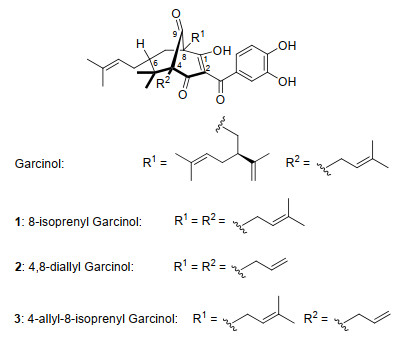
图 1.
山竹醇及其新类似物的化学结构
Figure 1.
Structures of garcinol and novel garcinol analogs

天然产物是新药的重要来源, 特别是在抗肿瘤和降血压领域[1].在印度, 乳香、姜黄、白牡丹、藤黄等天然植物或从中提取的化合物, 已经被证明具有非常高的药用价值[2, 3].山竹醇(Garcinol, 图 1), 黄色晶体化合物, 是从印度藤黄的干果皮中提取出来的一种多聚异戊二烯基苯甲酮[4].大量研究表明, 山竹醇具有广泛的生物学活性, 例如抗炎、抗氧化、抗增殖、诱导细胞凋亡等, 主要是由于山竹醇能抑制5-脂氧化酶(5-LOX)和环氧化酶-2 (COX-2)的活性[5~11].
近年来, 山竹醇及其衍生物对抑制口腔鳞癌细胞的活性引起了人们的研究兴趣[12~14].我们课题组前期通过计算机建模预测, 山竹醇与5-LOX的活性位点相结合, 从而抑制5-LOX[15].而山竹醇结构中C8位置上的过大支链可能会阻碍山竹醇与5-LOX的活性位点相结合, 导致不能有效抑制5-LOX.我们近期的研究表明, 将C8位置的侧链换成更小的甲基, 即8-甲基山竹醇[12], 抗肿瘤活性比山竹醇弱.而将C8位置侧链换成烯丙基时[13], 所生成8-烯丙基山竹醇在中低浓度(5-20 μmol•L-1)时, 抑制效果优于山竹醇.这些结果表明, C8位置的不饱和侧链是其活性必需基团.同时, 我们也证明了山竹醇13和14位两个羟基是其抗肿瘤活性的必需基团[14].在此基础上, 我们继续对山竹醇的结构进行修饰, 计划将C8位置的九碳取代基替换为异戊烯基, 简称为8-异戊烯基(代)山竹醇[8-isoprenyl garcinol, 全称: 8-异戊烯基-8-去-(2-异丙烯基-5-甲基-己-4-烯基)山竹醇](1)(图 1).并且, 我们还将山竹醇母体桥环结构中桥头的另一端进行修饰, 即将C4和C8位置的取代基替换成烯丙基, 简称4, 8-二烯丙基(代)山竹醇(4, 8-diallyl garcinol, 全称: 4, 8-双(烯丙基)-4-去-(异戊烯基)-8-去-(2-异丙烯基-5-甲基-己-4-烯基)-山竹醇)(2).将C4位置取代基替换为烯丙基, C8位置取代基替换为异戊烯基, 简称4-烯丙基-8-异戊烯基(代)山竹醇(4-allyl-8-isoprenyl garcinol, 全称: 4-烯丙基-4-去-(异戊烯基)-8-异戊烯基-8-去-(2-异丙烯基-5-甲基-己-4-烯基)-山竹醇)(3).
目前, 山竹醇及其衍生物的合成研究日渐引起人们的兴趣. Socolsky等[16]以乙酰丙酮作为起始原料, 经过Michael加成, Knoevenagel缩合, Dieckmann缩合等多步反应合成得到山竹醇. 2017年, Guttroff等[17]用类似的方法, 经过多步反应合成了多种山竹醇结构类似物.在本文中, 我们使用上述类似的合成方法, 成功地合成了三种未见报道的山竹醇类似物1~3, 并对其进行抗肿瘤活性筛选.
参照Biber等[18]的方法, 以乙酰丙酮(4)为起始原料合成了山竹醇的三个新类似物.如Scheme 1所示, 在NaH作用下, 4与3, 3-二甲基烯丙基溴进行烯丙基化反应, 随后进行α亚甲基化得到中间产物5[18], 然后, 5与1, 3-丙酮二羧酸二甲酯发生Michael加成和Knoevenagel缩合串联反应, 并进行环化, 以89%产率得到消旋化中间体6.随后甲基锂对中间体6进行1, 2-加成生成相应的甲基酮类化合物, 然后再进行羰基α位烯丙基化, 以较高的产率和高非对映选择性得到化合物7, 其高选择性主要原因是处于竖键的异戊烯基阻挡了亲电试剂从烯醇平面下方的进攻[18].此步反应要注意的是, 氢化钠需要分批加入, 温度要控制在0℃, 否则温度过高, 烷基化试剂会在其他位置发生取代反应.接着, 化合物7与甲基酮锂进行Michael加成, 得到双甲基化产物8.为了在8中两个羰基之间引入异戊烯基, 起初使用了第三步的条件, 即以NaH作为碱, 8与异戊烯基溴反应, 主要得到的却是O-烯丙基化产物.后来, 使用Plietker小组报道[17, 18]的含Fe催化剂Bu4N[Fe(CO)3(NO)][19]和配体SIMES*PF6[20]后, 则顺利得到了C-烯丙基化产物9.在叔丁醇钾的作用下, 中间体9进行Dieckmann缩合得到三环中间体10.为了得到目标化合物1, 需要对10进行酰化反应.根据本实验室前期的研究[13], 3, 4-二乙酰氧基苯甲酰氰比相应的酰氯更好地与10反应, 生成酰基化产物11.最后, 在K2CO3和甲醇溶液中, 化合物11发生水解得到目标化合物1.
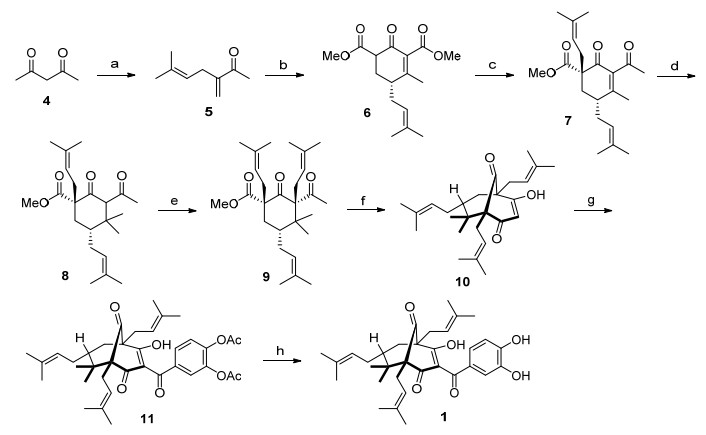
Reagents and conditions: (a) NaH, isoprenyl bromide, EtOH, 0 ℃ to r.t., 15 h, then K2CO3, formaldehyde, r.t., 15 h, 60% yield. (b) methylmagnesium chloride, dimethyl 1, 3-acetonedicarboxylate, MeOH, 0 to 60 ℃, 15 h, 89% yield. (c) NaH, THF, then MeLi, 0 ℃; NaH, isoprenyl bromide, THF, 0 ℃ to r.t., 63% yield. (d) LiCl, CuI, methylmagnesium bromide, Me3SiCl, THF, -78 ℃, 96% yield. (e) Potassium tert-amylate (KOtAm), 1, 3-dimesitylimidazolin-2-ylidene hexafluorophosphate (SIMES*PF6) (10 mol%), Bu4N[Fe(CO)3(NO)] (10 mol%), LiH, isobutyl (2-methylbut-3-en-2-yl) carbonate, THF/methyl-t-butyl ether, 0 to 80 ℃, 44% yield. (f) KOBut, THF, 0 ℃, 80% yield. (g) Et3N, 4-(cyanocar-bonyl)-1, 2-phenylene diacetate, THF, r.t., 16 h, 76% yield. (h) K2CO3, MeOH, r.t., 1 h, 64% yield.
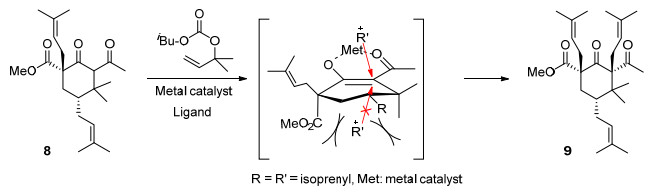
对于中间体9的形成过程, Scheme 2参照Biber等[18]的解释进行了说明.在含Fe催化剂Bu4N[Fe(CO)3-(NO)]和配体SIMES*PF6作用下, 8与2-甲基-3-丁烯-2-碳酸异丁酯反应, 在所形成的可能中间体中, 由于竖键位的羧酸甲酯(C1位)和甲基(C4位)阻挡了亲电试剂从烯醇平面下方的进攻, 新引入的异戊烯基选择性地从烯醇平面上方进攻2位碳原子(R=R'=异戊烯基, Met=金属离子), 从而得到β-构型为主的加成产物9.
山竹醇类似物2和3的合成如Scheme 3所示.为了在8和13中两个羰基之间引入烯丙基, 8和先与氯甲酸烯丙基酯反应, 分别得到区域异构体混合物8a~8b和13a~13b.随后在Pd催化剂作用下发生Tsuji-Trost反应[17], 以较高的产率得到主要产物14和17.接着, 中间体14和17发生Dieckmann缩合分别得到环化产物15和18, 然后酰基化得到中间体16和19, 最后水解得到山竹醇类似物2和3.
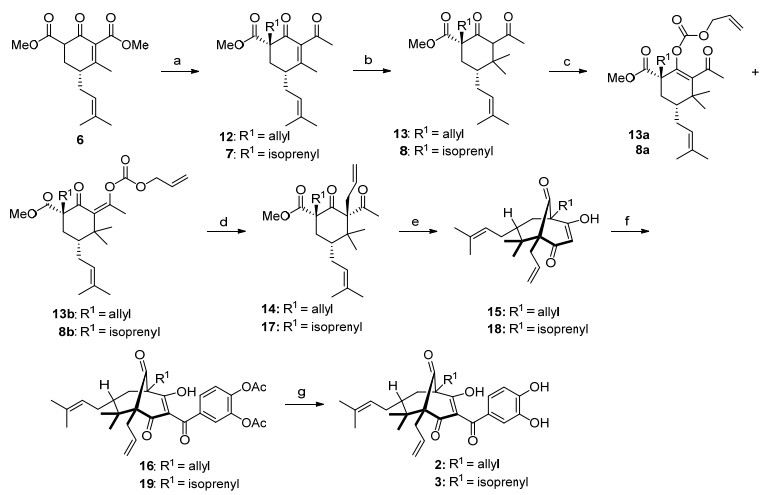
Reagents and conditions: (a) NaH, THF, then MeLi, 0 ℃; NaH, R1Br, 0 ℃ to r.t., for 7: 63% yield, for 12: 66% yield; (b) LiCl, CuI, methylmagnesium bromide, Me3SiCl, THF, -78 ℃. For 8: 96% yield; for 13: 91% yield; (c) NaH, allyl chloroformate, DMF, 0 ℃ to r.t., for 8a: 34% yield, 8b: 50% yield; for 13a: 32% yield, 13b: 63% yield; (d) Tris(dibenzylideneacetone)dipalladium(0)-chloroform [Pd2(dba)3-CHCl3] (5 mol%), tri(p-tolyl)phosphine, toluene, r.t., for 14: 73% yield, for 17: 75% yield. (e) KOBut, THF, 0 ℃, for 15: 81% yield, for 18: 72% yield; (f) Et3N, 4-(cyanocarbonyl)-1, 2-phenylene diacetate, THF, r.t., 16 h, for 16: 78% yield, for 19: 75% yield; (g) K2CO3, MeOH, r.t., 1 h, for 2: 69% yield, for 3: 58% yield.
我们利用MTT法, 测试了山竹醇和山竹醇新类似物(1, 2和3)对口腔鳞癌SCC-15和CAL-27细胞的抗增殖活性.结果表明, 与阴性对照组相比, 山竹醇和山竹醇新类似物均能够抑制口腔鳞癌细胞SCC-15和CAL-27的增殖, 差异具有统计学意义(P<0.01). 图 2中, 我们可以看到孵育时间不同, 不同化合物的活性大小顺序有变化, 而图 3中的活性大小顺序几乎无变化.这主要是因为化合物的活性与其分子量大小, 在细胞内代谢的速度以及对细胞膜的穿透能力都有很大关系, 并且细胞系的不同, 化合物的活性大小也会不同.总体来说, 随着山竹醇和山竹醇新类似物浓度的增高和作用时间延长, 对SCC-15和CAL-27细胞的抑制作用逐渐增强, 呈一定的浓度和时间依赖关系(图 2和3).但是, 与山竹醇对比, 山竹醇新类似物(1, 2和3)的抗增殖作用均不同程度的低于山竹醇.
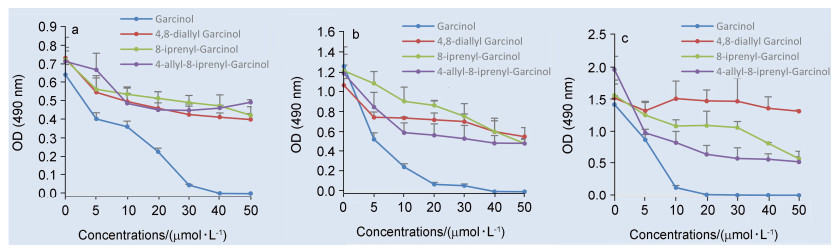
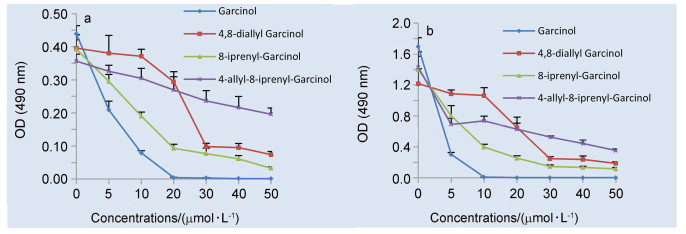
根据之前的研究[14], 8-烯丙基山竹醇的抑制作用在低至中浓度优于山竹醇.在8-烯丙基山竹醇的研究基础上, 将C8位置的烯丙基换成空间需求更大的异戊烯基时, 即8-异戊烯基山竹醇, 抑制作用较山竹醇明显降低.当把C4位置的异戊烯基替换成烯丙基时, 即4, 8-二烯丙基山竹醇, 同样, 抑制作用较山竹醇降低.接着, 将C4位置替换成烯丙基并且C8位置替换成异戊烯基, 即4-烯丙基-8-异戊烯基山竹醇, 抗增殖活性相较于山竹醇同样不理想.根据活性测试结果, 我们猜测C4和C8位置的侧链可能对山竹醇的抗增殖活性有影响.
为了进一步阐释山竹醇及其类似物的抗肿瘤活性结果, 我们通过Discovery Studio 2019 Client (DS 2019)软件, 将山竹醇及其类似物分别与5-LOX进行分子对接.分子对接结果显示, CDOCKER Energy的数值全部为负数, 表明分子内能较高.本实验中, 我们采用-CDOCKER Interaction Energy作为打分函数判定依据, 其数值分别为:山竹醇: 69.8055, 类似物1: 64.8402, 类似物2: 53.0178, 类似物3: 60.4108.分子模拟结果表明, 相比于山竹醇的三个类似物, 山竹醇与5-LOX结合位点相互作用时所需要的能量最低.
图 4是山竹醇及其类似物与5-LOX分子对接2D图.山竹醇与5-LOX结合位点处的Glu614、His550、His367、Leu607和Phe177等氨基酸残基产生非键作用: Glu614与苯环上两个羟基形成氢键; Phe555与苯环有π-π堆积作用; 连接苯环的C=O与金属受体FE2701结合, 同时与His550产生碳氢键作用; His367与C1位置的O产生氢键作用.类似物1与5-LOX结合位点处的Phe610、His372、His367和Leu607等氨基酸残基产生非键作用: Phe610与苯环上一个羟基发生氢键作用; 连接苯环的C=O与金属受体FE2701结合, 同时与His372产生碳氢键作用; His367与C1位置的O形成氢键作用.类似物2与5-LOX结合位点处的His367、Leu368、His372和Phe177等氨基酸残基产生非键作用: His367与C1位置的O形成氢键作用.类似物3与5-LOX结合位点处的Glu614、His550、His367、His372和Phe177等氨基酸残基产生非键作用: Glu614与苯环上一个羟基产生氢键作用; 连接苯环的C=O与金属受体FE2701结合, 同时与His550、His372产生氢键作用; His367与C1位置的O形成氢键作用.
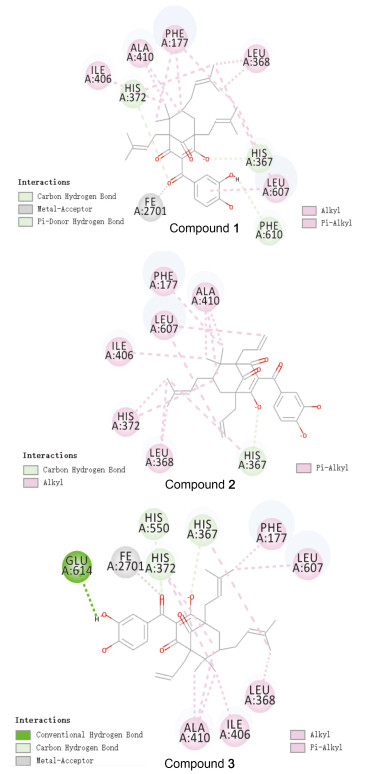
综上所述, DS模拟结果表明, 与山竹醇类似物相比, 山竹醇苯环上两个酚羟基分别与5-LOX结合位点处的Glu614形成两个氢键相互作用.因此, 山竹醇与5-LOX的相互作用能相比于新类似物(1, 2和3)都比较强.分子模拟结果和细胞抗增殖结果一致, 为山竹醇的结构优化提供了思路.
为了提高山竹醇对肿瘤细胞的抑制活性, 并研究其构效关系, 本文以乙酰丙酮为原料, 设计并合成了三个山竹醇的新类似物. MTT实验结果表明, 新合成的山竹醇类似物对人口腔鳞癌细胞的抗增殖活性均不同程度弱于山竹醇; 构效关系研究表明, 山竹醇C4和C8位的侧链对山竹醇的抗增殖活性具有重要作用, 这些结果对后续山竹醇的结构优化与改造研究具有重要的指导意义.
熔点使用上海申光公司的显微熔点仪, 型号为SGW X-4B; NMR用Bruker (400和500 MHz)型核磁共振仪测定(CDCl3, CD3OD作溶剂, TMS为内标); HRMS用Thermo Orbitrap Elite测定; 昆山市超声仪器有限公司SQ-5200型超声波清洗器; 分子对接模拟使用Discovery Studio 2019 Client (DS 2019)软件; 150~200目硅胶(青岛海洋化工厂生产); 薄层色谱使HSGF-254 (购自烟台江友硅胶开发有限公司).试剂均为分析纯. Fe催化剂Bu4N[Fe(CO)3(NO)]按照文献[19]方法合成, 配体SIMES*PF6按照文献[20]方法合成.
将乙酰丙酮(20 g, 199.7 mmol)溶于乙醇(140 mL)中, 冷却至0 ℃, 分批加入NaH (60% in mineral oil, 8.8 g, 219.7 mmol), 搅拌5 min后, 缓慢滴加3, 3-二甲基烯丙基溴(32.8 mL, 299.6 mmol), 然后将反应液升至室温, 搅拌过夜.随后将37%甲醛水溶液(50 mL)和K2CO3 (55.2 g, 399.4 mmol, 溶于200 mL水)加入反应液, 室温搅拌过夜, 薄层色谱(TLC)监测反应进程.反应毕, 向反应液加入280 mL水, 混合物用乙酸乙酯萃取(300 mL×3), 合并有机相, 再经过饱和食盐水洗涤, 无水硫酸钠干燥, 过滤浓缩, 并进行柱层析纯化, 得到无色液体5[18] (16.5 g, 产率60%). 1H NMR (400 MHz, CDCl3) δ: 5.97 (s, 1H), 5.71 (s, 1H), 5.11~5.07 (m, 1H), 2.90 (d, J=7.3Hz, 2H), 2.31 (s, 3H), 1.69 (s, 3H), 1.57 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 199.8, 148.1, 134.0 125.0, 120.8, 29.0, 26.0, 25.8, 17.6.
取115 mL甲醇于烧瓶中, 0 ℃下滴加甲基氯化镁(3 mol•L-1 in THF, 38.6 mL, 115.8 mmol)和1, 3-丙酮二甲酸二甲酯(4.7 g, 57.9 mmol), 并在该温度下搅拌1 h.随后将化合物2 (8 g, 57.9 mmol)滴加至反应液中, 60 ℃搅拌回流过夜, TLC监测.反应毕, 滴加2 mol/L HCl (42 mL), 减压浓缩有机相, 水相用乙酸乙酯萃取(50 mL×3), 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩, 粗产物通过柱层析分离化[洗脱剂: V(石油醚):V(乙酸乙酯)=8:1], 得到淡黄色油状物6 (15.1 g, 产率89%). 1H NMR (400 MHz, CDCl3) δ: 5.09~5.04 (m, 1H), 3.78 (s, 3H), 3.72 (s, 3H), 3.52~3.48 (m, 1H), 2.38~2.32 (m, 2H), 2.09~2.02 (m, 2H), 2.00 (s, 3H), 1.70~1.68 (m, 6H), 1.59 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 189.7, 170.3, 166.8, 162.8, 135.2, 132.2, 120.9, 52.5, 52.3, 49.2, 39.7, 29.8, 28.5, 25.9, 19.9, 18.0; HRMS calcd for C16H22O5Na [M+Na]+ 317.1359, found 317.1364.
将化合物6 (11.2 g, 38.1 mmol)溶于四氢呋喃(THF) (100 mL)中, 冷却至0 ℃, 分批加入NaH (60% in mineral oil, 1.7 g, 41.9 mmol), 同时保持反应液温度在5 ℃以下.在该温度下搅拌1 h后, 滴加甲基锂(1.6 mol•L-1 in Et2O, 47.6 mL, 76.2 mmol), 并将反应液在0 ℃下搅拌3 h, TLC监测.反应毕, 用饱和氯化铵溶液(12 mL)淬灭反应, 分液, 减压浓缩有机相, 水相用乙酸乙酯萃取(40 mL×3), 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 过滤减压浓缩, 粗产物无需进一步纯化, 直接投下一步.
将粗产物溶于THF (100 mL)中, 冷却至0 ℃, 分批加入NaH (60% in mineral oil, 1.7 g, 41.9 mmol), 在该温度下搅拌1 h后, 加入相应的溴代物(57.2 mmol).随后将反应液升至室温, 搅拌过夜, TLC监测.反应毕, 用饱和氯化铵溶液(30 mL)淬灭反应, 分液, 减压浓缩有机相, 水相用乙酸乙酯萃取(50 mL×3), 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 过滤减压浓缩, 并进行柱层析纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=10:1], 得到中间体7和12.
(±)-3-乙酰基-4-甲基-1, 5-双(3-甲基丁-2-烯-1-基)-2-氧亚基环己基-3-烯-1-羧酸甲酯(7):黄色油状物, 8.3 g, 产率63%. 1H NMR (400 MHz, CDCl3) δ: 5.08~5.01 (m, 2H), 3.72 (s, 3H), 2.63~2.56 (m, 1H), 2.49~2.34 (m, 3H), 2.29 (s, 3H), 2.27~2.21 (m, 1H), 2.11~2.05 (m, 2H), 1.92 (s, 3H), 1.71 (s, 3H), 1.68 (s, 3H), 1.61 (s, 6H); 13C NMR (100MHz, CDCl3) δ: 204.3, 194.8, 173.0, 160.6, 139.1, 135.5, 134.9, 120.6, 118.8, 56.6, 52.6, 38.6, 33.0, 31.9, 31.4, 30.5, 26.1, 26.0, 19.6, 18.2, 18.1; HRMS calcd for C21H30O4Na [M+Na]+ 369.2036, found 369.2041.
(±)-3-乙酰基-1-烯丙基-4-甲基-5-(3-甲基丁-2-烯- 1-基)-2-氧亚基环己基-3-烯-1-羧酸甲酯(12):淡黄色油状物, 8.0 g, 产率66%. 1H NMR (400 MHz, CDCl3) δ: 5.81~5.71 (m, 1H), 5.11~5.03 (m, 3H), 3.70 (s, 3H), 2.67~2.61 (m, 1H), 2.47~2.35 (m, 3H), 2.26 (s, 3H), 2.24~2.22 (m, 1H), 2.12~1.99 (m, 2H), 1.91 (s, 3H), 1.69 (s, 3H), 1.58 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 204.1, 194.5, 172.5, 160.6, 139.0, 135.0, 133.5, 120.4, 119.2, 56.3, 52.6, 38.0, 37.5, 33.0, 31.5, 30.5, 25.9, 19.5, 18.1; HRMS calcd for C19H26O4Na [M+Na]+ 341.1723, found 341.1727.
将干燥的LiCl (1.7 g, 39.3 mmol)和CuI (7.3 g, 38.4 mmol)溶于THF (70 mL)中, 室温搅拌5 min.随后将反应液冷却至-78 ℃, 滴加MeMgBr (3 mol•L-1 in Et2O, 12.8 mL, 38.4 mmol), TMSCl (4.9 mL, 38.4 mmol)和溶于THF (70 mL)的相应的中间体(19.2 mmol), -78 ℃下搅拌5 h, TLC监测.反应毕, 加入饱和NH4Cl:2 mol/L HCl (1:1, 350 mL)水解反应, 减压浓缩有机相, 水相用乙酸乙酯萃取(300 mL×3), 合并有机相, 有机相用NH4Cl:NH3•H2O (V:V=1:1, 直到有机层无色)洗, 饱和食盐水洗, 无水硫酸钠干燥, 过滤减压浓缩, 并进行柱层析纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=50:1], 得到中间体8和13.
(±)-3-乙酰基-4, 4-二甲基-1, 5-双(3-甲基丁-2-烯-1-基)-2-氧亚基环己基-1-甲酸甲酯(8):淡黄色油状物, 6.6 g, 产率96%. 1H NMR (400 MHz, CDCl3) δ: 5.10~5.04 (m, 2H), 3.77 (s, 3H), 3.53 (s, 1H), 2.51~2.40 (m, 2H), 2.33~2.26 (m, 2H), 2.09 (s, 3H), 2.02~1.91 (m, 2H), 1.72 (s, 12H), 1.30~1.20 (m, 1H), 1.11 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 205.2, 204.1, 172.3, 135.7, 133.0, 122.8, 118.6, 69.8, 61.1, 52.5, 43.0, 40.8, 33.3, 32.7, 32.2, 27.0, 26.1, 26.0, 24.1, 18.2, 16.1; HRMS calcd for C22H34O4Na [M+Na]+ 385.2349, found 385.2353.
(±)-3-乙酰基-1-烯丙基-4, 4-二甲基-5-(3-甲基丁-2-烯-1-基)-2-氧亚基环己基-1-甲酸甲酯(13):黄色油状物, 5.8 g, 产率91%. 1H NMR (400 MHz, CDCl3) δ: 5.78~5.70 (m, 1H), 5.05~4.99 (m, 3H), 3.73 (s, 3H), 3.51 (s, 1H), 2.54~2.39 (m, 2H), 2.31~2.23 (m, 2H), 2.08 (s, 3H), 1.99~1.87 (m, 2H), 1.61 (s, 6H), 1.29~1.24 (m, 1H), 1.10 (s, 3H), 1.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 204.8, 203.9, 172.1, 133.5, 133.2, 122.6, 118.9, 70.1, 60.7, 44.0, 41.9, 39.0, 34.8, 33.1, 27.1, 26.5, 24.9, 18.0, 16.2; HRMS calcd for C20H30O4Na [M+Na]+ 357.2036, found 357.2041.
将配体SIMES*PF6 (248 mg, 0.6 mmol)溶于甲基叔丁基醚(MTBE) (11 mL)中, 并加入KOtAm (25% in toluene, 276 mg, 0.6 mmol), 在60 ℃下搅拌1 h后, 冷却至室温, 加入Bu4N[Fe(CO)3(NO)] (247.2 mg, 0.6 mmol), 并将反应液在60 ℃下继续搅拌1 h (反应液1).同时, 将中间体6 (2.2 g, 6 mmol)溶于THF (11 mL)中, 冷却至0 ℃, 并加入LiH (52.8 mg, 6.6 mmol), 在该温度下搅拌30 min, 升至室温搅拌30 min后, 将此反应液和2-甲基-3-丁烯-2-碳酸异丁酯(2.2 g, 12 mmol)滴加至反应液1中, 100 ℃下搅拌过夜, TLC监测.反应毕, 将反应液通过二氧化硅垫过滤, 减压浓缩, 并进行柱层析纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=100:1], 得到淡黄色油状物9 (1.1 g, 产率44%). 1H NMR (400 MHz, CDCl3) δ: 5.16~5.11 (m, 1H), 5.05~5.02 (m, 1H), 4.66~4.61 (m, 1H), 3.75 (s, 3H), 3.06~2.99 (m, 1H), 2.37~2.25 (m, 3H), 2.22~2.16 (m, 1H), 2.12 (s, 3H), 2.03~1.99 (m, 2H), 1.81~1.77 (m, 2H), 1.70 (s, 6H), 1.61~1.57 (m, 12H), 1.04 (s, 3H), 0.99 (s, 3H); 13C NMR (100MHz, CDCl3) δ: 210.1, 206.9, 173.3, 135.5, 133.2, 132.9, 133.0, 119.7, 119.3, 73.1, 61.0, 52.6, 40.8, 38.3, 33.4, 33.0, 31.6, 31.3, 27.7, 26.1, 26.1, 22.2, 21.8, 18.4, 18.2, 18.0; HRMS calcd for C27H42O4Na [M+Na]+ 453.2975, found 453.2980.
将相应的环己酮中间体(4.5 mmol)溶于DMF (21 mL), 冷却至0 ℃, 分批加入NaH (60% in mineral oil, 216 mg, 5.4 mmol), 在该温度下搅拌1 h.然后滴加氯甲酸烯丙酯(0.56 mL, 5.4 mmol), 将反应液升至室温, 搅拌过夜, TLC监测.反应毕, 用饱和氯化铵溶液(20 mL)淬灭反应, 分液, 水相用乙酸乙酯萃取(80 mL×3), 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 过滤减压浓缩, 并进行柱层析纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=100:1], 得到中间体8a, 8b, 13a, 13b.
(±)-3-乙酰基-2-(((烯丙氧基)羰基)氧基)-4, 4-二甲基-1, 5-双(3-甲基丁-2-烯-1-基)环己基-2-烯-1-羧酸酯(8a):黄色油状物, 640 mg, 产率34%. 1H NMR (400 MHz, CDCl3) δ: 5.99~5.89 (m, 1H), 5.41~5.29 (m, 2H), 4.98~4.96 (m, 2H), 4.68~4.66 (m, 2H), 3.69 (s, 3H), 2.61~2.46 (m, 2H), 2.35~2.28 (m, 1H), 2.11~2.06 (m, 1H), 1.96 (s, 3H), 1.82~1.78 (m, 1H), 1.68~1.67 (d, J=3.1 Hz, 6H), 1.65~1.63 (m, 1H), 1.61 (s, 3H), 1.57 (s, 3H), 1.24~1.22 (m, 1H), 1.19 (s, 3H), 1.13 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 202.9, 172.1, 152.0, 150.1, 135.7, 135.5, 133.2, 131.2, 123.0, 119.6, 118.4, 69.2, 60.6, 52.6, 44.1, 41.9, 33.2, 32.3, 27.6, 26.5, 26.2, 26.1, 19.1, 18.5, 18.0, 18.0; HRMS calcd for C26H39O6 [M+H]+447.2741, found 447.2738.
(±)-3-(1-((((烯丙氧基)羰基)氧基)亚乙基)-4, 4-二甲基-1, 5-双(3-甲基丁-2-烯-1-基)-2-氧亚基环己烷-1-羧酸酯(8b):黄色油状物, 960 mg, 产率50%. 1H NMR (400 MHz, CDCl3) δ: 5.93~5.83 (m, 1H), 5.36~5.25 (m, 2H), 5.01~4.94 (m, 2H), 4.61~4.53 (m, 2H), 3.63 (s, 3H), 2.79~2.74 (m, 1H), 2.40~2.34 (m, 1H), 2.27 (s, 3H), 2.15~2.10 (m, 1H), 2.04~1.97 (m, 1H), 1.83~1.79 (m, 1H), 1.74~1.70 (m, 1H), 1.68 (s, 6H), 1.61 (s, 3H), 1.58 (s, 3H), 1.47~1.40 (m, 1H), 1.15 (s, 3H), 1.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 204.1, 173.3, 152.5, 144.2, 140.6, 135.1, 133.1, 131.2, 123.1, 119.5, 119.4, 69.3, 52.7, 50.5, 41.0, 38.1, 34.0, 32.4, 30.4, 27.8, 26.2, 26.0, 25.3, 21.2, 18.2, 18.0; HRMS calcd for C26H39O6 [M+H]+447.2471, found 447.2736.
(±)-3-乙酰基-1-烯丙基-2-(((烯丙氧基)羰基)氧基)-4, 4-二甲基-5-(3-甲基丁-2-烯-1-基)环己基-2-烯-1-羧酸酯(13a):黄色油状物, 567 mg, 产率32%. 1H NMR (400 MHz, CDCl3) δ: 5.99~5.89 (m, 1H), 5.72~5.62 (m, 1H), 5.41~5.28 (m, 2H), 5.09~4.99 (m, 3H), 4.68~4.66 (m, 2H), 3.69 (s, 3H), 2.70~2.65 (m, 1H), 2.47~2.42 (m, 1H), 2.34~2.27 (m, 1H), 2.11~2.07 (m, 1H), 1.96 (s, 3H), 1.90~1.85 (M, 1H), 1.73~1.70 (m, 1H), 1.67 (s, 3H), 1.57 (s, 3H), 1.25~1.20 (m, 1H), 1.18 (s, 3H), 1.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 202.5, 171.7, 151.9, 150.5, 135.4, 133.3, 133.0, 131.2, 122.9, 119.6, 119.4, 69.2, 60.0, 52.6, 44.0, 41.8, 39.1, 31.9, 27.5, 26.6, 25.9, 19.1, 18.5, 18.0; HRMS calcd for C24H34O6Na [M+ Na]+ 441.2248, found 441.2251.
(±)-1-烯丙基-3-(1-((((烯丙氧基)羰基)氧基)亚乙基)-4, 4-二甲基-5-(3-甲基丁-2-烯-1-基)-2-氧亚基环己烷- 1-羧酸酯(13b):黄色油状物, 1.1 g, 产率63%. 1H NMR (400 MHz, CDCl3) δ: 5.93~5.84 (m, 1H), 5.71~5.61 (m, 1H), 5.37~5.26 (m, 2H), 5.10~5.02 (m, 3H), 4.58~4.56 (m, 2H), 3.63 (s, 3H), 2.91~2.86 (m, 1H), 2.38~2.35 (m, 1H), 2.27 (s, 3H), 2.16~2.11 (m, 1H), 2.06~1.97 (m, 1H), 1.90~1.86 (m, 1H), 1.67 (s, 3H), 1.59 (s, 3H), 1.50~1.43 (m, 1H), 1.27~1.22 (m, 1H), 1.16 (s, 3H), 1.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 203.9, 172.9, 152.5, 144.0, 140.8, 133.5, 133.3, 131.1, 123.1, 119.6, 119.3, 69.4, 52.7, 50.2, 40.8, 39.7, 38.1, 32.4, 29.9, 27.6, 25.9, 25.2, 21.2, 18.0; HRMS calcd for C24H34O6Na [M+Na]+ 441.2248, found 441.2253.
将三(二亚苄基丙酮)二钯(0)-氯仿加合物(201 mg, 0.2 mmol)和三(对甲苯基)膦(293 mg, 1.0 mmol)溶于甲苯(11 mL)中, 室温搅拌1 h.随后滴加溶于甲苯的相应的中间体(4 mmol), 室温搅拌过夜, TLC监测.反应毕, 将反应液通过二氧化硅垫过滤, 减压浓缩, 并进行柱层析纯化[洗脱剂: V(石油醚):V(乙酸乙酯)=100:1], 得到中间体14和17.
(±)-3-乙酰基-1, 3-二烯丙基-4, 4-二甲基-5-(3-甲基丁-2-烯-1-基)-2-氧亚基环己烷-1-甲酸甲酯(14):淡黄色油状物, 1.1 g, 产率73%. 1H NMR (400 MHz, CDCl3) δ: 5.89~5.79 (m, 1H), 5.37~5.27 (m, 1H), 5.17~5.15 (m, 1H), 5.10~5.05 (m, 2H), 4.94~4.88 (m, 2H), 3.76 (s, 3H), 3.19~3.14 (m, 1H), 2.68~2.63 (m, 1H), 2.30~2.18 (m, 3H), 2.13 (s, 3H), 2.07~2.05 (m, 2H), 1.81~1.76 (m, 2H), 1.70 (s, 3H), 1.61 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 209.1, 206.3, 172.9, 134.0, 133.7, 133.5, 122.9, 199.8, 117.6, 73, 7, 59.8, 52.6, 40.6, 38.9, 37.7, 37.4, 32.9, 31.1, 27.4, 25.9, 22.3, 21.9, 18.1; HRMS calcd for C23H34O4Na [M+Na]+ 397.2349, found 397.2354.
(±)-3-乙酰基-3-烯丙基-4, 4-二甲基-1, 5-双(3-甲基丁-2-烯-1-基)-2-氧亚基环己烷-1-甲酸甲酯(17):淡黄色油状物, 1.2 g, 产率75%. 1H NMR (400 MHz, CDCl3) δ: 5.38~5.27 (m, 1H), 5.17~5.14 (m, 1H), 5.02~5.00 (m, 1H), 4.94~4.87 (m, 2H), 3.75 (s, 3H), 3.20~3.15 (m, 1H), 2.46~2.36 (m, 2H), 2.27~2.17 (m, 2H), 2.11 (s, 3H), 2.02~2.00 (m, 2H), 1.78~1.74 (m, 1H), 1.71 (s, 3H), 1.70 (s, 3H), 1.63 (s, 3H), 1.59 (s, 3H), 1.27~1.14 (m, 1H), 1.06 (s, 3H), 0.98 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 209.4, 206.5, 173.2, 135.7, 134.1, 133.3, 122.9, 119.6, 117.5, 73.6, 60.3, 52.6, 40.7, 38.2, 37.5, 33.1, 32.9, 31.3, 27.6, 26.1, 26.1, 22.3, 21.8, 18.4, 18.0; HRMS calcd for C25H38O4Na [M+Na]+ 425.2662, found 425.2658.
将相应的中间体(3.0 mmol)溶于THF (90 mL)中, 冷却至0 ℃, 加入叔丁醇钾(680 mg, 6 mmol), 在该温度下搅拌45 min, 随后升至室温, 用饱和氯化铵溶液淬灭反应, 减压浓缩有机相, 水相用乙酸乙酯萃取(50 mL×3), 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 过滤减压浓缩, 并进行柱层析纯化[洗脱剂: V(二氯甲烷):V(甲醇)=100:1], 得到中间体10, 15和18.
(±)-5, 5-二甲基-4, 6, 8-三(3-甲基丁-2-烯-1-基)双环[3.3.1]壬烷-1, 3, 9-三酮(10):白色固体, 956 mg, 产率80%. m.p. 116~119 ℃; 1H NMR (300 MHz, CDCl3) δ: 5.12~5.08 (m, 1H), 4.94~4.83 (m, 2H), 3.56 (d, J=17.2 Hz, 1H), 2.95 (d, J=17.2Hz, 1H), 2.56~2.54 (m, 2H), 2.49~2.47 (m, 2H), 2.19~2.14 (m, 2H), 2.07~2.00 (m, 1H), 1.66~1.65 (m, 9H), 1.61 (s, 6H), 1.51 (s, 3H), 1.48~1.39 (m, 1H), 1.33~1.27 (m, 1H), 1.25 (s, 3H), 0.96 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 210.6, 203.0, 202.8, 136.6, 135.7, 133.8, 122.7, 118.2, 117.6, 70.3, 64.9, 62.6, 51.3, 46.5, 40.6, 31.3, 29.3, 27.3, 26.5, 26.2, 26.2, 26.0, 23.0, 18.1, 18.0, 18.0; HRMS calcd for C26H38O3Na 421.2713 [M+Na]+, found 421.2719.
(±)-5, 5-二甲基-4, 8-二烯丙基-6-(3-甲基丁-2-烯-1-基)双环[3.3.1]壬烷-1, 3, 9-三酮(15):白色固体, 832 mg, 产率81%. m.p. 128~131 ℃; 1H NMR (400 MHz, CDCl3) δ: 5.85~5.75 (m, 1H), 5.69~5.59 (m, 1H), 5.10~5.01 (m, 4H), 4.86~4.83 (m, 1H), 3.61 (d, J=17.3 Hz, 1H), 2.89 (d, J=17.2 Hz, 1H), 2.76~2.72 (m, 1H), 2.60~2.55 (m, 1H), 2.48~2.42 (m, 2H), 2.24~2.15 (m, 2H), 2.09~2.04 (m, 1H), 1.66 (s, 3H), 1.51 (s, 3H), 1.48~1.40 (m, 1H), 1.37~1.32 (m, 1H), 1.24 (s, 3H), 0.97 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 210.0, 202.4, 202.3, 134.0, 132.5, 132.2, 122.5, 120.7, 119.9, 71.0, 64.7, 63.3, 51.5, 46.5, 41.0, 36.9, 32.6, 29.3, 26.4, 25.9, 23.1, 18.0; HRMS calcd for C22H30O3Na [M+Na]+ 365.2087, found 365.2092.
(±)-5, 5-二甲基-4-烯丙基-6, 8-二(3-甲基丁-2-烯-1-基)双环[3.3.1]壬烷-1, 3, 9-三酮(18):白色固体, 801 mg, 产率72%. m.p. 124~126 ℃; 1H NMR (400 MHz, CDCl3) δ: 5.72~5.60 (m, 1H), 5.13~5.01 (m, 3H), 4.90~4.85 (m, 1H), 3.74~3.56 (m, 1H), 2.92~2.88 (m, 1H), 2.77~2.72 (m, 1H), 2.47~2.41 (m, 2H), 2.22~2.15 (m, 2H), 2.11~2.02 (m, 2H), 1.69~1.63 (m, 9H), 1.51 (s, 3H), 1.35~1.30 (m, 1H), 1.23 (s, 3H), 1.10~1.08 (m, 1H), 0.96 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 210.2, 202.7, 202.5, 135.8, 133.8, 132.3, 122.6, 120.6, 118.0, 70.9, 64.8, 63.2, 51.5, 46.6, 40.9, 32.7, 31.5, 29.3, 26.4, 26.1, 25.9, 23.1, 18.1, 18.0; HRMS calcd for C24H34O3Na [M+Na]+ 393.2400, found 393.2399.
取相应的中间体(1.1 mmol)溶于无水THF中, 在氮气保护下缓慢加入三乙胺(0.4 mL, 3.3 mmol)和溶于THF的3, 4-二乙酰氧基苯甲酰氰(815 mg, 3.3 mmol), 搅拌过夜.反应毕, 加入饱和的氯化铵溶液, 减压浓缩有机相, 水相用乙酸乙酯萃取, 合并有机相, 有机相用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩, 粗产物经过柱层析分离纯化[洗脱剂: V(二氯甲烷):V(甲醇)=400:1], 得到中间体11, 16, 19.
(±)-4-(5, 5-二甲基-4, 6, 8-三(3-甲基丁-2-烯-1-基)-1, 3, 9-三氧亚基双环[3.3.1]壬烷-2-羰基)-1, 2-苯二乙酸酯(11):黄色油状物, 517 mg, 产率76%. 1H NMR (400 MHz, CDCl3) δ: 7.46~7.44 (m, 1H), 7.21~7.19 (m, 2H), 4.85~4.83 (m, 2H), 4.79~4.76 (m, 1H), 2.29 (s, 6H), 2.24~2.17 (m, 2H), 2.15~2.10 (m, 2H), 2.04~1.97 (m, 2H), 1.96~1.81 (m, 2H), 1.74 (s, 6H), 1.70 (s, 3H), 1.66 (s, 3H), 1.58 (s, 3H), 145~1.44 (m, 1H), 1.42 (s, 3H), 1.13 (s, 3H), 0.97 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.0, 197.4, 194.6, 193.6, 167.9, 167.6, 145.7, 141.6, 135.0, 134.8, 133.1, 128.0, 124.1, 123.5, 122.8, 119.8, 118.5, 115.6, 68.8, 65.6, 63.3, 58.5, 53.6, 48.5, 46.4, 39.4, 30.7, 29.0, 26.6, 26.2, 25.9, 22.6, 20.7, 18.2, 17.9; HRMS calcd for C37H46O8Na [M+Na]+ 641.3085, found 641.3083.
(±)-4-(5, 5-二甲基-4, 8-二烯丙基-6-(3-甲基丁-2-烯-1-基)-1, 3, 9-三氧亚基双环[3.3.1]壬烷-2-羰基)-1, 2-苯二乙酸酯(16):黄色油状物, 482 mg, 产率78%. 1H NMR (400 MHz, CDCl3) δ: 7.41~7.40 (m, 1H), 7.27~7.21 (m, 2H), 5.62~5.49 (m, 1H), 5.12~5.05 (m, 4H), 4.85~4.80 (m, 2H), 2.63~2.60 (m, 2H), 2.55~2.54 (m, 2H), 2.29 (s, 6H), 1.99~1.96 (m, 2H), 1.95~1.85 (m, 2H), 1.61 (s, 3H), 1.46~1.44 (m, 1H), 1.41 (s, 3H), 1.12 (s, 3H), 0.98 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.6, 196.2, 194.8, 192.9, 167.6, 145.9, 141.4, 134.8, 134.2, 133.3, 132.9, 128.0, 125.0, 123.4, 122.9, 119.7, 119.2, 116.0, 69.2, 65.7, 63.0, 58.1, 48.7, 46.5, 39.4, 36.0, 31.2, 29.0, 27.0, 25.8, 22.5, 20.7, 17.9; HRMS calcd for C33H38O8Na [M+Na]+ 585.2459, found 585.2455.
(±)-4-(5, 5-二甲基-4-烯丙基-6, 8-二(3-甲基丁-2-烯- 1-基)-1, 3, 9-三氧亚基双环[3.3.1]壬烷-2-羰基)-1, 2-苯二乙酸酯(19):黄色油状物, 487 mg, 产率75%. 1H NMR (500 MHz, CDCl3) δ: 7.60~7.59 (m, 1H), 7.24~7.20 (m, 2H), 5.62~5.54 (m, 1H), 5.08~5.00 (m, 2H), 4.87~4.82 (m, 2H), 2.61~2.54 (m, 2H), 2.52~2.39 (m, 2H), 2.29 (s, 6H), 2.04~1.96 (m, 2H), 1.93~1.84 (m, 2H), 1.74 (s, 3H), 1.70 (s, 3H), 1.62 (s, 3H), 1.42 (s, 3H), 1.26~1.25 (m, 1H), 1.12 (s, 3H), 0.98 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 207.8, 196.4, 194.6, 193.1, 167.9, 167.6, 145.8, 141.4, 135.1, 134.8, 133.2, 128.13, 124.55, 123.46, 122.84, 119.57, 116.16, 115.88, 69.0, 65.6, 63.2, 58.3, 48.7, 46.5, 39.4, 31.3, 30.8, 29.0, 26.2, 25.9, 22.5, 20.8, 20.6, 18.2, 17.9; HRMS calcd for C35H42O8Na [M+ Na]+ 613.2772, found 613.2769.
取相应的中间体(0.9 mmol)和K2CO3 (496 mg, 3.6 mmol)于烧瓶中, 抽空换氮, 随后加入甲醇(15 mL), 室温搅拌1 h.反应毕, 加入饱和的氯化铵溶液, 用乙酸乙酯萃取三次, 合并有机相, 有机相用饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩, 粗产物柱层析分离纯化[洗脱剂: V(二氯甲烷):V(甲醇)=50:1], 得到最终产物.
8-异戊烯基(代)山竹醇(1):黄色粘稠物, 342.2 mg, 产率64%. 1H NMR (400 MHz, CD3OD) δ: 7.18~7.17 (m, 1H), 6.98~6.96 (m, 1H), 6.71~6.69 (m, 1H), 5.17~5.15 (m, 1H), 4.94 (m, 2H), 2.75~2.70 (m, 1H), 2.59~2.49 (m, 3H), 2.18~2.00 (m, 4H), 1.72 (s, 3H), 1.68 (s, 6H), 1.65 (s, 6H), 1.48 (s, 3H), 1.34~1.29 (m, 1H), 1.24 (s, 3H), 1.01 (s, 3H); 13C NMR (100 MHz, CD3OD) δ: 209.9, 195.9, 152.5, 146.2, 135.6, 135.5, 133.7, 129.5, 125.4, 125.1, 120.8, 117.8, 117.4, 115.1, 47.8, 32.1, 30.1, 27.3, 27.1, 26.3, 26.0, 23.2, 18.3, 18.2, 18.1; HRMS calcd for C33H42O6Na [M+Na]+ 557.2874, found 557.2871.
4, 8-二烯丙基(代)山竹醇(2):黄色粘稠物, 297.2 mg, 产率69%. 1H NMR (400 MHz, CD3OD) δ: 6.98~6.83 (m, 2H), 6.68~6.58 (m, 1H), 5.94~5.84 (m, 1H), 5.67~5.66 (m, 1H), 5.12~4.99 (m, 5H), 2.71~2.64 (m, 2H), 2.51~2.45 (m, 2H), 2.18~2.01 (m, 4H), 1.63 (s, 3H), 1.48 (s, 3H), 1.29 (m, 1H), 1.23 (s, 3H), 0.99 (s, 3H); 13C NMR (125 MHz, CD3OD) δ: 209.4, 196.3, 152.6, 146.0, 135.0, 133.8, 129.8, 125.4, 125.3, 119.7, 119.5, 118.3, 117.4, 115.1, 52.2, 47.7, 40.7, 37.5, 32.3, 30.1, 27.2, 26.0, 23.2, 18.1; HRMS calcd for C29H34O6Na [M+Na]+ 501.2248, found 501.2246.
4-烯丙基-8-异戊烯基(代)山竹醇(3):黄色粘稠物, 264.4 mg, 产率58%. 1H NMR (400 MHz, CD3OD) δ: 7.16~6.98 (m, 2H), 6.68~6.59 (m, 1H), 5.67~5.65 (m, 1H), 5.19~4.99 (m, 4H), 2.72~2.64 (m, 2H), 2.50~2.45 (m, 2H), 2.18~2.01 (m, 4H), 1.73 (s, 3H), 1.66 (s, 3H), 1.63 (s, 3H), 1.47 (s, 3H), 1.29 (m, 1H), 1.24 (s, 3H), 0.99 (s, 3H); 13C NMR (125 MHz, CD3OD) δ: 209.7, 196.0, 194.1, 152.5, 146.1, 135.5, 134.9, 133.7, 129.6, 125.3, 125.2, 120.8, 119.7, 118.1, 117.4, 115.1, 58.3, 54.8, 47.7, 41.0, 32.2, 30.0, 27.2, 26.3, 26.0, 23.3, 18.2, 18.1; HRMS calcd for C29H34O6Na [M+Na]+ 529.2561, found 529.2560.
MTT法检测细胞增殖:将对数生长期的SCC-15细胞悬液调整浓度至1×104/mL和CAL-27调至3×104/mL后, 接种到96孔培养板, 每孔200 μL, 细胞贴壁后, 根据预实验结果, 分别加入不同浓度的山竹醇和山竹醇类似物(5、10、20、30、40、50 μmol•L-1).每个浓度设置6个复孔, 同时设置不加细胞的调零孔和不加药物的阴性对照孔.在37 ℃ 5% CO2分别孵育24、48、72 h, 每孔加入20 μL MTT溶液(5 mg/mL), 继续培养4 h后终止培养, 吸去孔内培养液, 每孔加入200 μL二甲基亚砜, 置摇床上低速振荡10 min, 使结晶物充分溶解.在酶联免疫检测仪波长OD490 nm处测量各孔的吸光值.应用SPSS 17.0 for windows统计软件进行统计分析.采用ANOVA检验, 以α=0.05作为检验水准.
首先在PDB (http://www.rcsb.org)蛋白质数据库下载5-LOX(PDB_ID: 3V99)的晶体结构.借助软件DS 2019对其进行去除水分子, 去除蛋白质多构象, 补充非完整的氨基酸残基, 为蛋白加氢等预处理.经过处理的蛋白, 可进行后续的对接等操作.接着借助软件DS 2019, 对小分子配体进行预处理.最后利用处理好的蛋白和配体进行分子对接, 使用DS 2019软件中的CDOCKER模块, 设置相应的流程参数, 运行分子对接模拟.
辅助材料(Supporting Information) 目标化合物的1H NMR, 13C NMR谱图.这些材料可以免费从本刊网站(http://siocjournal.cn/)上下载.
Newman, D. J.; Cragg, G. M.; Snader, K. M. J. Nat. Prod. 2003, 66, 1022. doi: 10.1021/np030096l
Aggarwal, B. B.; Kunnumakkara, A. B. Molecular Targets and Therapeutic Use of Spices, World Scientific Publishing Co. Pte. Ltd., 2009, pp. 281~309.
Schobert, R.; Biersack, B. Chem. Biodiversity. 2019, 16, e1900366.
Aggarwal, S.; Das, S. N. Tumor. Biol. 2016, 37, 7175. doi: 10.1007/s13277-015-4583-8
Ranjbarnejad, T.; Saidijam, M.; Tafakh, M. S.; Pourjafar, M.; Talebzadeh, F.; Najafi, R. Hum. Exp. Toxicol. 2017, 36, 692. doi: 10.1177/0960327116660865
Li, F.; Shanmugam, M. K.; Chen, L. X.; Chatterjee, S.; Basha, J.; Kumar, A. P.; Kundu, T. K.; Sethi, G. Cancer Prev. Res. (Phila) 2013, 6, 843. doi: 10.1158/1940-6207.CAPR-13-0070
Wang, J. H.; Wang, L. W..; Ho, C. T.; Zhang, K. S.; Liu, Q.; Zhao, H. J Agric. Food Chem. 2017, 65, 3675. doi: 10.1021/acs.jafc.7b00346
Ahmad, A.; Wang, Z. W.; Wojewoda, C.; Ali, R.; Kong, D.; Maitah, M. Y.; Banerjee, S.; Bao, B.; Padhye, S.; Sarkar, F. H. Front. Biosci. 1492, 3, 1483.
Liu, C.; Ho, P. C.; Wong, F. C.; Sethi, G.; Wang, L. Z.; Goh, C. Cancer Lett. 2015, 362, 8. doi: 10.1016/j.canlet.2015.03.019
Liao, C. H.; Ho, C. T.; Lin, J. K. Biochem. Biophys. Res. Commun. 2005, 329, 1306. doi: 10.1016/j.bbrc.2005.02.110
Liao, C. H.; Sang, S. M.; Liang, Y. C.; Ho, C. T.; Lin, J. K. Mol. Carcinog. 2004, 41, 140. doi: 10.1002/mc.20050
Zhou, X. Y.; Cao, J.; Han, C. M.; Li, S. W.; Zhang, C.; Du, Y, D.; Zhou, Q. Q.; Zhang, X. Y.; Chen, X. Bioorg. Chem. 2017, 71, 74. doi: 10.1016/j.bioorg.2017.01.013
曹菁, 韩超明, 张桂莲, 周新莹, 李舒雯, 杜银端, 赵帅, 张辛燕, 陈新, 有机化学, 2017, 37, 8. doi: 10.6023/cjoc201701017Cao, J.; Han, C. M.; Zhang, G. L.; Zhou, X. Y.; Li, S. W.; Du, Y. D.; Zhao, S.; Zhang, X. Y.; Chen, X. Chin. J. Org. Chem. 2017, 37, 8(in Chinese). doi: 10.6023/cjoc201701017
Han, C. M.; Zhou, X. Y.; Cao, J.; Zhang, X. Y.; Chen, X. Bioorg. Chem. 2015, 60, 123. doi: 10.1016/j.bioorg.2015.04.010
Chen, X.; Zhang, X. Y.; Lu, Y.; Shim, J. Y.; Sang, S. M.; Sun, Z.; Chen, X. X. Nutr. Cancer. 2012, 64, 1211. doi: 10.1080/01635581.2012.718032
Socolsky, C.; Plietker, B. Chem.-Eur. J. 2015, 21, 3053. doi: 10.1002/chem.201406077
Guttroff, C.; Baykal, A.; Wang, H. H.; Popella, P.; Kraus, F.; Biber, N.; Krauss, S.; Gotz, F.; Plietker, B. Angew. Chem., Int. Ed. 2017, 56, 15852. doi: 10.1002/anie.201707069
Biber, N.; Mows, K.; Plietker, B. Nat. Chem. 2011, 3, 938. doi: 10.1038/nchem.1170
Holzwarth, M.; Dieskau, A.; Tabassam, M.; Plietker, B. Angew. Chem. Int. Ed. 2009, 48, 7251. doi: 10.1002/anie.200901930
Saba, S.; Brescia, A.; Kaloustian, M. Tetrahedron Lett. 1991, 32, 5031. doi: 10.1016/S0040-4039(00)93420-8

图式 1 山竹醇类似物1的合成
Scheme 1 Synthesis of garcinol analog 1
Reagents and conditions: (a) NaH, isoprenyl bromide, EtOH, 0 ℃ to r.t., 15 h, then K2CO3, formaldehyde, r.t., 15 h, 60% yield. (b) methylmagnesium chloride, dimethyl 1, 3-acetonedicarboxylate, MeOH, 0 to 60 ℃, 15 h, 89% yield. (c) NaH, THF, then MeLi, 0 ℃; NaH, isoprenyl bromide, THF, 0 ℃ to r.t., 63% yield. (d) LiCl, CuI, methylmagnesium bromide, Me3SiCl, THF, -78 ℃, 96% yield. (e) Potassium tert-amylate (KOtAm), 1, 3-dimesitylimidazolin-2-ylidene hexafluorophosphate (SIMES*PF6) (10 mol%), Bu4N[Fe(CO)3(NO)] (10 mol%), LiH, isobutyl (2-methylbut-3-en-2-yl) carbonate, THF/methyl-t-butyl ether, 0 to 80 ℃, 44% yield. (f) KOBut, THF, 0 ℃, 80% yield. (g) Et3N, 4-(cyanocar-bonyl)-1, 2-phenylene diacetate, THF, r.t., 16 h, 76% yield. (h) K2CO3, MeOH, r.t., 1 h, 64% yield.

图式 3 山竹醇类似物2和3的合成
Scheme 3 Synthetic of garcinol analogs 2 and 3
Reagents and conditions: (a) NaH, THF, then MeLi, 0 ℃; NaH, R1Br, 0 ℃ to r.t., for 7: 63% yield, for 12: 66% yield; (b) LiCl, CuI, methylmagnesium bromide, Me3SiCl, THF, -78 ℃. For 8: 96% yield; for 13: 91% yield; (c) NaH, allyl chloroformate, DMF, 0 ℃ to r.t., for 8a: 34% yield, 8b: 50% yield; for 13a: 32% yield, 13b: 63% yield; (d) Tris(dibenzylideneacetone)dipalladium(0)-chloroform [Pd2(dba)3-CHCl3] (5 mol%), tri(p-tolyl)phosphine, toluene, r.t., for 14: 73% yield, for 17: 75% yield. (e) KOBut, THF, 0 ℃, for 15: 81% yield, for 18: 72% yield; (f) Et3N, 4-(cyanocarbonyl)-1, 2-phenylene diacetate, THF, r.t., 16 h, for 16: 78% yield, for 19: 75% yield; (g) K2CO3, MeOH, r.t., 1 h, for 2: 69% yield, for 3: 58% yield.

图 2 不同浓度山竹醇和山竹醇类似物处理CAL-27细胞24 (a)、48 (b)和72 h (c)的OD值
Figure 2 OD values of CAL-27 cells treated with different concentrations of garcinol and garcinol analogs at 24 (a), 48 (b) and 72 h (c)

图 3 不同浓度山竹醇和山竹醇类似物处理SCC-15细胞24 (a)和72 h (b)的OD值
Figure 3 OD values of SCC-15 cells treated with different concentrations of garcinol and garcinol analogs at 24 and 72 h

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:


 下载:
下载: