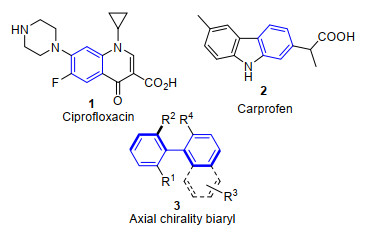
图 1.
典型含苯环结构的分子
Figure 1.
Selected compounds containing benzene rings

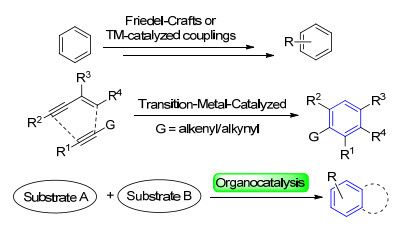
多取代苯等芳香环是有机分子中最基本的母核结构之一, 在200种最畅销的药物里接近80%的分子结构中都至少含有1个苯环结构[1].例如环丙沙星化合物1作为第三代喹诺酮类抗菌药物, 对革兰阴性菌作用效果明显, 同时具有广谱的抗菌活性, 是广泛应用于临床治疗的抗菌药物[2]; 非甾体抗炎药物卡洛芬2中则以咔唑结构为主要骨架[3]; 轴手性联芳基化合物3作为手性配体或催化剂被广泛应用于催化反应中, 并表现出非常好的性能[4](图 1).传统合成多取代苯类化合物的方法主要有: (1)在已有苯环结构上引入新的官能团, 典型方法包括芳环的亲电或亲核取代反应[5]或过渡金属催化的偶联反应(Scheme 1)[6]; (2)由过渡金属催化的非环状炔烃或烯烃的组合构建苯环结构(Scheme 1)[7].然而这些合成方法往往需要多步反应且收率较低, 或需要导向基团的存在才能实现较好的区域选择性, 效率较低.另一方面, 反应的经济性和环境友好性也是需要考虑的因素.有机催化的概念由MacMillan在21世纪初提出并得到充分的发展, 成为继金属催化和酶催化化学反应的主要类型之一.有机催化反应具有条件温和、操作简单等优点, 为高效构建多取代苯环结构化合物提供了一条新的合成路径并得到广泛关注.通过对底物的预先官能化, 经有机催化可以实现构建苯环骨架的同时导入不同官能团, 完成芳香环的多样性合成(Scheme 1).本文基于不同种类小分子催化剂催化合成多取代苯环结构的方法, 简述不同种类催化剂的催化特点及其应用.由化学计量的催化剂参与的反应本文不讨论.
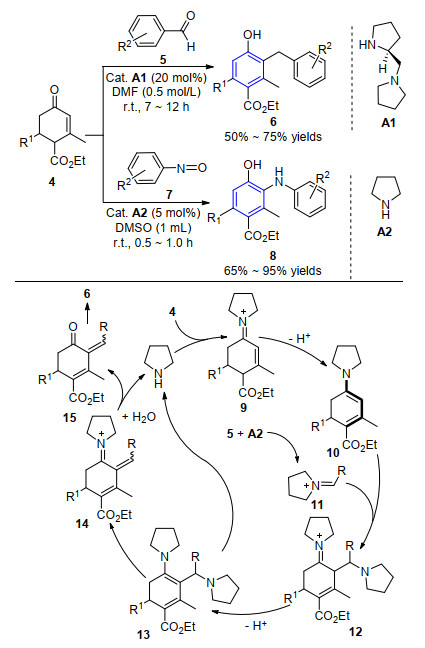
2005年, Ramachary课题组[8]报道了首例有机催化Hagemann酯和醛的串级Claisen-Schmidt异构/芳构化反应, 得到一系列多取代的苯酚结构化合物.该反应在小分子催化剂A1催化作用下与Hagemann酯4经原位生成的二烯胺中间体9得到2-芳叉Hagemann酯10, 而醛与催化剂作用生成的亚胺正离子11能很好地与二烯胺中间体10发生Mannich反应, 进一步经逆-Mannich反应、异构/芳构化生成目标产物6.紧接着, 该小组[9]利用同样的策略, 实现了吡咯烷A2催化的Hagemann酯4与亚硝基苯7的串级反应, 以较高产率得到二芳基胺类化合物8 (Scheme 2). 2007年, Hong小组[10]报道了吡咯烷或脯氨酸催化的α, β-不饱和醛的分子间环加成反应, 通过[3+3]、[4+2]形式实现了多取代芳香醛的合成.反应采用不同的催化剂组合, 在一些反应中需要加入过量的氧化剂如二氧化锰(MnO2)或2, 3-二氯-5, 6-二氰基-1, 4-苯醌(DDQ)来实现产物的芳构化.同时, 该反应还能实现不饱和醛的分子内环加成反应, 为具有生物活性小分子的构建提供了一条简单、高效的合成方法.
2010年, 许鹏飞课题组[11]发展了一例吡咯烷A2催化2-(2-氧乙基)-苯甲醛16与硝基烯烃17的串级环化反应, 历经Michael/Henry反应、脱水和芳构化得到多取代萘环化合物18.反应对芳香或杂环取代的硝基烯烃底物有很好的适应性, 以中等收率得到目标产物, 而烷基取代的硝基烯烃则不能发生反应(Eq. 1).
|
|
(1) |
2012年, 史达清小组[12]报道了靛红20与3-氨基取代环己烯酮19的反应, 当底物3-氨基取代环己烯酮环上取代基为氢原子时, 该反应在催化量的L-脯氨酸(A3)作用下实现吖啶类衍生物21的高效合成.反应在1~3 h即可完成, 能以60%~82%的收率得到芳构化产物(Eq. 2).
|
|
(2) |
王卫课题组[13]报道了有机催化Michael/Aldol芳构化反应, 实现了β-烷基取代烯醛22的二聚以及与烯醛23的环合反应(Eq. 3), 以高产率及非常好的区域/化学选择性地得到多取代芳香醛类化合物24.
|
|
(3) |
具有轴手性的芳香化合物广泛存在于天然产物、药物分子、手性催化剂和功能分子中, 其化学合成备受关注.这类分子的合成方法主要是对反应底物中已存在的芳香基团进行不对称的取代或修饰, 从而构建出手性轴.但在多取代芳环结构中, 逐步取代的方法往往会受到区域选择性的限制而需要较长的合成步骤.而且当手性轴两边空间位阻相差不大的时候, 通过不对称取代的方法就很难能得到高的立体选择性.因此, 通过直接构建芳香环结构来制备轴手性化合物的方法则具有独特的优势.
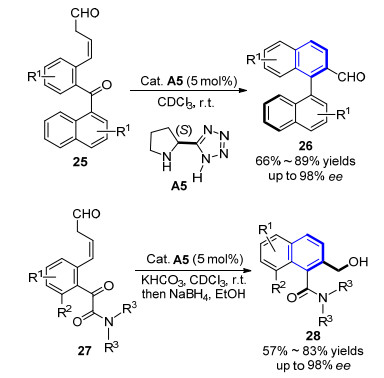
2014年, Sparr小组[14]首次报道了在有机催化Aldol反应合成芳香环的同时, 实现轴手性的构建.反应以5 mol%的吡咯烷基-四氮唑(A5)为催化剂, 室温下完成底物25的高效转化, 经由烯胺活化底物、中心手性向轴手性的转移及芳构化过程, 以中等到良好的收率和高达98%的非对映体过量值(ee)得到联萘甲醛类化合物26 (Scheme 3).随后, 该小组[15]利用同样的策略, 在相同催化剂A5作用下, 将底物结构更换为27时, 能以高对映选择性得到轴手性联芳基酰胺类化合物.反应在30 min~2 h即可完成, 再经硼氢化钠还原得到更稳定的羟甲基取代的芳基酰胺产物28 (Scheme 3).
2015年, Lee小组[16]报道了由L-脯氨酸(A3)催化的1, 4-萘醌或1, 4-蒽醌29与α, β-不饱和醛30的[4+2]环加成反应, 合成了一系列蒽醌类或四苯并二酮类化合物31.机理研究显示脯氨酸与α, β-不饱和醛作用形成二烯胺中间体, 当底物结构不同或催化剂结构中羧基与底物之间存在氢键作用时, 可能得到两种区域选择性产物.该反应为蒽醌或四苯并二酮类化合物的合成提供一条简单高效的合成方法(Eq. 4).
|
|
(4) |
2015年, Vitale和Michelet课题组[17]报道了三氟甲磺酰亚胺(A6)催化炔-醛偶联苯环化合成多取代萘的方法.传统方法由炔烃参与萘环中苯环构建的[4+2]环化过程主要是金属或路易斯酸催化完成的, 而该小组首次采用三氟甲磺酰亚胺作为催化剂实现多取代萘的构建.苯环上和α位有不同取代基的苯乙醛底物32均能很好地与各种取代炔烃底物33发生反应, 同时该反应还能在炔烃与环氧化物或二甲基乙缩醛类底物中顺利发生, 同样得到多取代萘环产物(Eq. 5).
|
|
(5) |
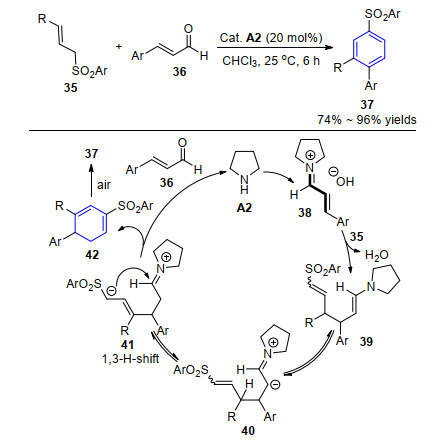
2018年, 蒋琳和袁明龙等[18]报道了含烯丙基砜取代的1, 3-双亲核试剂35与不饱和醛36的[3+3]苯环化反应.以20 mol%的吡咯烷(A2)作催化剂, 与不饱和醛作用生成亚胺正离子中间体38, 经Michael加成得到烯胺中间体39, 再经异构化、1, 3-氢迁移、分子内的亲核加成环合反应得到产物42并脱除催化剂实现循环过程, 最后经空气氧化得到目标产物37.该反应很好地避免了过渡金属以及化学计量的强碱的使用, 在绿色、温和条件下实现多取代苯化合物的多样性合成(Scheme 4).
2019年, 许鹏飞课题组[19]成功利用环己二胺衍生的硫脲类手性氢键催化剂, 通过芳香化反应在吲哚的碳环上构筑了轴手性, 得到的含有吲哚骨架的轴手性联芳基二酚可以作为一类具有较高催化活性的不对称催化剂.通过对底物结构和催化剂的筛选, 作者选定5-羟基吲哚43和氮杂对苯醌类底物44作为起始原料, 经条件优化及底物扩展可以定量的产率、高达99%的ee值得到最终轴手性产物45 (Eq. 6).进一步考察了反应机理, 通过核磁实验发现催化剂A7与底物44之间存在较强的氢键作用, 同时底物5-羟基吲哚43中的羟基与催化剂的硫原子也存在弱相互作用, 并通过羟基保护实验证明底物中羟基的存在对反应起到了至关重要的作用.另外, 作者将得到的轴手性联芳基二酚产物作为催化剂用于催化酮的不对称烯丙基化反应.结果表明, 不论是催化活性还是立体选择性的控制能力都优于传统轴手性联二萘酚(BINOL), 反应效果甚至比已有报道的最优催化剂还要好.该实验充分说明新合成的这类轴手性骨架在不对称催化领域有着巨大的开发潜力.最后, 作者对手性氢键催化苯环化反应进行了放大反应和催化剂回收利用实验, 均取得令人满意的结果.这一方法的建立, 实现了由有机催化-有机催化剂的完美转化, 为轴手性吲哚类化合物的合成提供了开拓性的思路, 也拓展了氢键催化的应用范围.
|
|
(6) |
有机碱是一类结构简单、便宜易得的化合物, 如吡啶、1, 8-二氮杂二环十一碳-7-烯(DBU)和金鸡纳碱等都是一些常见的有机碱.利用有机碱作为小分子催化剂催化反应, 可以通过分子中所含的氮原子等富电子中心与底物作用形成活化中间体来启动化学反应, 并利用自身的结构因素来控制反应的立体选择性, 且催化剂容易从产物中脱离完成催化循环.
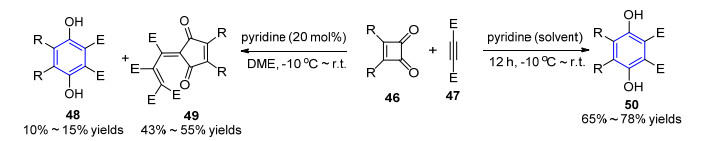
2005年, Nair小组[20]报道了吡啶介导的丁炔二酸二酯47与环丁烯1, 2-二酮46的反应, 通过调节吡啶的量可以选择性地实现六取代苯环衍生物48或环戊烯二酮类化合物49的简捷合成.当反应以吡啶为溶剂时, 可以中等到良好的收率得到全取代的对苯二酚产物50; 当使用催化量的吡啶时, 反应主要生成环戊烯二酮产物, 仅有10%~15%的苯环化产物(Scheme 5).紧接着, 该课题组报道了催化量的二甲氨基吡啶催化β-酮酯与丁炔二酸二甲酯的反应, 同样得到多取代苯环产物和联芳基化合物.但该反应的底物适用范围比较受限, 底物47中酯基的存在对反应的顺利发生起到关键性作用.
基于同样的策略, 薛松小组[21]进一步将Nair报道的反应中的底物扩展到β-二酮51与丙炔酸乙酯52或丁炔二酸二甲酯54, 分别在20 mol%的4-二甲氨基吡啶(DMAP)或25 mol%吡啶/叔丁醇钾催化条件下得到多取代苯环产物(Scheme 6).

Hamdi和Silva小组[22]报道了一锅法合成苯并香豆素58的方法, 由催化量的4-吡咯取代吡啶B1催化化合物56与57的反应, 经1, 4-共轭加成、脱羧、芳构化得到目的产物.其中底物57中3-羧基的存在对反应的发生起到决定性作用(Eq. 7).
|
|
(7) |
2014年, Babu等[23]报道了一例简单高效一锅法合成杂芳基取代苯环产物的方法, 反应以5 mol%的1, 4-二氮杂二环[2.2.2]辛烷(B2, DABCO)为催化剂, 乙烯基丙二腈59与杂芳基硝基烯烃60经Michael加成反应, 在碱性条件下进一步环合和异构化, 最后经氧化得到杂芳基取代的苯环结构化合物61.值得注意的是, 该小组对合成的产物进行了活性测试, 结果显示部分化合物在抗结核和抗菌方面表现出很好的广谱活性(Eq. 8).
|
|
(8) |
Salvio和Bella课题组[24]实现金鸡纳碱B3催化不对称合成2', 2'-联萘酚类轴手性化合物67的高效合成.通过条件优化, 该反应在15 mol%的金鸡纳碱催化下, 以四氢呋喃为溶剂, 4 ℃下反应足够长时间, 能以几乎定量的产率得到联萘酚类轴手性产物.同时, 该反应同样能以高收率实现克级规模的制备, 反应产物通过重结晶能得到高达98%的ee值(Eq. 9).尽管反应的对映选择性不太理想, 但仍能为探索新型有机催化构建轴手性的研究奠定基础.
|
|
(9) |
2017年, 徐显秀小组[25]报道了一锅法合成菲啶衍生物70的简便方法, 在30 mol%的DBU催化作用下, 一系列2-异氰基查耳酮68与活泼亚甲基酮69经Michael加成、分子内异腈亲电环化、Aldol缩合, 再经脱水、脱酰基并被氧气氧化和芳构化得到结构多样的菲啶产物.该反应通过多步多米诺转化可以一次性实现连续双环及三个C—C键的构建, 为合成三环骨架结构化合物提供了有效的合成方法(Eq. 10).
|
|
(10) |
氮杂卡宾(NHC)作为有机小分子催化剂中的一员, 具有很好的亲核性,其对羰基化合物的催化反应表现出独特的活化模式和高效的催化活性及立体选择性[26].通过NHC催化活化不同的反应类型和底物可以高效地合成出各种结构新颖的化合物, 这些化合物为筛选高活性药物小分子提供了基础.利用氮杂卡宾催化反应构建环化产物及与氧化环境兼容性好的特点, 一方面通过NHC催化剂实现环化反应的动力学调控, 确保芳环构建时的区域选择性和化学选择性控制; 另一方面, 通过引入串联氧化芳香化过程, 为催化环化反应提供额外的热力学推动力, 完成热力学稳定的芳香体系的构建.两方面相辅相成, 为多取代芳环化产物的合成提供了一条非常高效的捷径.
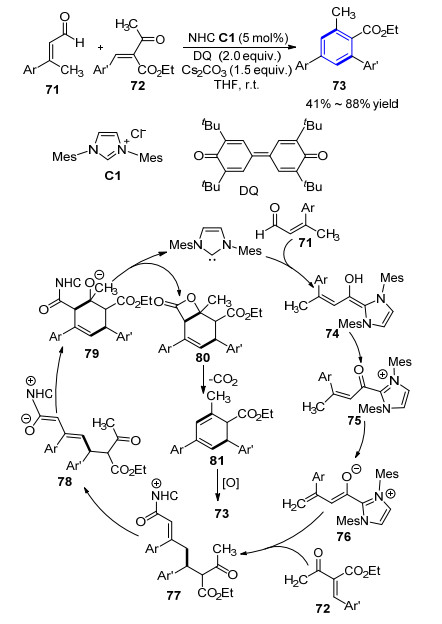
2014年, 池永贵课题组[27]首次实现利用氮杂卡宾催化[3+3]环加成构建苯环的反应.以简单易得的烯醛与烯酮为起始原料, 活化后的氮杂卡宾首先对烯醛71发生加成及质子化得到Breslow中间体74, 再在苯醌氧化剂(DQ)作用下得到α, β-不饱和酰基唑中间体75, 在碱性条件下质子化生成乙烯基烯醇式中间体76, 该中间体的γ-碳对烯酮72发生Michael加成反应得到中间体77, 接着发生分子内的Aldol缩合、内酯化生成产物80并脱除卡宾催化剂实现催化循环.产物80再经脱羧、氧化得到最终四取代苯环产物73.底物普适性考察显示, 其中烯醛和烯酮底物的E/Z异构体对反应结果影响不大, 均能很好的得到苯环化产物(Scheme 7).同时, 反应产物也能进一步发生转化, 通过一步反应即能得到一些天然产物结构中的核心骨架如芴酮和异吲哚酮等结构.该方法高区域选择性地一步快速构建了多取代芳环, 为相应复杂结构化合物的合成提供了全新的合成思路.
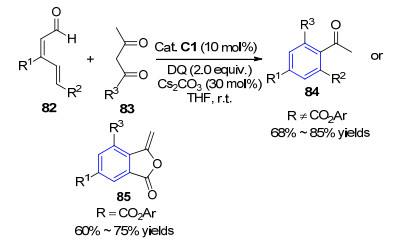
不久, 该小组[28]再次利用氮杂卡宾催化实现α, β-γ, δ-不饱和醛82的δ位活化, 与1, 3-二酮83反应得到多取代芳烃化合物84.当底物82中δ位被苯酚酯单元取代时, 反应则按另一途径得到3-亚基茚苯酞产物85.该反应最大亮点在于在α, β-γ, δ-不饱和醛的β位引入取代基, 从而限制β碳的反应活性, 实现远程δ位的活化, 通过底物结构控制产物的化学/区域选择性得到多取代芳烃化合物(Scheme 8).
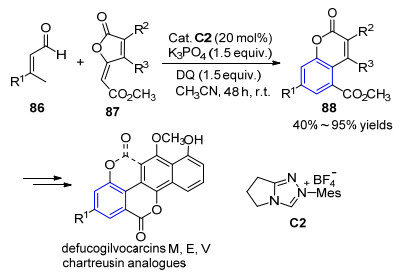
2017年, 池永贵等[29]报道了卡宾催化β-甲基-α, β-不饱和醛86与呋喃酮87的[5+5]反应, 成功合成香豆素结构化合物88, 并成功将该策略应用于具有重要生物活性的天然产物的合成中(Scheme 9).最近, 该小组[30]进一步将反应底物扩展到β-甲基-α, β-不饱和酯类化合物, 与氧杂二烯底物在卡宾催化下, 采用更廉价易得的四甲基哌啶氮氧化物(TEMPO)作为氧化剂实现了苯环化反应, 高产率地得到了2, 4, 6-三取代的苯乙酮类产物.
2019年, 池永贵课题组[31]和王兴旺小组[32]相继报道了卡宾催化环戊烯二酮89的去对称化, 直接构建苯环结构得到了二氢化茚产物90, 通过远程手性控制实现全季碳手性中心的构建(Eq. 11).基于该策略, 可以完成部分螺环分子或天然产物的结构片段的高效合成, 同时还可以对产物进行多种结构转化.两个小组的工作类似, 区别仅仅在于催化剂中负离子和碱的使用不同, 而王兴旺小组则可以实现产物的一锅法甲基化过程.
|
|
(11) |
2015年, Lupton小组[33]报道了氮杂卡宾催化酯91的氧化还原异构化反应, 合成了官能化苯甲醛类化合物92.相比之前卡宾催化酯的反应生成β-内酯或环己二烯产物, 该反应经过对催化剂结构、溶剂和添加物的筛选, 可以43%~91%的收率得到多取代的苯甲醛产物(Eq. 12).
|
|
(12) |
2016年, 汪舰课题组[34]利用α-氰基-β-甲基烯酮93与α, β-不饱和醛94为底物, 在卡宾催化下实现[4+2]苯环化反应, 构建了一系列多取代的苯甲腈类化合物95.随后, 叶松小组[35]也报道了同样的反应, 仅仅是反应条件略有不同, 反应均能以良好到优秀的收率得到苯甲腈结构.由于氰基的存在, 使得合成的产物可以进行多样性转化(Eq. 13).随后, 该小组[36]将α, β-不饱和醛更换为α-溴代烯醛, 与α-氰基-β-甲基烯酮93在卡宾催化下得到1, 3, 5-三取代苯结构化合物, 反应采用碳酸铯和DBU的组合, 分别用于活化卡宾前体和消除氰基芳构化过程, 高产率地得到了不对称的1, 3, 5-三取代苯.
|
|
(13) |
房新强等[37]在研究氮杂卡宾催化β, γ-不饱和1, 2-二酮底物96的极性反转反应过程中, 得到多官能化的双环环己烯-β-内酯产物97或1, 2, 4-三芳基取代苯环产物98, 反应机理揭示亲核O-酰基化高烯醇式中间体的生成是反应成功进行的关键(Eq. 14).同时, 反应产物可以实现多种合成转化如胺化、还原或氧化等过程.
|
|
(14) |
咔唑骨架广泛存在于众多活性分子和天然产物结构中并展现出独特的生物活性.付振乾和黄维等[38]利用卡宾催化化合物99与烯醛100的[4+2]芳环化反应成功合成了一系列多取代咔唑类化合物101 (Eq. 15).该反应具有广泛的底物适用范围、条件温和及无金属参与等优点, 同时也可以放大到克级规模, 为探索其他芳烃的绿色合成方法起到很好的参考作用.
|
|
(15) |
2019年, 叶松小组[39]报道了氮杂卡宾催化β-甲基-α, β-不饱和醛86与橙酮类化合物102的反应, 合成了2, 2'-二羟基二苯甲酮103 (Eq. 16).机理研究显示卡宾催化剂C7对烯醛86加成、氧化后, 在碱作用下得到乙烯基烯醇式中间体, 再对橙酮发生加成反应及分子内的C-酰基化生成螺环产物, 该螺环产物在碱性条件下发生C—O键断裂并经逆Michael加成, 最终芳构化得到2, 2'-二羟基二苯甲酮类产物.
|
|
(16) |
最近, 朱庭顺课题组[40]通过氮杂卡宾催化共轭二烯醛105与α-芳基酮104的[4+2]苯环化实现了位阻选择性反应, 一步构建1, 2, 3, 5-四取代苯环, 同时在2-位形成轴手性(Eq. 17).该反应的底物适应性较好, 底物α-芳基酮104中有卤素、杂环、氰基、三氟甲基、磷氧化合物等都能在体系中兼容, 给出较好的产率及90%以上的ee值.而反应产物106通过简单的转化, 可以高效合成轴手性的各类催化剂与配体.同时, 该反应的还能与电化学反应体系兼容, 通过氧化剂的电极循环, 可以将原来2.5~3.5 equiv.的氧化剂减少至催化量.该工作拓展了氮杂卡宾作为有机小分子催化剂在催化合成方面的应用, 为轴手性化合物的合成提供了一种具有潜力的合成手段.
|
|
(17) |
手性磷酸是一类高效、高对映选择性的Brønsted酸类有机催化剂, 已被成功应用于众多不对称催化反应中.磷酸分子结构中磷原子上所连的羟基可作为Brønsted酸的酸性位点提供质子, 或与底物形成氢键作用对亲电试剂进行活化, 而磷原子上的双键氧又可以作为Lewis碱性位点提供孤对电子来活化亲核试剂, 因此磷酸类化合物具有双功能催化剂的特点, 既能提高反应的催化活性, 又能有效地控制反应的立体选择性, 从而完成高对映选择性合成.近年来, 手性磷酸作为催化剂被用于构建联芳基轴手性的研究成为一大热点.其中轴手性联萘二酚类化合物作为配体或独立催化剂使用已被广泛关注, 其核心骨架结构也存在于众多具有生物活性的天然产物中[41].但该类化合物的合成方法却鲜有报道, 目前仅有的对映选择性合成方法主要是金属催化的不对称氧化偶联与动力学拆分.因此, 发展高效构建轴手性联芳基二醇类化合物的方法仍然是有必要的.
2015年, 谭斌小组[42]报道了有机催化2-萘酚108的芳基化反应, 成功实现了轴手性联萘二醇109的制备.化合物2-萘酚108与苯醌107作为起始反应底物在磷酸类催化剂D1作用下能有效完成轴手性联萘二醇的合成(Scheme 10).通过条件优化, 该过程很好地解决了以下关键问题: (1)选择合适的催化剂来有效控制2-萘酚底物中碳/氧的化学选择性; (2)催化剂能对共轭加成过程进行有效的立体控制; (3)在温和的条件下实现由中心手性到轴手性的转移控制.底物扩展显示2-萘酚及苯醌107中2-位酯基或卤原子取代基的存在对反应的进行起到重要作用.值得注意的是, 合成的轴手性联萘二醇产物109可以作为非常高效的配体用于二乙基锌对醛的加成反应中, 并取得高达98%的ee值, 充分显示了该方法的适用性及轴手性联萘二醇产物的重要性.在此研究基础之上, 该小组继续探索有机催化在构建轴手性领域的应用研究. 2017年, 该小组[43]首次实现了2-萘胺111作为亲核试剂对亚胺苯醌底物110的加成, 在相同手性磷酸D1催化下得到轴手性的联芳基氨基醇类产物112 (Scheme 10).反应机理显示手性磷酸作为双官能化催化剂, 同时对底物苯醌110和2-萘酚或2-萘胺活化并促进对映选择性共轭加成反应的发生, 进而由中心手性到轴手性的转移和芳构化得到轴手性联芳基产物.
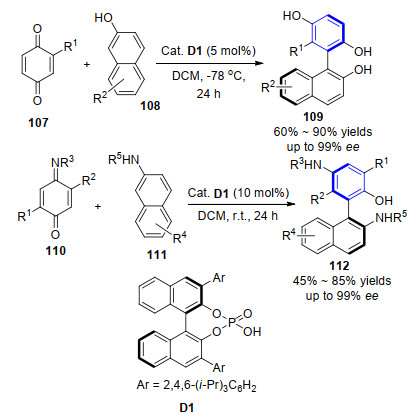
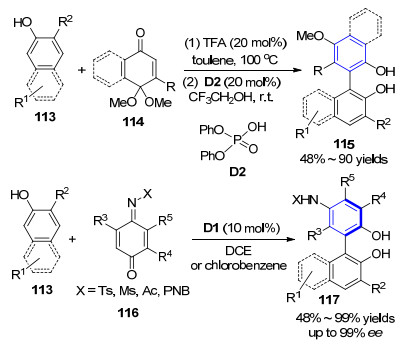
另外, Kürti小组[44]同样报道了类似的工作, 选取苯醌单缩醛底物114与2-萘酚108为底物, 在Brønsted酸催化作用下得到官能化的非C2对称的联芳基结构产物115 (Scheme 11).该反应以20 mol%的三氟乙酸(TFA)或二苯基磷酸D2为催化剂即可实现目的产物的高效合成, 并指出反应经由乙缩醛交换、[3,3]-σ重排和芳构化路径完成.遗憾的是通过手性磷酸催化剂的引入也没能实现该产物的对映选择性过程.但随后不久, 该小组就实现突破, 报道了与Tan小组类似的工作, 即完成了手性磷酸D1催化2-萘酚113与不同取代的亚胺苯醌116的反应, 实现了构建非C2对称的联萘二酚117的轴手性过程.通过底物结构控制实验, 作者指出该反应可能经过一步缩醛胺生成的步骤, 再经[3,3]重排、苯环化得到联芳基产物(Scheme 11)[45].
2018年, Maji课题组[46]报道了Brønsted酸催化2-烯基吲哚118与1, 3-二羰基化合物119的一锅法苯环化反应, 合成咔唑120或吲哚咔唑类产物(Eq. 18).该反应由25 mol%的磷酸催化剂D2催化, 经6π-电环化、空气氧化过程得到目标产物, 同时反应产物经过简单几步反应即可实现重要药物分子的高效合成.该方法具备了无金属参与、底物普适性广和可规模化制备等特点, 并通过空气直接氧化的简单操作即可实现咔唑类化合物的绿色合成.
|
|
(18) |
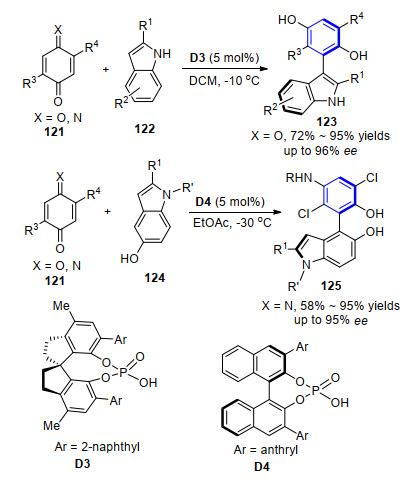
最近, 李绍玉和谭斌小组[47]报道了手性磷酸催化吲哚与苯醌的位阻异构化构建轴手性芳基吲哚骨架的合成方法.当采用2-取代吲哚122与苯醌作为起始原料时, 反应在磷酸催化剂D3催化下能很好地得到轴手性苯基吲哚产物123, 经过条件优化和底物扩展发现, 该反应能以72%~95%的产率和高达96%的对映选择性得到目的产物.而将苯醌底物替换成亚胺苯醌, 在磷酸催化剂D4催化下则能很好地控制5-羟基吲哚124中C-4的亲核性, 完成轴手性苯基吲哚产物125的合成.值得注意的是, 底物5-羟基吲哚124中N被保护后对反应的顺利进行没有影响.通过控制实验和机理研究发现, 底物中吲哚的N—H或5-位羟基均可能与催化剂之间形成氢键作用参与到反应的立体控制过程中, 进而提高反应的对映选择性(Scheme 12).
2010, 张俊良小组[48]报道了三氟甲磺酸钪或三氟甲磺酸催化化合物127的分子内苯环化合成多取代萘128和苯并芴醇类化合物129的方法(Eq. 19).其中使用5 mol%的三氟甲磺酸, 在二氯甲烷溶剂、室温下仅5 min即能以高达98%的收率得到多取代萘产物128.当底物中取代基变为苯环时, 在三氟甲磺酸催化下以产物苯并芴醇129为主; 而以三氟甲磺酸钪为催化剂时, 则能保持以多取代萘产物为主.因此, 在合成多取代萘和苯并芴醇的过程中使用三氟甲磺酸和三氟甲磺酸钪能很好地互补.
|
|
(19) |
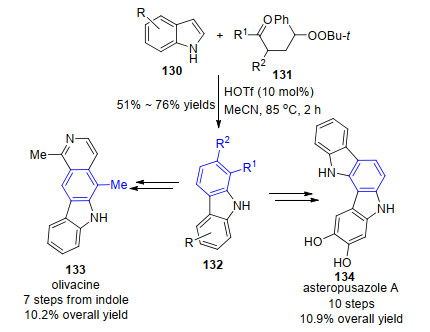
2014年, 李志平小组[49]报道了10% mol的三氟甲磺酸催化吲哚130与γ-羰基叔丁基过氧化物131的苯环化反应合成咔唑类化合物132 (Scheme 13).而当吲哚上N—H有甲基取代时, 则有两种异构产物生成.可能原因是有螺环亚胺正离子中间体原位生成, 由于位阻导致两类不同1, 2-烷基迁移方式形成不同产物.值得注意的是, 该方法能直接应用于具有重要生理活性的药物分子Olivacine和Asteropusazole A的快速合成.
2016年, 蔡春小组[50]利用抗坏血酸E1作为催化剂成功实现了邻氨基联苯135与炔烃136合成菲类多环芳烃结构化合物137 (Eq. 20).该反应在亚硝酸叔丁酯和催化量的抗坏血酸作用下生成重氮盐并经自由基过程与端炔底物环化、芳构化得到目标产物.用简单易得的起始原料在无金属参与的温和条件下简单操作即可实现菲类多芳烃结构的构建.
|
|
(20) |
2017年, Reddy等[51]报道了一锅法实现多环芳烃的多步合成反应, 在5 mol%的对甲苯磺酸催化作用下, 1, 1-二芳基乙醇138经脱水生成烯烃与丙炔醇139发生亲核加成得到烯炔中间体, 接着在碱DBU存在下异构、电环化并芳构化得到烯基取代的萘环产物140 (Eq. 21).值得注意的是, 该方法能扩展到多芳烃结构䓛、苉和苯并苉等化合物的高效合成, 极大地拓展了该方法的实用性.
|
|
(21) |
2012年, 周磊等[52]报道了曙红Y在光作用下催化联芳基重氮盐141与炔烃142的[4+2]苯环化反应, 成功合成了9-或9, 10-二取代菲产物143 (Eq. 22).可能的反应机理认为联芳基重氮盐在光敏剂曙红Y催化下生成联芳基自由基, 进而与炔烃发生自由基加成、环化等串级过程.值得注意的是该方法同样能应用于非端炔类底物参与的反应, 并以中等收率得到目的产物.
|
|
(22) |
有机小分子催化经过多年的研究积累, 取得了长足的发展.建立小分子催化活化模式实现多取代苯类化合物的高效快速合成方法, 成为当下有机催化领域的一个热门课题.通过总结发现:第一, 有机催化合成芳香结构化合物的新方法区别于传统合成路径, 不仅满足有机催化的绿色、无毒、无污染及反应条件温和等优点, 还为进一步实现复杂多样的取代苯类化合物的合成提供了路线支持.第二, 由有机催化构建苯及多取代苯类化合物的方法可以完成取代苯类化合物的多样性合成与转化, 即通过改变底物中取代基的结构和位置, 即可实现苯环上取代基团的种类和位置变化, 为合成特定某类取代苯类化合物提供了快速、简便的方法.因此, 有机催化芳香化反应的实现对反应底物的结构也存在一定的依赖性.第三, 发展新的催化剂骨架、设计结构新颖的候选化合物和减少催化剂的用量等问题一直是有机催化过程面临的挑战, 同时需要考虑在构建苯环的同时如何实现产物的手性诱导.最后, 对于新的反应模式和机理研究还有待于进一步探索.相信随着有机催化的发展, 更多的有机催化反应类型和结构多样的芳香结构化合物会被合成和应用到生活的各个领域中.
(a) McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348.
(b) Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N. Beilstein J. Org. Chem. 2011, 7, 442.
Davis, R.; Markham, A.; Balfour, J. A. Drugs 1996, 51, 1019. doi: 10.2165/00003495-199651060-00010
(a) O'Brien, W. M.; Bagby, G. F. Pharmacotherapy 1987, 7, 16.
(b) Ricketts, A. P.; Lundy, K. M.; Seibel, S. B. Am. J. Vet. Res. 1998, 59, 1441.
(c) Thau-Zuchman, O.; Shohami, E.; Alexandrovich, A. G.; Trembovler, V.; Leker, R. R. J. Neurotraum. 2012, 29, 375.
(a) Chen, Y.; Yekta, S.; Yudin, A. K. Chem. Rev. 2003, 103, 3155.
(b) Brunel, J. M. Chem. Rev. 2005, 105, 857.
(c) Schenker, S.; Zamfir, A.; Freund, M.; Tsogoeva, S. B. Eur. J. Org. Chem. 2011, 2011, 2209.
(a) Calloway, N. O. Chem. Rev. 1935, 17, 327.
(b) Tanaka, K. Transition-metal-mediated Aromatic Ring Construction, Wiley, Hoboken, NJ, 2013.
(c) Sunke, R.; Nallapati, S. B.; Kumar, J. S.; Kumarb, K. S.; Pal, M. Org. Biomol. Chem. 2017, 15, 4042.
(a) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
(b) Saito, S.; Yamamoto, Y. Chem. Rev. 2000, 100, 2901.
(c) Kotha, S.; Misra, S.; Halder, S. Tetrahedron 2008, 64, 10775.
(d) Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4442.
(a) van Otterlo, W. A.; De Koning, C. B. Chem. Rev. 2009, 109, 3743.
(b) Saito, S.; Salter, M. M.; Gevorgyan, V.; Tsuboya, N.; Tando, K.; Yamamoto, Y. J. Am. Chem. Soc. 1996, 118, 3970.
(c) Gevorgyan, V.; Takeda, A.; Yamamoto, Y. J. Am. Chem. Soc. 1997, 119, 11313.
(d) Gevorgyan, V.; Sadayori, N.; Yamamoto, Y. Tetrahedron Lett. 1997, 38, 8603.
Ramachary, D. B.; Ramakumar, K.; Kishor, M. Tetrahedron Lett. 2005, 46, 7037. doi: 10.1016/j.tetlet.2005.08.051
Ramachary, D. B.; Ramakumar, K.; Narayana, V. V. J. Org. Chem. 2007, 72, 1458. doi: 10.1021/jo0623639
Hong, B. C.; Tseng, H. C.; Chen, S. H. Tetrahedron 2007, 63, 2840. doi: 10.1016/j.tet.2007.01.039
Li, S. G.; Hu, X. Q.; Jia, Z. X.; Xu, P. F. Tetrahedron 2010, 66, 8557. doi: 10.1016/j.tet.2010.08.069
Wang, H.; Li, L.; Lin, W.; Xu, P.; Huang, Z.; Shi, D. Org. Lett. 2012, 14, 4598. doi: 10.1021/ol302058g
Song, X.; Zhang, X.; Zhang, S.; Li, H.; Wang, W. Chem.-Eur. J. 2012, 18, 9770. doi: 10.1002/chem.201201709
Link, A.; Sparr, C. Angew. Chem., Int. Ed. 2014, 53, 1. doi: 10.1002/anie.201310509
Fäseke, V. C.; Sparr, C. Angew. Chem., Int. Ed. 2016, 55, 7261. doi: 10.1002/anie.201602689
Magar, K. B. S.; Xia, L.; Lee, Y. R. Chem. Commun. 2015, 51, 8592. doi: 10.1039/C5CC00623F
Ponra, S.; Vitale, M. R.; Michelet, V.; Ratovelomanana-Vidal, V. J. Org. Chem. 2015, 80, 3250. doi: 10.1021/acs.joc.5b00353
Jiang, L.; Li, H.; Zhou, J. F.; Yuan, M. W.; Li, H. L.; Chuan, Y. M.; Yuan, M. L. Synth. Commun. 2018, 48, 336. doi: 10.1080/00397911.2017.1402351
Liu, J. Y.; Yang, X. C.; Liu, Z.; Luo, Y. C.; Lu, H.; Gu, Y. C.; Fang, R.; Xu, P. F. Org. Lett. 2019, 21, 5219. doi: 10.1021/acs.orglett.9b01828
(a) Nair, V.; Pillai, A. N.; Beneesh, P. B.; Suresh, E. Org. Lett. 2005, 7, 4625.
(b) Nair, V.; Vidya, N.; Biju, A. T.; Deepthi, A.; Abhilash, K. G.; Suresh, E. Tetrahedron 2006, 62, 10136.
(a) Zhou, Q. F.; Yang, F.; Guo, Q. X.; Xue, S. Synlett 2007, 2073.
(b) Hu, B.; Meng, L. G.; Liu, Y. L.; Liang, M.; Xue, S. Synthesis 2009, 24, 4137.
Talhi, O.; Makhloufi-Chebli, M.; Pinto, D. C.; Hamdi, M.; Silva, A. M. Synlett 2013, 24, 2559. doi: 10.1055/s-0033-1339895
Babu, G. N.; Ayalew, H. M.; Jain, S. Med. Chem. Res. 2014, 23, 2608. doi: 10.1007/s00044-013-0857-0
Moliterno, M.; Cari, R.; Puglisi, A.; Antenucci, A.; Sperandio, C.; Moretti, E.; Di Sabato, A.; Salvio, R.; Bella, M. Angew. Chem., Int. Ed. 2016, 55, 6525. doi: 10.1002/anie.201601660
Hu, Z.; Dong, J.; Men, Y.; Li, Y.; Xu, X. Chem. Commun. 2017, 53, 1739. doi: 10.1039/C6CC09430A
(a) Enders, D.; Balensiefer, T. Acc. Chem. Res. 2004, 37, 534.
(b) Marion, N.; Diez-Gonzalez, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988.
(c) Nair, V.; Vellalath, S.; Babu, B. P. Chem. Soc. Rev. 2008, 37, 2691.
(d) Bugaut, X.; Glorius, F. Chem. Soc. Rev. 2012, 41, 3511.
(e) Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485.
(f) Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307.
(g) Zhang, C.; Hooper, J. F.; Lupton, D. W. ACS Catal. 2017, 7, 2583.
(h) Zhao, M.; Zhang, Y.-T.; Chen, J.; Zhou, L. Asian J. Org. Chem. 2018, 7, 54.
Zhu, T. S.; Zheng, P. C.; Mou, C. L.; Yang, S.; Song, B. A.; Chi, Y. R. Nat. Commun. 2014, 5, 6.
Zhu, T. S.; Mou, C. L.; Li, B. S.; Smetankova, M.; Song, B. A.; Chi, Y. R. J. Am. Chem. Soc. 2015, 137, 5658. doi: 10.1021/jacs.5b02219
Huang, X.; Zhu, T.; Huang, Z.; Zhang, Y.; Jin, Z.; Zanoni, G.; Chi, Y. R. Org. Lett. 2017, 19, 6188. doi: 10.1021/acs.orglett.7b03102
Wu, J.; Mou, C.; Chi, Y. R. Chin. J. Chem. 2018, 36, 333. doi: 10.1002/cjoc.201700773
Zhu, T.; Liu, Y.; Smetankova, M.; Zhuo, S.; Mou, C.; Chai, H.; Jin, Z.; Chi, Y. R. Angew. Chem., Int. Ed. 2019, 58, 15778. doi: 10.1002/anie.201910183
Hu, J. M.; Zhang, J. Q.; Sun, B. B.; Chen, J. B.; Yu, J. Q.; Yang, X. P.; Lv, H. P.; Wang, Z.; Wang, X. W. Org. Lett. 2019, 21, 8582. doi: 10.1021/acs.orglett.9b03178
Candish, L.; Levensa, A.; Lupton, D. W. Chem. Sci. 2015, 6, 2366. doi: 10.1039/C4SC03726J
Jia, Q.; Wang, J. Org. Lett. 2016, 18, 2212. doi: 10.1021/acs.orglett.6b00844
Zhang, C. L.; Gao, Z. H.; Liang, Z. Q.; Ye, S. Adv. Synth. Catal. 2016, 358, 2862. doi: 10.1002/adsc.201600531
Zhang, C. L.; Ye, S. Org. Lett. 2016, 18, 6408. doi: 10.1021/acs.orglett.6b03306
Liu, J.; Das, D. K.; Zhang, G.; Yang, S.; Zhang, H.; Fang, X. Org. Lett. 2018, 20, 64. doi: 10.1021/acs.orglett.7b03358
Liu, D.; Gao, Y.; Huang, J.; Fu, Z.; Huang, W. J. Org. Chem. 2018, 83, 14210. doi: 10.1021/acs.joc.8b02532
Chen, K. Q.; Luo, Z.; Gao, Z. H.; Ye, S. Chem.-Eur. J. 2019, 25, 3253.
Xu, K.; Li, W.; Zhu, S.; Zhu, T. Angew. Chem., Int. Ed. 2019, 58, 17625. doi: 10.1002/anie.201910049
(a) Chen, Y.; Yekta, S.; Yudin, A. K. Chem. Rev. 2003, 103, 3155.
(b) Brunel, J. M. Chem. Rev. 2005, 105, 857.
(c) Renzi, P. Org. Biomol. Chem. 2017, 15, 4506.
(d) Witzig, R. M.; Lotter, D.; Fäseke, V. C.; Sparr, C. Chem.-Eur. J. 2017, 23, 12960.
(e) Link, A.; Sparr, C. Chem. Soc. Rev. 2018, 47, 3804.
Chen, Y. H.; Cheng, D. J.; Zhang, J.; Wang, Y.; Liu, X. Y.; Tan, B. J. Am. Chem. Soc. 2015, 137, 15062. doi: 10.1021/jacs.5b10152
Chen, Y. H.; Qi, L. W.; Fang, F.; Tan, B. Angew. Chem., Int. Ed. 2017, 56, 16308. doi: 10.1002/anie.201710537
Gao, H.; Xu, Q. L.; Keene, C.; Yousufuddin, M.; Ess, D. H.; Kürti, L. Angew. Chem., Int. Ed. 2016, 55, 566. doi: 10.1002/anie.201508419
Wang, J. Z.; Zhou, J.; Xu, C.; Sun, H.; Kürti, L.; Xu, Q. L. J. Am. Chem. Soc. 2016, 138, 5202. doi: 10.1021/jacs.6b01458
Saha, S.; Banerjee, A.; Maji, M. S. Org. Lett. 2018, 20, 6920. doi: 10.1021/acs.orglett.8b03063
Lu, D. L.; Chen, Y. H.; Xiang, S. H.; Yu, P.; Tan, B.; Li, S. Org. Lett. 2019, 21, 6000. doi: 10.1021/acs.orglett.9b02143
Liu, L.; Wei, L.; Zhang, J. Adv. Synth. Catal. 2010, 352, 1920. doi: 10.1002/adsc.201000286
Zheng, X.; Lv, L.; Lu, S.; Wang, W.; Li, Z. Org. Lett. 2014, 16, 5156. doi: 10.1021/ol5025053
Bu, M. J.; Lu, G. P.; Cai, C. Org. Chem. Front. 2016, 3, 630. doi: 10.1039/C6QO00020G
Reddy, C. R.; Dilipkumar, U.; Shravya, R. Chem. Commun. 2017, 53, 1904. doi: 10.1039/C6CC09108C
Xiao, T.; Dong, X.; Tang, Y.; Zhou, L. Adv. Synth. Catal. 2012, 354, 3195. doi: 10.1002/adsc.201200569

图式 2 基于推挽式二烯胺中间体合成多取代苯环化合物
Scheme 2 Synthesis of highly substituted benzenes based on push-pull dienamine intermediate

图式 3 有机催化芳环化合成轴手性联芳基化合物及芳香酰胺化合物
Scheme 3 Organocatalytic synthesis of axially chiral biaryls and aromatic amides by arene formation

图式 4 二级胺催化[3+3]苯环化合成多取代苯化合物
Scheme 4 Secondary amine-catalyzed [3+3] benzannulation to access polysubstituted benzenes

图式 5 吡啶催化丁炔二酸二甲酯(DMAD)与环丁烯1, 2-二酮的反应
Scheme 5 Pyridine catalyzed reaction of DMAD with cyclobutene-1, 2-diones

图式 6 DMAP或吡啶/叔丁醇钾催化苯环化反应
Scheme 6 DMAP or pyridine/potassium tert-butoxide catalyzed benzannulation

图式 7 有机催化[3+3]环加成构建苯环的反应及机理
Scheme 7 Benzene construction via organocatalytic formal [3+3] cycloaddition reaction and mechanism

图式 8 氮杂卡宾催化不饱和醛的δ‑碳LUMO活化.
Scheme 8 N-Heterocyclic carbene-catalyzed δ-carbon LUMO activation of unsaturated aldehydes.

图式 9 卡宾催化[5+5]反应合成香豆素结构
Scheme 9 Carbene-catalyzed formal [5+5] reaction for coumarin construction

图式 10 手性磷酸催化轴手性联芳基化合物的合成
Scheme 10 Synthesis of axially chiral biaryls by chiral phosphoric acid

图式 11 有机催化2-萘酚与苯醌单缩醛或亚胺苯醌构建轴手性的反应
Scheme 11 Organocatalytic atroposelective arylation of 2-naphthol with quinone monoacetals or iminoquinones

图式 12 手性磷酸催化吲哚与苯醌的位阻异构化构建轴手性芳基吲哚骨架
Scheme 12 Atroposelective construction of arylindoles by chiral phosphoric acid-catalyzed cross-coupling of indoles and quinones

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:























 下载:
下载:


 下载:
下载: