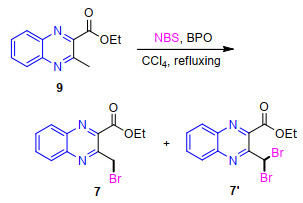
After these unsuccessful trials, we decided to circumvented the problem by designing an alternative approach. As well known, halomethyl-functionalized aromatic heterocycles have been widely served as versatile building blocks for the construction of various intriguing and complex small molecules.[37] For example, Wang's group have reported the use of 2-chloromethyl-pyrido[1, 2-a]pyrimi- dins as important building blocks for the construction various bioactive polycyclic-fused heterocycle systems.[38] In this context, our research group has gained substantial ability in the application of 2-(halomethyl)quinoline derivatives as the key intermediates for the flexible synthesis of diversely substituted quinoline-based hybridds.[39-40] Building on these evolving expertise and further extending the applications of our previously described methodology to new substrates, we envisioned that if the functionalization of 9 at its 2-methyl position with bromomethyl moiety could be achieved, the resulting 2-(bromomethyl)quinoxa- line-3-carboxylate might open opportunity for the construction of the title compounds. Thus, at the beginning of our investigation, we devoted our efforts to establish the synthesis of ethyl 3-(bromomethyl)quinoxaline-2-carboxy- late (7), which was successfully synthesized by radical bromination reaction of 2-methylquinoxaline 9 with 1.2 equiv. of NBS in refluxing CCl4 with the presence of catalytic amount of benzoyl peroxide (BPO) as the initiator as described in Scheme 2.
It is worthy to note that the order of addition of NBS was found to have an obvious influence on the product yield. When 1.2 equiv. of NBS was added simultaneously together with 9 to CCl4 solution, the desired monobrominated product 7 was obtained only in 49% yield, along with 30% dibromide byproduct 7', 12% unreacted 9 and small quantities of tarry products. Moreover, the chromatographic separation was very tedious due to the very similar polarities of three compounds 9, 7 and 7ʹ. This is common to numerous radical bromination protocols.[41] It was found that if 1.2 equiv. of NBS was added in batches in the amount of 1/3 portions every 1.5 h to the gently refluxing CCl4 solution, 22% increase in the product 71% yield was achieved after the reaction was complete with a small amount of byproduct. Presumably, the resulting Br2 from NBS could maintain in a low concentration throughout the course of the bromination reaction, thereby restraining the side reaction and leading to the formation of the desired product in a higher yield. The newly resulting monobrominated product 7 represents a valuable intermediate and would be highly useful in the field of synthetic organic chemistry. Its structure was easily confirmed by spectral (1H NMR and 13C NMR) data. In 1H NMR spectrum, no signal attributable to methyl protons of its precursor was observed, but instead a two-proton singlet at δ 5.16 was found, readily recognizable as arising from bromomethyl protons, supporting the signal of 13C NMR spectrum at δ 62.98 for the methylene carbon. In addition, the structure of the dibromide byproduct ethyl 3-(dibromomethyl)- quinoxaline-2-carboxylate (7'), obtained by careful column chromatography over silica gel, was readily established according to its spectral data.
Next, the newly-synthesized monobrominated quinoxaline 7 was employed as substrate for the construction of 3-arylvinylquinoxaline-2-carboxylic acids. Herein, we devised a simple and facile one-pot three-step protocol involving the successive Michaelis-Arbuzov reaction followed by Horner-Wadsworth-Emmons (HWE) olefination reaction and subsequent ester hydrolysis reaction procedure as shown in Scheme 3.
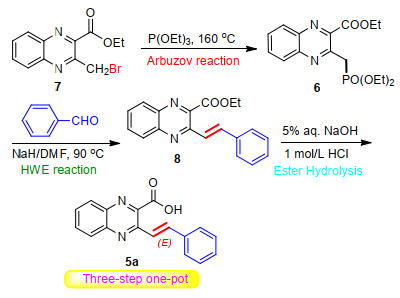
The substrate 7 was first subjected to the Michaelis-Arbuzov reaction. It was found that by only heating the mixture of 7 and excessive triethyl phosphate at 160 ℃ without added catalyst, the reaction proceeded very smoothly with nearly quantitative transformation into the corresponding (quinoxalinylmethyl)phosphonate 6 within 2 h. As the reaction did not require use of any organic solvents and catalysts, product 6 was very pure. It was speculated that isolation and purification at this stage were unnecessary and the subsequent HWE olefination reaction might be conducted successively in one-pot manner. Accordingly, the excess triethylphosphite was evaporated to dryness under reduced pressure condition, followed by the direct addition of 1.1 equiv. of benzaldehyde in N, N'- dimethylformamide (DMF) along with 1.1 equiv. of NaH into the residue to carry out in situ the HWE olefination reaction by heating the resulting mixture at 90 ℃ (Scheme 3). To our delight, the sequential Horner-Emmons olefination proceeded very cleanly and thin-layer chromatography (TLC) analysis revealed nearly quantitative conversion to the corresponding olefination intermediate 8 within 4 h. At this stage, it is important to mention that the use of NaH was found to be optimal to complete the reaction and other bases, such as NaOH, K2CO3, Et3N, EtONa, t-BuOK are inferior. Finally, the resulting 8 was underwent in situ ester hydrolysis reaction by adding 10% aqueous NaOH solution directly to the reaction mixture. After refluxing for 2 h (TLC) followed by an acidic work-up, the corresponding quinoxaline carboxylic acid 5a was obtained in 76% overall yield. The beauty of this reaction procedure is that the three chemical transformations take place in one pot, obtaining the product in high yield with simple synthetic operation and workup.
Thereafter, a range of aromatic aldehydes available in our laboratory containing mono- or di-substituents with differing electronic properties were subjected to the reaction sequence in a similar fashion. As expected, these aromatic aldehydes were equally amenable to the reaction process without any experimental difficulties, successfully furnishing the corresponding 5b~5j in satisfactory yields of 62%~79%. Encouraged by these results and with the aim of diversifying our work, we further attempted the reaction with 2-chloroquinoline-3-carbaldehyde and 2- chloro-N-substituted-1H-indole-3-carbaldehydes under the same reaction conditions. To our delight, these species were also viable substrates for this one-pot transformation, invariably giving the expected products 5k~5o in satisfactory yields (Entries 11 and 12). Their structures were explicitly characterized based on the spectral and analytical data. Particularly, characteristic was the resonances of the arising two vinylic protons CH=CH in their 1H NMR spectra appearing as two doublets at δ 7.64 and 8.33 with the value of spin-spin coupling constant Jab in the range 15~16 Hz, which is indicative of the E-configuration of arylvinyl moiety. The yields of the products were listed in Table 1, from which it appeared that the nature and site of substituent present in the aromatic aldehydes had little impact on the success of the transformation, neither in product yield nor in reaction rate. Moreover, the strong withdrawing groups such as cyanogen and nitro groups, which were sensitive to bases, were well tolerated in the one-pot process without any experimental difficulties.
 下载:
导出CSV
下载:
导出CSV
| Entry | Product | 5 | Yielda/% | IC50/(μmol•L-1) | MIC/(μg•mL-1) | |
| 1 |  |
5a | 77 | 31.4 | 125 | |
| 2 |  |
5b | 74 | 33.7 | 250 | |
| 3 |  |
5c | 71 | 46.3 | 125 | |
| 4 |  |
5d | 65 | 48.1 | 250 | |
| 5 |  |
5e | 73 | 54.3 | 250 | |
| 6 |  |
5f | 76 | 27.4 | 62.5 | |
| 7 |  |
5g | 79 | 27.1 | 62.5 | |
| 8 |  |
5h | 78 | 24.4 | 31.25 | |
| 9 |  |
5i | 62 | 22.6 | 15.6 | |
| 10 |  |
5j | 67 | 14.7 | 15.6 | |
| 11 |  |
5k | 63 | 25.2 | 31.25 | |
| 12 |  |
R=Me | 5l | 69 | 27.4 | 62.5 |
| R=Et | 5m | 64 | 24.3 | 62.5 | ||
| R=Bn | 5n | 71 | 28.1 | 62.5 | ||
| R=4-ClBn | 5o | 62 | 22.8 | 31.25 | ||
| 13 | AN2679 | — | — | — | 21.7 | — |
| 14 | Rifampicin | — | — | — | — | 15.6 |
| a Isolated yield. | ||||||
Having the newly-synthesized quinoxaline derivatives in hand, a preliminary evaluation for their in vitro inhibitory activity against Mycobacterium smegmatis (M. smegmatis) LeuRS was assayed. The results, as recorded in Table 1, revealed that these synthesized compounds displayed moderate to good inhibitory effects against M. smegmatis LeuRS. The structure-activity relationship study suggested that among (E)-3-styrylquinoxaline-2-carboxylic acid derivatives (5a~5j) the electron nature of the substituents appeared to play a role for the inhibitory activity of these compounds. For example, the introduction of an electron-donating methyl group (5b) gave a slightly lower activity comparable to compound 5a (Entry 2), and especially, the presence of methoxyl or ethoxyl substituent (5c~5e) resulted in much lower activities (Entries 3~5). This observation might indicate that electron-donating substituents disfavoured the inhibitory activities for M. tuberculosis LeuRS. Interestingly, the introduction of electron withdrawing chloro group (5f) gave an improvement of the inhibitory activity (Entry 6). However, it is found that the activity was not further potentiated with the introduction of a second chloro atom (5g) (Entry 7). Similarly, bromo- and cyano-substituted compounds 5h and 5i also exhibited a higher activity than 5a with the IC50 values of 24.4 and 22.6 μmol•L-1, respectively (Entries 8 and 9), being equipotent with the reference AN2679 (Entry 13). It was found that the introduction of stronger electron withdrawing nitro group (5j) exhibited the highest inhibitory activity with IC50 value of 14.7 μmol•L-1 (Entry 10), which was much superior to the reference drug and might be interesting and promising candidates for further biological research. In addition, the vinyl-linked bisheterocyclic compounds 5k~5o with the intriguing quinoline and indole ring moiety also exhibit the satisfactory inhibition of M. smegmatis LeuRS with the IC50 values of 25.2, 27.4, 24.3 28.1 and 22.8 μmol•L-1, respectively (Entries 11 and 12), having the potential to further exploitation in new drug discovery.
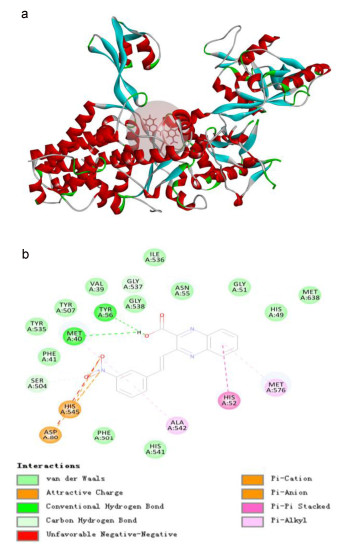
In order to explore the possible mode of binding to M. smegmatis LeuRS, the most active compound 5j in this study was selected to make a docking analysis through computer simulation method. In this work, the CDOCKER docking programs of Discovery Studio (DS) software equipped with the available PharmaDB pharmacophore database was used to perform the receptor-ligand flexible docking.[42] As the LeuRS gene from M. smegmatis has not been reported in PDB database (http://www.rcsb.org/), a homology model of Thermus thermophilus LeuRS (PDB ID 1OBH) was employed as a template. The docking model (Figure 2) showed that the compound inhibited the LeuRS seemingly by interacting with the amino acid residues such as Met576, His52, Ala542, His545, Asp80 and forming hydrogen bonds with Met40, Tyr56 and Gly537 in the leucyl-binding region of LeuRS active site. We proposed that the m-NO2 substituted analog of compound 5j may mimic the binding pose of LeuRS more closely and consequently gave improved affinity.
Basing from the above results, we further investigated if these newly-synthesized compounds could effectively inhibit the M. smegmatis strains growth in vitro. Thus, all of these compounds were evaluated in vitro using a 96-well microtiter plate and a serial dilution method to obtain their minimum inhibitory concentration (MIC) values as listed in Table 1. The antitubercular activity demonstrated that in general, compounds 5f~5j (Entries 6~10) with electron-withdrawing groups and vinyl-linked bisheterocycles 5k~5o (Entries 11 and 12) exhibited good inhibitory activity against M. smegmatis, while the inhibitory activities of unsubstituted 5a and electron-donating substituted 5b~5e were low, probably due to their poor bonding mode with the amino acid residues of M. smegmatis. Especially, the cyano- and nitro-substituted 5i and 5j showed the best anti-tubercular activity with the MIC value of 15.6 μg/mL (Entries 9 and 10), being comparable with the reference rifampicin (Entry 14).
Overall, we have achieved the synthesis of new type of quinoxaline derivatives for inhibitors targeting M. smegmatis LeuRS, which could be an attractive template for further study from both a biological as well as a building block perspective. Our next effort will mainly focus on the mechanisms of Mtb LeuRS inhibition, structural optimization and target specificity towards the ultimate goal of providing intriguing lead compounds for the development of new anti-tubercular agents.
A facile synthesis of a new series of structurally intriguing (E)-3-arvylvinylquinoxaline-2-carboxylic acids has been achieved through the radical bromination reaction followed by the one-pot sequential Arbuzov/ HWE/hydro- lysis reaction procedure by using readily available benzene-1, 2-diamine and ethyl 3-oxobutanoate as the starting materials. Experimental simplicity, inexpensive reagents and satisfactory yields would contribute to the usefulness of this method. The in vitro evaluation of their inhibitory activity against M. smegmatis LeuRS revealed that compounds 5f~5j with electron withdrawing groups exerted excellent activity, among which nitro-substituted 5j showed the best activity with the IC50 values of 14.7 μmol• L-1. The molecular docking analyses using CDOCKER module of Discovery Studio (DS) software suggested that compound 5j could bind well to the active site of Mtb LeuRS. The antitubercular activities of these compounds against M. smegmatis strains also demonstrated that electron-withdrawing substituted 5f~5j exhibited moderate to good inhibitory activity with the MIC values up to 15.6 μg/mL, being comparable with the reference rifampicin. These results might provide valuable information for designing novel inhibitors against Mtb LeuRS. Work is currently ongoing in our laboratory, mainly focusing on the mechanisms of Mtb LeuRS inhibition, the structural optimization and further application of this method for structurally diverse derivatives, which will be communicated in due course.
The chemicals used in this work were obtained from Energy Chemical and were used without purification. Melting points were determined by use of a WRS-1B melting point apparatus and were uncorrected. The 1H NMR and 13C NMR spectra were recorded on an Agilent 400-MR spectrometer using DMSO-d6 as the solvent. The reported chemical shifts (δ values) are given from tetramethylsilane (TMS) as the internal standard. HRMS (ESI) data were acquired on a Bruker Customer micrOTOF-Q 125 highresolution mass spectrometer with ESI. Elemental analyses were carried out on an EA 2400II elemental analyzer (PerkinElmer, Waltham, MA). The progress of reactions was monitored by TLC on silica gel GF254 using ethyl acetate/petroleum ether (V:V=1:8) as the eluent.
Ethyl 2-methylquinoxaline-3-carboxylate (9) (10.812 g, 50 mmol) was added to CCl4 (500 mL) and heated with stirring until refluxed gently. Benzoyl peroxide (BPO) (0.302 g, 1.25 mmol) as initiator was then added to the reaction mixture. After that, a slightly excessive amount of NBS (10.678 g, 60 mmol) was added carefully in three batches to the gently refluxing CCl4 solution every 1.5 h, i.e., 3.56 g (20 mmol) portions of 10.678 g (60 mmol) NBS were added every 1.5 h. After the addition in batches was complete, the mixture continued to reflux gently till the disappearance of 9 (as monitored by TLC). The reaction mixture was cooled to room temperature and the precipitated succinimide was filtered off. The obtained filtrate was washed with water and dried over Na2SO4. Evaporation of the solvent under reduced pressure afforded a crude solid product, which was subjected to column chromatography over silica gel (200~400 mesh) using petroleum ether/ethyl acetate mixture (V:V=10:1) as eluent to give 10.46 g of product 7and 3.37 g of 7'.
Ethyl 3-(bromomethyl)quinoxaline-2-carboxylate (7): 71% yield. White solid, m.p. 96~98 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.54 (t, J=7.2 Hz, 3H, OCH2CH3), 4.63 (q, J=7.2 Hz, 2H, OCH2CH3), 5.16 (s, 2H, CH2Br), 7.86~7.91 (m, 2H, Quin-H), 8.12 (d, J=7.2 Hz, 1H, Quin-H), 8.25 (d, J=7.2 Hz, 1H, Quin-H); 13C NMR (100 MHz, CDCl3) δ: 14.2, 31.5, 63.0, 129.0, 129.9, 131.3, 132.4, 140.6, 141.9, 143.6, 151.2, 164.8. Anal calcd for C12H11BrN2O2: C 48.84, H 3.76, N 9.49; found C 48.56, H 3.87, N 9.78.
Ethyl 3-(dibromomethyl)quinoxaline-2-carboxylate (7'): 18% yield. White solid, m.p. 91~92 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.46 (t, J=7.2 Hz, 3H, OCH2CH3), 4.54 (q, J=7.2 Hz, 2H, OCH2CH3), 7.81~7.91 (m, 2H, Quin- H), 7.87 (s, 1H, CHBr2), 8.20 (d, J=8.4 Hz, 2H, Quinoxa- H); 13C NMR (100 MHz, CDCl3) δ: 14.2, 39.0, 63.3, 129.4, 129.8, 132.1, 133.0, 138.9, 141.1, 142.3, 151.1, 164.5. Anal calcd for C12H10Br2N2O2: C 38.53, H 2.69, N 7.49; found C 38.24, H 2.93, N 7.28.
Ethyl 3-(bromomethyl)quinoxaline-2-carboxylate (7) (0.295 g, 1 mmol) was added in triethyl phosphate (8 mL). The resulting reaction mixture was stirred at 160 ℃ for 5~6 h. The conversion was monitored by TLC. After the reaction was complete, the excessive triethyl phosphate was removed in vacuo, and a solution of respective aromatic aldehydes, 2-chloroquinoline-3-carbaldehyde or 2- chloro-N-substituted-1H-indole-3-carbaldehydes (1.2 mmol) in DMF (15 mL) and NaH (0.036 g, 1.5 mmol) were added to the residue, respectively. The mixture was stirred for about 0.5 h at room temperature and then stirred at 90 ℃ for 2 h. After completion of the reaction (TLC), 10% aqueous solution of sodium hydroxide (10 mL) was added to the reaction mixture and continued to stir at 90 ℃ for 2 h. The reaction mixture was then cooled to room temperature, followed by acidification with 1 mol• L-1 HCl. The precipitated crude product was purified by recrystallization from ethanol to give the desired products 5a~5o.
(E)-3-Styrylquinoxaline-2-carboxylic acid (5a): 88% yield. Yellow solid, m.p. 145~146 ℃; 1H NMR δ: 7.37 (t, J=7.6 Hz, 1H, Ben-H), 7.44 (t, J=7.6 Hz, 2H, Ben-H), 7.69 (d, J=7.6 Hz, 2H, Ben-H), 7.77 (d, J=15.6 Hz, 1H, CH=CH), 7.85 (td, J=8.4, 1.2 Hz, 1H, Quin-H), 7.92 (td, J=8.0, 1.2 Hz, 1H, Quin-H), 8.00 (d, J=15.6 Hz, 1H, CH=CH), 8.10 (dd, J=8.4, 1.2 Hz, 1H, Quin-H), 8.12 (dd, J=8.4, 1.2 Hz, 1H, Quin-H), 14.19 (s br, 1H, COOH); 13C NMR δ: 122.6, 127.3, 128.4, 128.8, 128.8, 129.1, 130.3, 131.9, 135.5, 136.4, 139.1, 141.6, 145.0, 147.4, 166.6. HRMS calcd for C17H13N2O2 [M+H]+: 277.0972, found 277.0941.
(E)-3-(4-Methylstyryl)quinoxaline-2-carboxylic acid (5b): 76% yield. Yellow solid, m.p. 151~152 ℃; 1H NMR δ: 2.38 (s, 3H, Me), 7.30 (d, J=8.0 Hz, 2H, Ben-H), 7.65 (d, J=8.0 Hz, 2H, Ben-H), 7.76 (d, J=16.0 Hz, 1H, CH=CH), 7.90 (t, J=7.6 Hz, 1H, Quin-H), 7.98 (t, J=7.6 Hz, 1H, Quin-H), 8.03 (d, J=16.0 Hz, 1H, CH=CH), 8.15 (d, J=8.0 Hz, 1H, Quin-H), 8.16 (d, J=8.4 Hz, 1H, Quin-H), 14.20 (s br, 1H, COOH); 13C NMR δ: 20.7, 121.4, 127.3, 128.3, 128.8, 129.4, 130.1, 131.9, 132.7, 136.4, 138.9, 139.0, 141.6, 145.0, 147.5, 166.7. Anal. calcd for C18H14N2O2: C 74.47, H 4.86, N 9.65; found C 74.62, H 4.91, N 9.49.
(E)-3-(4-Methoxystyryl)quinoxaline-2-carboxylic acid (5c): 80% yield. Yellow solid, m.p. 175~176 ℃; 1H NMR δ: 3.84 (s, 3H, OMe), 7.06 (d, J=8.0 Hz, 2H, Ben-H), 7.65 (d, J=15.6 Hz, 1H, CH=CH), 7.71 (d, J=8.4 Hz, 2H, Ben-H), 7.88 (t, J=7.6 Hz, 1H, Quin-H), 7.97 (t, J=8.0 Hz, 1H, Quin-H), 8.03 (d, J=15.6 Hz, 1H, CH=CH), 8.14 (d, J=8.4 Hz, 2H, Quin-H), 14.19 (s br, 1H, COOH); 13C NMR δ: 55.7, 114.2, 119.9, 128.1, 128.3, 128.8, 128.9, 130.0, 131.8, 136.3, 138.9, 141.6, 145.0, 147.6, 160.1, 166.7. Anal. calcd for C18H14N2O3: C 70.58, H 4.61, N 9.15; found C 70.29, H 4.85, N 8.94.
(E)-3-(2, 4-Dimethoxystyryl)quinoxaline-2-carboxylic acid (5d): 73% yield. Yellow solid, m.p. 164~165 ℃; 1H NMR δ: 3.86 (s, 3H, OMe), 3.94 (s, 3H, OMe), 6.67 (dd, J=8.4, 2.0 Hz, 1H, Ben-H), 6.69 (d, J=2.0 Hz, 1H, Ben-H), 7.65 (d, J=8.4 Hz, 1H, Ben-H), 7.72 (d, J=15.6 Hz, 1H, CH=CH), 7.85 (t, J=7.6 Hz, 1H, Quin-H), 7.94 (t, J=8.0 Hz, 1H, Quin-H), 8.11 (d, J=8.4 Hz, 1H, Quin-H), 8.13 (d, J=8.4 Hz, 1H, Quin-H), 8.24 (d, J=16.0 Hz, 1H, CH=CH), 14.15 (s br, 1H, COOH); 13C NMR δ: 55.1, 55.4, 98.2, 105.9, 117.0, 120.0, 128.3, 128.7, 129.0, 129.7, 131.6, 131.8, 138.8, 141.7, 145.4, 147.9, 158.8, 161.6, 166.8. Anal. calcd for C19H16N2O4: C 67.85, H 4.79, N 8.33; found C 68.11, H 4.72, N 8.61.
(E)-3-(4-Ethoxystyryl)quinoxaline-2-carboxylic acid (5e): 77% yield. Yellow solid, m.p. 165~166 ℃; 1H NMR δ: 1.37 (t, J=7.2 Hz, 3H, OCH2CH3), 4.10 (q, J=7.2 Hz, 2H, OCH2CH3), 7.03 (d, J=8.4 Hz, 2H, Ben-H), 7.64 (d, J=15.6 Hz, 1H, CH=CH), 7.69 (d, J=8.4 Hz, 2H, Ben-H), 7.88 (t, J=7.6 Hz, 1H, Quin-H), 7.96 (t, J=8.0 Hz, 1H, Quin-H), 8.02 (d, J=15.6 Hz, 1H, CH=CH), 8.13 (d, J=8.4 Hz, 2H, Quin-H), 14.20 (s br, 1H, COOH); 13C NMR δ: 14.3, 62.9, 114.6, 119.8, 128.0, 128.2, 128.8, 128.9, 129.9, 131.8, 136.3, 138.9, 141.6, 145.0, 147.6, 159.3, 166.7. Anal. calcd for C19H16- N2O3: C 71.24, H 5.03, N 8.74; found C 71.08, H 4.97, N 8.88.
(E)-3-(2-Chlorostyryl)quinoxaline-2-carboxylic acid (5f): 65% yield. Yellow solid, m.p. 161~162 ℃; 1H NMR δ: 7.44~7.49 (m, 2H, Ben-H), 7.60 (dd, J=7.6, 2.0 Hz, 1H, Ben-H), 7.91 (d, J=16.0 Hz, 1H, CH=CH), 7.95~8.02 (m, 3H, ArH), 8.18 (d, J=8.4 Hz, 1H, Quin-H), 8.21 (d, J=8.0 Hz, 1H, Quin-H), 8.33 (d, J=16.0 Hz, 1H, CH=CH), 14.23 (s br, 1H, COOH); 13C NMR δ: 125.9, 127.3, 127.6, 128.6, 128.9, 129.7, 130.5, 130.6, 131.3, 132.1, 133.0, 133.3, 139.3, 141.5, 144.8, 147.1, 166.5. HRMS calcd for C17H12ClN2O2 [M+H]+: 311.0582, found 311.0555.
(E)-3-(2, 6-Dichlorostyryl)quinoxaline-2-carboxylic acid (5g): 47% yield. Brown solid, m.p. 189~190 ℃; 1H NMR δ: 7.42 (t, J=8.0 Hz, 1H, Ben-H), 7.61 (d, J=8.0 Hz, 2H, Ben-H), 7.83~7.92 (m, 3H, CH=CH and Quin-H), 8.03 (d, J=16.4 Hz, 1H, CH=CH), 8.09 (d, J=7.6 Hz, 1H, Quin-H), 8.14 (d, J=7.6 Hz, 1H, Quin-H), 14.27 (br s, 1H, COOH); 13C NMR δ: 128.5, 128.6, 128.7, 128.9, 129.9, 130.2, 130.6, 131.7, 132.7, 133.6, 139.9, 140.8, 146.0, 150.2, 167.8. Anal. calcd for C17H10Cl2N2O2: C 59.15, H 2.92, N 8.12; found C 59.50, H 2.69, N 7.88.
(E)-3-(3-Bromostyryl)quinoxaline-2-carboxylic acid (5h): 82% yield. Yellow solid, m.p. 154~156 ℃; 1H NMR δ: 7.45 (t, J=7.6 Hz, 1H, Ben-H), 7.62 (d, J=7.6 Hz, 1H, Ben-H), 7.77 (d, J=7.6 Hz, 1H, Ben-H), 7.89 (d, J=15.6 Hz, 1H, CH=CH), 7.93 (t, J=8.0 Hz, 1H, Quin-H), 7.96~8.02 (m, 3H, CH=CH and Ar-H), 8.17 (d, J=8.0 Hz, 2H, Quin-H), 14.22 (s br, 1H, COOH); 13C NMR δ: 122.1, 124.5, 126.3, 128.4, 128.9, 129.7, 130.5, 130.8, 131.5, 132.0, 134.5, 138.1, 139.2, 141.5, 144.9, 147.2, 166.5. Anal. calcd for C17H11BrN2O2: C 57.49, H 3.12, N 7.89; found C 57.78, H 2.92, N 7.78.
(E)-3-(2-Cyanostyryl)quinoxaline-2-carboxylic acid (5i): 61% yield. Brown solid, m.p. 162~164 ℃; 1H NMR δ: 7.62 (t, J=7.6 Hz, 1H, Ben-H), 7.84 (t, J=8.0 Hz, 1H, Ben-H), 7.94~8.03 (m, 3H, ArH), 8.10 (d, J=8.0 Hz, 1H, Quin-H), 8.13 (d, J=15.6 Hz, 1H, CH=CH), 8.18~8.23 (m, 2H, Quin-H), 8.25 (d, J=15.6 Hz, 1H, CH=CH), 14.27 (s br, 1H, COOH); 13C NMR δ: 110.8, 117.3, 126.3, 127.7, 128.7, 128.9, 129.4, 130.8, 130.9, 132.1, 133.3, 133.5, 138.2, 139.5, 141.5, 144.9, 146.8, 166.4. Anal. calcd for C18H11N3O2: C 71.75, H 3.68, N 13.95; found C 71.92, H 3.54, N 14.18.
(E)-3-(3-Nitrostyryl)quinoxaline-2-carboxylic acid (5j): 55% yield. Brown solid, m.p. 169~171 ℃; 1H NMR δ: 7.74~7.77 (m, 1H, ArH), 7.93 (d, J=8.0 Hz, 1H, ArH), 7.98~8.02 (m, 2H, ArH and CH=CH), 8.07~8.23 (m, 5H, ArH and CH=CH), 8.54 (d, J=2.0 Hz, 1H, Ben-H), 14.14 (s br, 1H, COOH); 13C NMR δ: 121.4, 123.2, 125.7, 128.4, 128.9, 130.2, 130.6, 132.0, 133.5, 133.7, 137.3, 139.3, 141.5, 149.9, 147.0, 148.0, 166.5. Anal. calcd for C17H11N3O4: C 63.55, H 3.45, N 13.08; found C 63.37, H 3.39, N 13.30.
(E)-3-(2-(2-Chloroquinolin-3-yl)vinyl)quinoxaline-2-car-boxylic acid (5k): 63% yield. Yellow solid, m.p. 193~195 ℃; 1H NMR δ: 7.66 (d, J=8.8 Hz, 1H, ArH), 7.85 (d, J=8.4 Hz, 1H, ArH), 8.00 (d, J=16.0 Hz, 1H, CH=CH), 8.15 (d, J=8.8 Hz, 1H, ArH), 8.18 (d, J=8.4 Hz, 1H, ArH), 8.30 (d, J=15.6 Hz, 1H, CH=CH), 8.82 (s, 1H, ArH), 14.22 (s br, 1H, COOH); 13C NMR δ: 127.6, 127.6, 127.8, 128.0, 128.8, 129.4, 129.6, 131.3, 131.5, 132.9, 134.0, 135.9, 137.9, 140.1, 142.3, 145.6, 145.8, 147.5, 148.9, 167.2. Anal. calcd for C20H12ClN3O2: C 66.40, H 3.34, N 11.61; found C 66.17, H 3.62, N 11.45.
(E)-3-(2-(2-Chloro-1-methyl-1H-indol-3-yl)vinyl)quino-xaline-2-carboxylic acid (5l): 69% yield. Yellow solid, m.p. 208~210 ℃; 1H NMR δ: 3.82 (s, 3H, CH3), 7.30 (t, J=8.0 Hz, 1H, ArH), 7.34 (t, J=7.6 Hz, 1H, ArH), 7.62 (d, J=7.6 Hz, 1H, ArH), 7.81 (t, J=7.6 Hz, 1H, ArH), 7.89~7.94 (m, 3H, ArH and CH=CH), 8.08 (d, J=8.4 Hz, 1H, ArH), 8.10 (d, J=8.0 Hz, 1H, ArH), 8.21 (d, J=16.0 Hz, 1H, CH=CH), 14.14 (s br, 1H, COOH); 13C NMR δ: 30.9, 109.4, 111.4, 119.4, 119.4, 122.2, 123.5, 124.2, 128.2, 128.9, 129.5, 129.7, 130.3, 132.5, 136.7, 139.4, 142.5, 145.4, 148.9, 167.7. Anal. calcd for C20H14ClN3O2: C 66.03, H 3.88, N 11.55; found C 66.19, H 3.98, N 11.26.
(E)-3-(2-(2-Chloro-1-ethyl-1H-indol-3-yl)vinyl)quino-xaline-2-carboxylic acid (5m): 64% yield. Yellow solid, m.p. 187~188 ℃; 1H NMR δ: 1.30 (t, J=7.6 Hz, 3H, CH2CH3), 4.35 (q, J=7.6 Hz, 2H, CH2CH3), 7.30 (t, J=7.6 Hz, 1H, ArH), 7.34 (t, J=8.0 Hz, 1H, ArH), 7.66 (d, J=8.0 Hz, 1H, ArH), 7.82 (t, J=7.6 Hz, 1H, ArH), 7.89~7.95 (m, 3H, ArH and CH=CH), 8.09 (d, J=8.4 Hz, 1H, ArH), 8.12 (d, J=8.8 Hz, 1H, ArH), 8.21 (d, J=16.0 Hz, 1H, CH=CH), 14.17 (s br, 1H, COOH); 13C NMR δ: 15.1, 39.1, 109.6, 111.3, 119.6, 122.2, 123.6, 124.4, 128.1, 128.7, 129.0, 129.5, 130.3, 132.5, 135.6, 139.5, 142.5, 145.4, 148.9, 167.7. Anal. calcd for C21H16ClN3O2: C 66.76, H 4.27, N 11.12; found C 66.51, H 4.10, N 10.97.
(E)-3-(2-(1-Benzyl-2-chloro-1H-indol-3-yl)vinyl)quino-xaline-2-carboxylic acid (5n): 71% yield. Yellow solid, m.p. 222~224 ℃; 1H NMR δ: 5.60 (s, 2H, CH2), 7.14 (d, J=7.6 Hz, 2H, ArH), 7.25 (t, J=7.2 Hz, 1H, ArH), 7.30~7.34 (m, 4H, ArH), 7.65 (d, J=8.4 Hz, 1H, ArH), 7.83 (t, J=7.6 Hz, 1H, ArH), 7.92 (t, J=7.6 Hz, 1H, ArH), 7.94 (d, J=8.0 Hz, 1H, ArH), 7.97 (d, J=15.6 Hz, 1H, CH=CH), 8.09 (d, J=8.4 Hz, 1H, ArH), 8.12 (d, J=8.4 Hz, 1H, ArH), 8.23 (d, J=16.0 Hz, 1H, CH=CH), 14.21 (s br, 1H, COOH); 13C NMR δ: 47.08, 110.11, 111.75, 119.65, 120.21, 122.47, 123.82, 124.43, 126.94, 127.96, 128.02, 129.00, 129.22, 129.55, 130.40, 132.53, 136.39, 137.08, 139.52, 142.47, 145.43, 148.83, 167.67. Anal. calcd for C26H18ClN3O2: C 70.99, H 4.12, N 9.55; found C 71.23, H 4.32, N 9.33.
(E)-3-(2-(2-Chloro-1-(4-chlorobenzyl)-1H-indol-3-yl)-vinyl)quinoxaline-2-carboxylic acid (5o): 62% yield. Yellow solid, m.p. 210~211 ℃; 1H NMR δ: 5.59 (s, 2H, CH2), 7.14 (d, J=8.4 Hz, 2H, ArH), 7.26~7.29 (m, 2H, ArH), 7.38 (d, J=8.4 Hz, 2H, ArH), 7.60~7.63 (m, 1H, ArH), 7.74 (t, J=7.2 Hz, 1H, ArH), 7.80 (t, J=7.2 Hz, 1H, ArH), 7.93 (d, J=16.0 Hz, 1H, CH=CH), 7.97~8.01 (m, 2H, ArH), 8.04 (d, J=8.0 Hz, 1H, ArH), 8.16 (d, J=15.6 Hz, 1H, CH=CH), 14.28 (s br, 1H, COOH); 13C NMR δ: 46.4, 110.4, 111.5, 120.0, 120.7, 121.5, 122.4, 123.8, 124.5, 126.9, 128.6, 128.8, 129.2, 129.7, 131.0, 132.6, 136.2, 138.1, 140.0, 141.8, 148.5, 151.0, 168.7. Anal. calcd for C26H17Cl2N3O2: C 65.83, H 3.61, N 8.86; found C 65.60, H 3.79, N 8.70.
The purification of Mycobacterium tuberculosis LeuRS and the standard in vitro aminoacylation assays were performed mainly based on the recently reported procedure described in literature.[32] The inhibitory activity experiment was performed in a solution containing 100 mmol•L-1 Tris-HCl (pH 8.0), 10 mol•L-1 MgCl2, 2 mol•L-1 dithiothreitol (DTT), 5 mmol•L-1 ATP, 10 mmol•L-1 KCl, 90 μmol•L-1 L-[14C]leucine (238 mCi/mmol), 4 mg•mL-1 E. coli tRNA and 25 nmol•L-1 M. tuberculosis LeuRS. The reaction was incubated at 37 ℃ and aliquots were quenched by 10% trichloracetic acid (TCA). For the inhibitory studies in aminoacylation reaction 20 μL of solution containing 25 nmol•L-1 M. tuberculosis LeuRS, 100 mmol•L-1 Tris-HCl (pH 8.0), 10 mmol•L-1 MgCl2, 90 μmol•L-1 L-[14C]leucine (238 mCi/mmol), 2 mmol•L-1 DTT, 4 mg/mL E. coli tRNA and appropriate concentrations of inhibitor (dissolved in DMSO) were incubated for 5 min at 37 ℃. Reactions were initiated by addition of ATP to final concentration of 2 mmol•L-1. The inhibitory of aminoacylation of M. smegmatis LeuRS was determined by liquid scintillation counting. To determine the IC50 values, at least triplicates were averaged to generate an IC50 value using Origin 7.0.
All the synthesized compounds 5a~5o herein were screened for their potential in vitro anti-tubercular activity against M. smegmatis [CGMCC 1.2621], which was obtained from the National Center for Medical Culture Collection, China. The MIC value was determined according to our previously described method.[44] Each of the test compounds was dissolved in DMSO and then was serially diluted in five concentrations at twofold dilutions (250, 125, 62.5, 31.25, 15.6, 7.81, 3.91 μg/mL). The diluted solutions in Mueller Hinton Broth were dispensed into the wells of a microtiter plate, and then an aliquot of 5×105 c.f.u/mL of bacterial suspension was added to each well. The value of MIC was determined after 24 h of incubation at 37 ℃. Rifampicin was used as the reference standards.
Supporting Information 1H NMR and 13C NMR spectra of compounds 7, 7' and5a~5o are available free of charge via the Internet at http://sioc-journal.cn/.
World Health Organization Global Tuberculosis Report 2019, World Health Organization, Geneva, 2019, http://www.who.int/tb/publications/global_report/en/
Chan, E. D.; Iseman, M. D. Curr. Opin. Infect. Dis. 2008, 21, 587. doi: 10.1097/QCO.0b013e328319bce6
Gudzera, O. I.; Golub, A. G.; Bdzhola, V. G.; Volynets, G. P.; Kovalenko, O. P.; Boyarshin, K. S.; Yaremchuk, A. D.; Protopopov, M. V.; Yarmoluk, S. M.; Tukalo, M. A. J. Enzyme Inhib. Med. Chem. Chem. 2016, 31, 201. doi: 10.1080/14756366.2016.1190712
Lee, S. W.; Choi, E. C.; Choi, S. Y. Appl. Microbiol. Biotechnol. 2003, 61, 278. doi: 10.1007/s00253-003-1243-5
Ding, D.; Meng, Q.; Gao, G.; Zhao, Y.; Wang, Q.; Nare, B.; Jacobs, R.; Rock, F.; Alley, M. R. K.; Plattner, J. J.; Chen, G.; Li, D.; Zhou, H. J. Med. Chem. 2011, 54, 1276. doi: 10.1021/jm101225g
Zhang, F.; Du, J.; Wang, Q.; Hu, Q.; Zhang, J.; Ding, D.; Zhao, Y.; Yang, F.; Wang, E.; Zhou, H. Org. Biomol. Chem. 2013, 11, 5310. doi: 10.1039/c3ob40236c
Bernier, S.; Akochy, P.-M.; Lapointe, J.; Chênevert, R. Bioorg. Med. Chem. 2005, 13, 69. doi: 10.1016/j.bmc.2004.09.055
Li, X.; Hernandez, V.; Rock, F. L.; Choi, W.; Mak, Y. S. L.; Mohan, M.; Mao, W.; Zhou, Y.; Easom, E. E.; Plattner, J. J.; Zou, W.; Pérez-Herrán, E.; Giordano, I.; Mendoza-Losana, A.; Alemparte, C.; Rullas, J.; Angulo-Barturen, I.; Crouch, S.; Ortega, F.; Barros, D.; Alley, M. R. K. J. Med. Chem. 2017, 60, 8011. doi: 10.1021/acs.jmedchem.7b00631
Gudzera, O. I.; Golub, A. G.; Bdzhola, V. G.; Volynets, G. P.; Lukashov, S. S.; Kovalenko, O. P.; Kriklivyi, I. A.; Yaremchuk, A. D.; Starosyla, S. A.; Yarmoluk, S. M.; Tukalo, M. A. Bioorg. Med. Chem. 2016, 24, 1023. doi: 10.1016/j.bmc.2016.01.028
Zhang, P.; Ma, S. Med. Chem. Commun. 2019, 10, 1329. doi: 10.1039/C9MD00139E
Montana, M.; Mathias, F.; Terme, T.; Vanelle, P. Eur. J. Med. Chem. 2019, 163, 136. doi: 10.1016/j.ejmech.2018.11.059
丛文霞, 王莉, 于福强, 李纪兴, 有机化学, 2018, 38, 2866.Cong, W. X.; Wang, L.; Yu, F. Q.; Li, J. X. Chin. J. Org. Chem. 2018, 38, 2866(in Chinese).
Ajani, O. O. Eur. J. Med. Chem. 2014, 85, 688. doi: 10.1016/j.ejmech.2014.08.034
Tariq, S.; Somakala, K.; Amir, M. Eur. J. Med. Chem. 2018, 143, 542. doi: 10.1016/j.ejmech.2017.11.064
Santivañez-Veliz, M.; Pérez-Silanes, S.; Torres, E.; Moreno-Viguri, E. Bioorg. Med. Chem. Lett. 2016, 26, 2188. doi: 10.1016/j.bmcl.2016.03.066
Ancizu, S.; Moreno, E.; Solano, B.; Villar, R.; Burguete, A.; Torres, E.; Pérez-Silanes, S.; Aldana, I.; Monge, A. Bioorg. Med. Chem. 2010, 18, 2713. doi: 10.1016/j.bmc.2010.02.024
Puratchikody, A.; Natarajan, R.; Jayapal, M.; Doble, M. Chem. Biol. Drug Des. 2011, 78, 988. doi: 10.1111/j.1747-0285.2011.01246.x
Kumar, K. S.; Rambabu, D.; Sandra, S.; Kapavarapu, R.; Krishna, G. R.; Rao, M. V. B.; Chatti, K.; Reddy, C. M.; Misra, P.; Pal, M. Bioorg. Med. Chem. 2012, 20, 1711. doi: 10.1016/j.bmc.2012.01.012
Seitz, L. E.; Suling, W. J.; Reynolds, R. C. J. Med. Chem. 2002, 45, 5604. doi: 10.1021/jm020310n
Magnet, S.; Hartkoorn, R. C.; Székely, R.; Pató, J.; Triccas, J. A.; Schneider, P.; Szántai-Kis, C.; Orfi, L.; Chambon, M.; Banfi, D.; Bueno, M.; Turcatti, G.; Kéri, G.; Cole, S. T. Tuberculosis 2010, 90, 354. doi: 10.1016/j.tube.2010.09.001
Mekouar, K.; Mouscadet, J.-F.; Desmaële, D.; Subra, F.; Leh, H.; Savouré, D.; Auclair, C.; d'Angelo, J. J. Med. Chem. 1998, 41, 2846. doi: 10.1021/jm980043e
戴红, 丁颖, 杜显超, 姚炜, 陈庆文, 王祥龙, 仲苏林, 曹雄飞, 石玉军, 有机化学, 2018, 38, 1755.Dai, H.; Ding, Y.; Du, X. C.; Yao, W.; Chen, Q. W.; Wang, X. L.; Zhong, S. L.; Cao, X. F.; Shi, Y. J. Chin. J. Org. Chem. 2018, 38, 1755(in Chinese).
Wei, Q.; Li, J.; Tang, F.; Yin, Y.; Zhao, Y.; Yao, Q. Eur. J. Med. Chem. 2018, 144, 504. doi: 10.1016/j.ejmech.2017.12.008
He, H.; Ge, Y.; Dai, H.; Cui, S.; Ye, F.; Jin, J.; Shi, Y. Molecules 2016, 21, 1722. doi: 10.3390/molecules21121722
Shingalapur, R. V.; Hosamani, K. M.; Keri, R. S. Eur. J. Med. Chem. 2009, 44, 4244. doi: 10.1016/j.ejmech.2009.05.021
Jadhavar, P. S.; Dhameliya, T. M.; Vaja, M. D.; Kumar, D.; Sridevi, J. P.; Yogeeswari, P.; Sriram, D.; Chakraborti, A. K. Bioorg. Med. Chem. Lett. 2016, 26, 2663. doi: 10.1016/j.bmcl.2016.04.012
De, P.; Veau, D.; Belval, F. B.; Chassaing, S.; Baltas, M. Cinnamic Derivatives in Tuberculosis, In Understanding Tuberculosis-New Approaches to Fighting Against Drug Resistance, Ed.: Cardona, P.-J., ISBN: 978-953-307-948-6, In Tech., 2012.
Fan, Y.; Wu, J.; Cheng, X.; Zhang, F.; Feng, L. Eur. J. Med. Chem. 2018, 146, 554. doi: 10.1016/j.ejmech.2018.01.080
Lu, X.; Wan, B.; Franzblau, S. G.; You, Q. Eur. J. Med. Chem. 2011, 46, 3551. doi: 10.1016/j.ejmech.2011.05.018
Maresca, A.; Vullo, D.; Scozzafava, A.; Manole, G.; Supuran, C. T. J. Enzym. Inhib. Med. Chem. 2013, 28, 392. doi: 10.3109/14756366.2011.650168
Chang, F.-S.; Chen, W.; Wang, C.; Tzeng, C.-C.; Chen, Y.-L. Bioorg. Med. Chem. 2010, 18, 124. doi: 10.1016/j.bmc.2009.11.012
Kumar, B. S. P. A.; Madhav, B.; Reddy, K. H. V.; Nageswar, Y. V. D. Tetrahedron Lett. 2011, 52, 2862. doi: 10.1016/j.tetlet.2011.03.110
Gong, L.; Xing, L. J.; Xu, T.; Zhu, X. P.; Zhou, W.; Kang, N.; Wang, B. Org. Biomol. Chem. 2014, 12, 6557. doi: 10.1039/C4OB01025F
Kamal, A.; Rahim, A.; Riyaz, S.; Poornachandra, Y.; Balakrishna, M.; Kumar, C. G.; Hussaini, S. M. A.; Sridhar, B.; Machiraju, P. K. Org. Biomol. Chem. 2015, 13, 1347. doi: 10.1039/C4OB02277G
Sharma, R.; Abdullaha, M.; Bharate, S. B. J. Org. Chem. 2017, 82, 9786. doi: 10.1021/acs.joc.7b00856
Achelle, S.; Barsella, A.; Baudequin, C.; Caro, B.; Guen, F. R. J. Org. Chem. 2012, 77, 4087. doi: 10.1021/jo3004919
Carbon, J. A. J. Org. Chem. 1960, 25, 1731. doi: 10.1021/jo01080a013
徐姣, 张丽宏, 张美琦, 刘秀波, 马伟, 唐益鑫, 王道林, 有机化学, 2019, 39, 2808.Xu, J.; Zhang, L. H.; Zhang, M. Q.; Liu, X. B.; Ma, W.; Tang, Y. X.; Wang, D. L. Chin. J. Org. Chem. 2019, 39, 2808(in Chinese).
Li, Y.; Tang, B. Y.; Dong, S. Y.; Gao, W. T.; Jiang, W. T.; Chen, Y. ChemistrySelect 2020, 5, 2746. doi: 10.1002/slct.201904434
Li, Y.; Xu, Q. Q.; Li, Z. Y.; Gao, W. T.; Chen, Y. Mol. Diversity 2020, 24, 167. doi: 10.1007/s11030-019-09938-3
Thapa, R.; Brown, J.; Balestri, T.; Brown, J.; Taylor, R. T. Tetrahedron Lett. 2014, 55, 6743. doi: 10.1016/j.tetlet.2014.08.069
钟克利, 赵杰, 李秋莹, 侯淑华, 汤轶伟, 边延江, 汤立军, 有机化学, 2018, 38, 1786.Zhong, K. L.; Zhao, J.; Li, Q. Y.; Hou, S. H.; Tang, Y. W.; Bian, Y. J.; Tang, L. J. Chin. J. Org. Chem. 2018, 38, 1786(in Chinese).

Figure 1 Anti-tubercular quinoxaline, arylvinyl heterocyclic derivatives and the designed structure of targeted compounds

Scheme 3 Three-step one-pot synthesis of (E)-3-styrylquin- oxaline-2-carboxylic acid (5a)

Figure 2 3D docking modes (a) and 2D docking modes (b) for 5j into homology model of T. brucei LeuRS
Table 1. Yields and in vitro inhibitory activity of compounds 5a~5o toward Mtb LeuRS and M. smegmatis
| Entry | Product | 5 | Yielda/% | IC50/(μmol•L-1) | MIC/(μg•mL-1) | |
| 1 | |
5a | 77 | 31.4 | 125 | |
| 2 | |
5b | 74 | 33.7 | 250 | |
| 3 | |
5c | 71 | 46.3 | 125 | |
| 4 | |
5d | 65 | 48.1 | 250 | |
| 5 | |
5e | 73 | 54.3 | 250 | |
| 6 | |
5f | 76 | 27.4 | 62.5 | |
| 7 | |
5g | 79 | 27.1 | 62.5 | |
| 8 | |
5h | 78 | 24.4 | 31.25 | |
| 9 | |
5i | 62 | 22.6 | 15.6 | |
| 10 | |
5j | 67 | 14.7 | 15.6 | |
| 11 | |
5k | 63 | 25.2 | 31.25 | |
| 12 | |
R=Me | 5l | 69 | 27.4 | 62.5 |
| R=Et | 5m | 64 | 24.3 | 62.5 | ||
| R=Bn | 5n | 71 | 28.1 | 62.5 | ||
| R=4-ClBn | 5o | 62 | 22.8 | 31.25 | ||
| 13 | AN2679 | — | — | — | 21.7 | — |
| 14 | Rifampicin | — | — | — | — | 15.6 |
| a Isolated yield. | ||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: