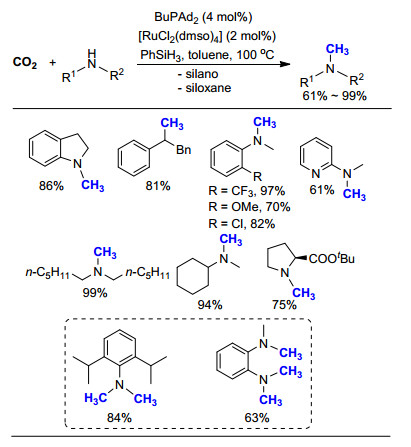
图式 1.
以苯硅烷为还原剂钌催化的氮甲基化反应
Scheme 1.
Ru-catalyzed N-methylation of amines with phen- ylsilane

甲基取代的胺是一类重要的化合物[1], 胺和亚胺的氮甲基化反应一直是人们研究的热点, 反应所形成的甲基胺、二甲基胺和甲酰胺除了作为实验室的反应试剂以外, 还可以作为原料应用于药物、染料、农药、杀菌剂、人造皮革和高分子等的合成[1-4].工业上多是使用具有毒性的甲醛作为C1来源, 通过Eschweiler-Clarke反应实现, 而实验室中则多是采用活性的甲基化合物, 如碘甲烷、硫酸二甲酯、三氟甲磺酸甲酯和重氮甲烷等.这些试剂除了具有毒性外, 在反应结束后也会产生大量的废弃物.因此, 利用环境友好的C1来源, 开发符合绿色环保需求的胺和亚胺氮甲基化反应的新方法, 成为近年来该研究领域重要的发展方向.基于这一思路, 一系列毒性低、廉价易得的化合物, 如CO2、HCOOH、甲醇和碳酸二甲酯等, 作为C1来源被成功应用于胺和亚胺的氮甲基化反应中.本文将从氮甲基化反应中不同的C1来源(二氧化碳、甲酸、甲醇、其它碳源四类)介绍近年来胺和亚胺氮甲基化反应的研究进展.
当前, 全球性的能源短缺问题和日益增加的二氧化碳排放量, 使得人们越来越关注二氧化碳的开发和利用. CO2作为自然界中来源丰富的碳源, 具有经济、环保、可再生等诸多优点, 广泛应用于生产甲醇、聚碳酸酯、甲酸以及其它重要化学品[5-7], 此外, CO2分子也可直接作为燃料或氢气能源的载体.在早期的研究中, 主要集中于将二氧化碳还原为甲酸和甲醇.近年来, 以CO2作为原料, 制备更多类型的复杂化合物已成为有机化学研究的重要领域.随着方法的不断改进, 目前已成功应用于杂环类化合物、羧酸、酰胺以及甲基胺等的合成中[8-11].其中, 以二氧化碳为碳源, 高效选择性地制备胺和亚胺的氮甲基化产物, 取得的重要研究进展尤为引人注目.
2013年, Beller课题组[12-13]报道了利用CO2作为碳源、苯硅烷(PhSiH3)为还原剂的氮甲基化反应(Scheme 1).作者使用二氯四(二甲基亚砜)钌(Ⅱ) [RuCl2(dmso)4]为催化剂, 在磷配体BuPAd2的作用下, 高效地实现了芳香胺与脂肪胺, 一级胺与二级胺的氮甲基化反应, 均具有较好的产率.在较温和的条件下实现了CO2的转化和利用, C1来源更加廉价易得.对比其他方法, 此项工作取得了该领域的突破性进展, 极大地促进了该研究领域的迅速发展.
Beller课题组[14]将还原剂苯硅烷更换为H2, 也实现了一级胺与二级胺的高效氮甲基化反应(Scheme 2).在反应条件筛选后, 作者采用醋酸钌[Ru(acac)3]作为催化剂, 1, 1, 1-三(二苯基膦甲基)乙烷(triphos)为配体, 通过添加甲基磺酸或氯化锂, 以较高产率得到一系列氮甲基化产物.同时, 作者进一步利用该方法成功制备出含13C标记的氮甲基化药物分子丙咪嗪(Imipramine, 1)和阿米替林(Amitriptyline, 2), 产率可以达到95%以上, 具有较好的应用前景.通过将还原剂苯硅烷替换为氢气, 不仅提高了该反应的原子经济型, 而且使得该转化方法更加绿色环保, 为甲胺类化合物的工业化生产提供了切实可行的解决方案.
2013年, Cantat课题组[15]在使用苯硅烷为还原剂时, 将金属更换为更加廉价易得的氯化锌, 使用其与卡宾的络合物IPrZnCl2作为催化剂, 以CO2为碳源实现了胺的氮甲基化反应(Scheme 3).作者在研究底物范围时发现, 当选用RNH2为底物时, 经过20 h的反应, 主要产物为RNHCH3, 有少量副产物RNHCHO和RN(CH3)3生成; 如果将反应时间延长至72 h, 所得到的产物则以N, N-二甲基胺为主.虽然该方法的反应活性、底物适用范围, 尤其是选择性都有待进一步提高, 但是该方法避免了使用昂贵的过渡金属, 为今后锌催化剂的发展奠定了基础.
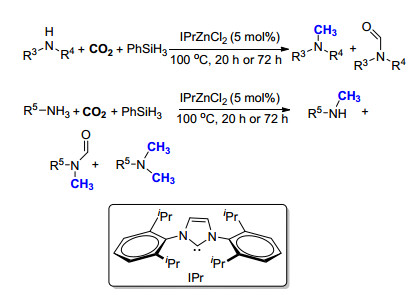
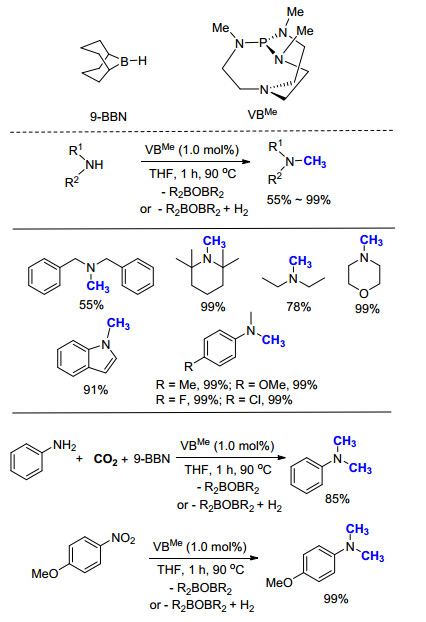
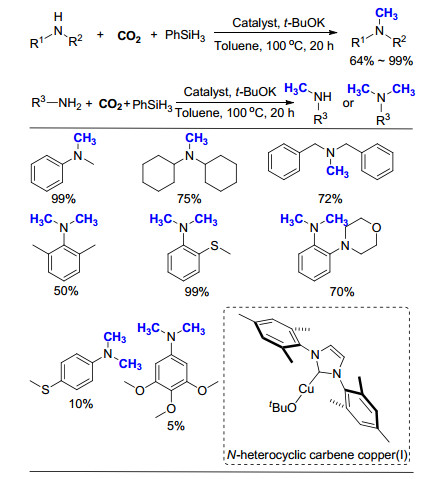
2014年, Cantat课题组[16]继续报道了有机小分子催化的氮甲基化反应, 以9-BBN为还原剂, Verkade超强碱(VBMe)为催化剂, 100 kPa CO2气体为C1来源, 以较高产率实现了一级胺和二级胺的氮甲基化反应(Scheme 4).该方法具有较高的反应活性, 仅需1 mol%的催化剂即可完成CO2到甲基的转化, 且具有很高的选择性, 在以一级胺为反应原料时, 产物均为N, N-二甲基胺.值得一提的是, 该方法以硝基苯为原料时, 可以利用一锅法的策略, 经过两步连续的转化过程, 以55%的产率得到N, N-二甲基苯胺.
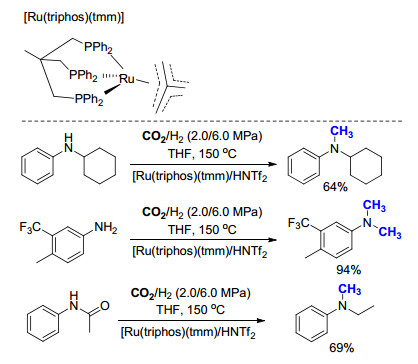
2013年, Klankermayer课题组[17]报道了以H2为还原剂、CO2为碳源, 钌催化芳香胺的氮甲基化反应(Scheme 5).作者使用triphos为配体, 其形成的钌络合物[Ru(triphos)(tmm)]为催化剂, 三氟甲烷磺酰亚胺(HNTf2)为添加剂, 成功催化了一系列一级和二级芳香胺的氮甲基化反应.若使用乙酰苯胺(PhNHAc)为起始原料时, 在最优反应条件下, 可发生连续的氢化反应和氮甲基化反应, 以69%的收率得到N-甲基-N-乙基苯胺[PhNH(CH3)(C2H5)].但是, 该方法的底物适用范围仅局限于各类芳香胺, 对于脂肪胺等其他类型的底物并未涉及.
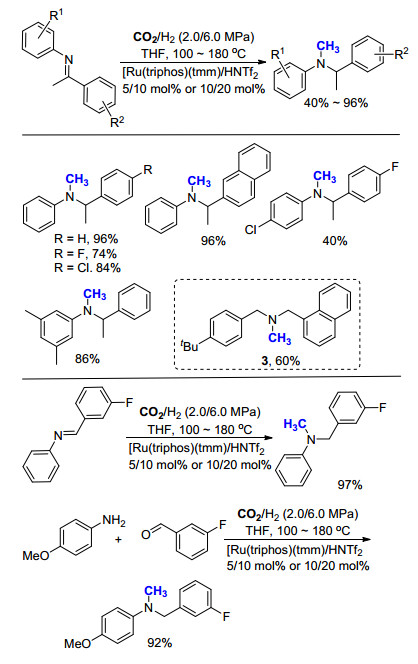
2014年, Klankermayer课题组[18]利用所发展的方法[17], 成功实现一系列亚胺的氮甲基化反应, 进一步扩大了底物的适用范围(Scheme 6).与芳香胺相比, 亚胺甲基化反应所需要的催化剂用量有所增加(2.5 mol%→5.0 mol%), 反应温度有所下降(150→100 ℃), 可适用于各种含有吸电子基团或推电子基团的亚胺类底物.为了实现该方法的应用, 作者将此方法应用于布替萘芬(butenafine, 3)的合成中, 其盐酸盐是一类常用的抗真菌类药物.传统的合成方法中, 以对叔丁基苯甲酸为起始原料, 经由四步转化制得, 产生大量的无机盐副产物, 同时需要使用大量溶剂.与之相比, 作者采用1-萘醛和对叔丁基苄胺为起始原料, 首先生成亚胺, 之后通过氮甲基化反应, 仅需两步即可得到布替萘芬(3), 产率高达88%.反应中需要用到的CO2和H2均为便宜易得的原料, 反应过程中所产生的副产物仅为H2O, 原子经济性达到85%, 为布替萘芬的高效合成提供了一种绿色环保的新方法.
2014年, 石峰课题组[19]利用硝酸铜和硝酸铝制备出CuAlOx作为催化剂, 可以有效催化各类一级胺和二级胺的氮甲基化反应(Scheme 7), 其中脂肪族的胺相比于芳香胺和环胺, 反应活性偏低.在考察一级胺底物时, 当使用气体的压力为3.0 MPa CO2, 6.0 MPa H2, 反应24 h后可选择性地生成单一甲基胺, 而当使用气体的压力为3.0 MPa CO2, 7.0 MPa H2, 反应48 h后则选择性地生成N, N-二甲基胺.通过改变H2的压力和反应时间, 可以改变产物结构的选择性, 是这项工作的一大亮点.此外, 利用相同的反应条件, 作者同样实现了对硝基苯和苯甲腈类化合物的还原甲基化, 得到的产物以N, N-二甲基胺为主.
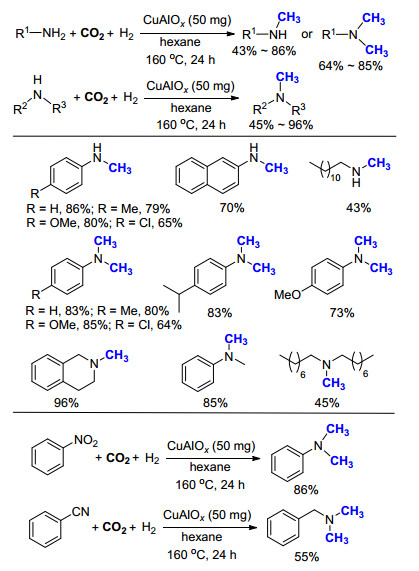
由于上述反应存在条件过于苛刻, 使用的CO2和H2压力过高的缺点, 不便于操作和工业化应用.该课题组继续将三种金属复合, 制备出Pd/CuZrOx非均相催化剂[20], 成功地将气体的压力降为1.0 MPa CO2, 2.5 MPa H2 (Scheme 8).在更温和的反应条件下, 得到了和之前相类似的底物适用范围.该体系的一个显著优点是无需添加任何配体或酸, 单一的催化剂即可高效地催化氮甲基化反应, 同时后处理简单, 产物易分离.他们还成功地将此方法应用于药物分子丙咪嗪(Imipramine, 4)的合成中, 产率达到85%.
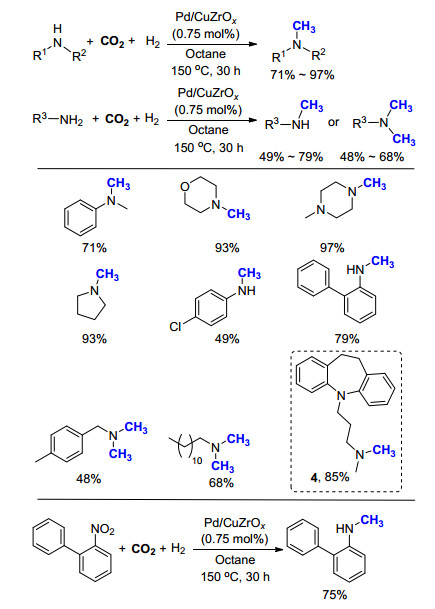
2014年, Shimizu课题组[21]将Pt与MoOx共同负载于TiO2上, 作为催化二级胺氮甲基化的催化剂(Scheme 9).通过条件筛选, 金属Pt的效果最好, 可达85%转化率, 其他金属如Pd、Ru、Ni、Cu几乎没有催化活性, 而且氧化物的成分对反应活性也有很大影响, 若将氧化物换为VOx、WOx或VOx, 反应活性明显大大降低.在此反应体系中, 复合型多金属催化剂起到关键作用.若只有单一金属负载, 如Pt/TiO2, Pt/Al2O3, 仅有极少量的产物生成, 而商品化Pt/C则完全不能催化该反应.该反应的优势在于使用的非均相催化剂在反应结束后, 可以通过回收再活化, 继续循环使用.在反应10次后, 依然可以保持较高的产率(80%~90%), TON为433, 大于已报道的均相钌催化体系(TON=80[14]和TON=40[17]).但是, 该反应底物范围有限, 仅给出10例二级胺的反应结果, 底物拓展也未涉及具有吸电子官能团的胺, 而且反应温度高达170~230 ℃, 反应时间需24 h, 因此此非均相催化体系的反应活性有待进一步提高.
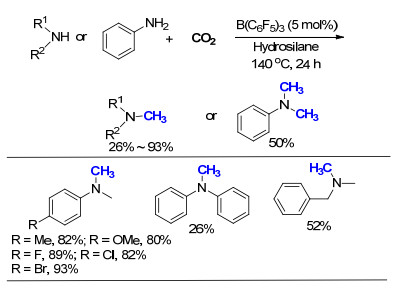
2015年, 刘志敏课题组[22]报道了一例无需金属催化的氮甲基化反应, 采用三(五氟苯基)硼烷[B(C6F5)3]作为催化剂, 以CO2为碳源, 在温和的条件下实现了二级胺的氮甲基化过程(Scheme 10).作者通过核磁共振1H NMR、11B NMR、19F NMR, 证明B(C6F5)3确实能够起到活化甲基苯胺和苯硅烷的作用, 之后通过条件筛选, 成功将CO2的压力降至0.5 MPa, 使得整个反应更加容易操作.底物拓展结果表明, N-甲基苯胺(PhNHCH3)的苯环上无论是接有吸电子基团还是推电子基团, 该反应都能顺利进行.作者认为N-甲酰苯胺(PhNHCHO)是该反应的中间产物, 而在动力学实验中, 甲酰苯胺的反应速度要大大高于甲基苯胺, 因此, 作者认为甲酰苯胺的形成是整个氮甲基化反应的决速步(rate-determine step).
先前的报道已经证实Zn与氮杂环卡宾配体(NHC)的络合物可以催化一级胺的氮甲基化反应[15]. 2015年, 刘志敏课题组[23]制备出含氟的氮杂环卡宾与锌的聚合物(F-PNHC-Zn) (Scheme 11), 并对氮甲基化反应的活性进行了考察.以N-甲基苯胺(PhNHCH3)为例, 虽然转化率达到100%, 但是产物中含有71%的甲酰化产物PhNH(CH3)CHO和27%的N, N-二甲基化产物PhNH- (CH3)2, 而且实验表明温度对该反应的化学选择性影响较大.这项工作证明含NHC配体的高分子骨架固载金属锌后, 可成功地应用于胺的氮甲基化反应, 所考察的底物主要为芳香二级胺.与其他方法相比, 该方法的反应活性和选择性仍有待进一步提高.

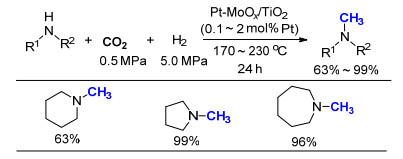
2016年, 在已有的工作基础上, 该课题组[24]继续合成了氮杂三苯基膦聚合物poly(PPh3)-azo (Scheme 12), 与Ag或Ru形成络合物后, 应用于CO2的转化和利用.其中, poly(PPh3)-azo-Ru在芳香二级胺的氮甲基化反应中展现了很高的催化活性, 仅需1 mol% Ru即可以99%的产率将N-甲基苯胺(PhNHCH3)转化为N, N-二甲基苯胺[PhNH(CH3)2].实验表明, 如果单独使用氯化钌(RuCl3)和三苯基膦(PPh3), 反应的产率降为64%, 说明高分子的骨架对于提高Ru的催化活性起着至关重要的作用.扫描电子显微镜(TEM)和X射线光电子能谱(XPS)测试表明, poly(PPh3)-azo-Ru催化剂在反应前后性质没有太大的改变, 具有很好的稳定性, 同时感应耦合等离子体(ICP)测试也表明, 在反应过程中没有金属Ru的脱除.将配体嵌入高分子骨架中, 作为金属载体制备催化剂, 这一策略为发展可应用于氮甲基化反应的非均相催化剂提供了研究方向和思路.
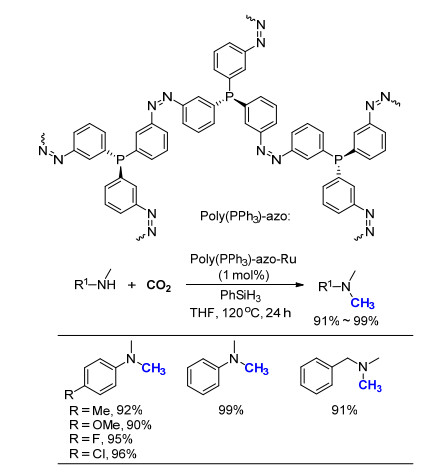
利用Cantat所发展的方法[15], 以锌的卡宾络合物IPrZnCl2为催化剂, Billard课题组[25]于2015年利用11CO2为C1来源, 通过氮甲基化反应制备了一系列含11CH3的芳香胺化合物(Scheme 13).由于11C (t1/2=20.4 min)半衰期比18F要短, 其使用范围没有18F广泛, 但是含有11C的化合物仍具有重要的医学和研究意义.近期所发展的氮甲基化反应为在结构中引入11C提供了直接有效的解决方案, 成功制备出了含11C标记的药物分子丙咪嗪(Imipramine, 5)、麻黄素(Ephedrine, 6)和β-蛋白放射示踪剂PIB 7.相较于CO2的反应, 11C标记的产物胺普遍产率偏低, 但是这一工作为氮甲基化反应的方法提供了重要的应用实例, 为合成11C标记的药物分子开辟了新的途径.
2015年, 王建强课题组[26]以γ-Al2O3为载体, 制备出直径约为2.0 nm的金纳米粒子, 成功应用于一系列胺的氮甲基化反应中(Scheme 14).该方法采用CO2为C1来源, H2为还原剂, 高效地将一系列芳香胺和脂肪胺转化为相应的甲基化产物.体系中使用的非均相催化剂具有很高的反应活性, 仅使用0.5 mol%的Au反应7 h即可完成转化, 但是转化过程中所需要的气体压力略高(CO2 2 MPa, H2 6 MPa).
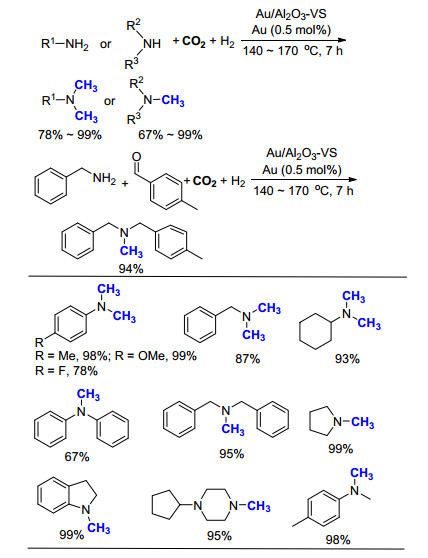
此外, Juventino等[27]研究发现, 零价镍的络合物[(dippe)-Ni(μ-H)]2和已商品化的Ni(COD)2/dcype在一级和二级脂肪胺的氮甲基化反应中都表现出非常高的反应活性(Scheme 15).在此条件下, 当以一级脂肪胺为底物时, 可以选择性地得到甲基胺产物, 同时伴随着酰胺副产物的生成.该反应仅需要使用101 kPa的CO2即可实现转化, 但是反应的产率(33%~69%)和选择性有待进一步提高.
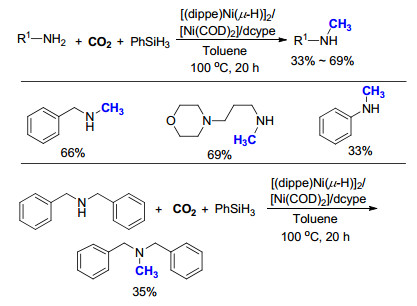
相比于贵金属, 金属铜更加廉价易得, 更具有工业化应用前景. 2015年, Cazin等[28]使用氮杂环卡宾为配体, 与一价铜形成络合物.如Scheme 16所示, 在同样使用CO2为C1来源, 苯硅烷(PhSiH3)为还原剂的情况下, 一级胺和二级胺的氮甲基化产物均可以在温和的条件下生成.在研究中, 作者发现, 如果向体系中加入10 mol%的强碱KOtBu, 一些底物的反应性会有显著提高.但是, 当使用一级胺作为底物时, 某些底物的选择性并不是非常好.例如以2, 6-二甲基苯胺为底物时, N, N-二甲基胺的产率50%, 为主要产物, 同时也检测到有19%的甲基胺生成.此外, 该反应底物适用范围仍需进一步拓展, 对于一些特定底物, 如对硫甲基苯胺、3, 4, 5-三甲氧基苯胺, 反应的产率较低, 可能因为取代基和金属铜之间存在着一定的配位作用.因此, 铜催化氮甲基化反应的催化活性和底物适用范围等仍需进一步完善.
在以往的报道中, 氮甲基化反应往往需要金属催化剂的参与.与之相对, 2015年, Ong课题组[29]制备出一种非对称结构的碳二卡宾(Scheme 17), 并进一步成功将其应用于催化以硼氢化物(9-BBN)为还原剂的氮甲基化反应.该反应在使用一级胺为底物时, 可选择性得到单一氮甲基化的产物.机理研究表明, 该催化体系的活性成分为碳二卡宾与硼烷原位形成的加合物.碳二卡宾由于其强供电性, 中心碳原子可被双质子化, 且可与过渡金属形成双配位, 被广泛应用于二氧化碳的活化和催化转化中.
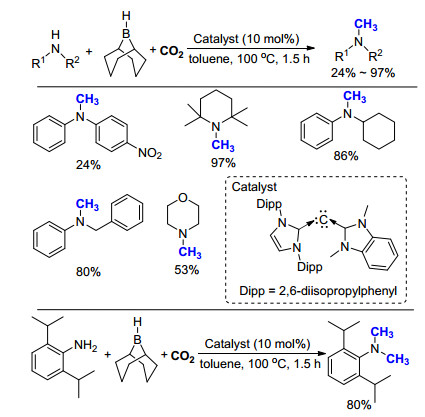
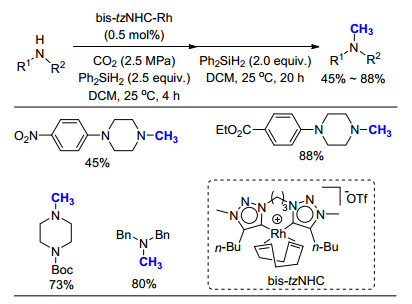
Kobayashi课题组长期致力于开发新型氮杂环卡宾与金属铑的络合物. 2016年, 他们[30]报道了一种以三氮唑卡宾(tzNHC, tz=1, 2, 3-triazol-5-ylidene)为配体的金属铑络合物, 由于配体具有非常强的给电子效应, 会增加铑氢中间体的亲核性, 加速从中间体向产物的氢转移, 从而提高反应活性.实验表明, 当使用2.53 MPa CO2时, 二级胺的氮甲基化反应可以顺利的进行(Scheme 18), 产率为45%~88%.基于类似的反应机理, 他们利用苯硅烷作为还原剂, 在相同的催化条件下, 成功实现了对一系列酰胺和羧酸底物羰基的还原.
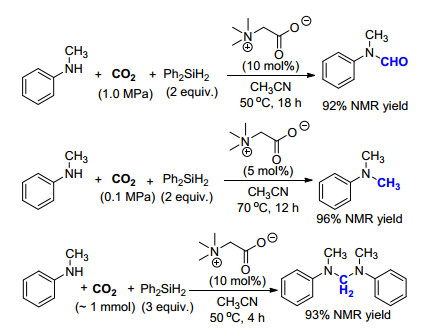
除超强碱[16]以外, 2017年何良年课题组[31]报道了一例有机小分子催化的氮甲基化反应.作者使用甜菜碱作为催化剂, 无需使用任何金属, 成功地实现了二级胺的氮甲基化反应, 该方法更加绿色环保, 也简化了后处理步骤.值得注意的是, 当采用不同的反应条件, 如CO2的用量及压力、反应温度等因素时, 会极大地影响反应中间体的转化程度和最终的产物结构.以甲基苯胺为原料时, 在1.0 MPa CO2, 50 ℃下反应主要得到甲酰化的产物, 在0.1 MPa CO2, 70 ℃下反应主要得到氮甲基化的产物, 而当减少CO2用量至1 mmol, 50 ℃下反应则主要得到二胺产物(Scheme 19).基于这一实验结果, 作者推断反应的机理如下:当CO2压力较大时, 主要生成的中间体为HCOOSiHPh2, 与甲基苯胺反应得到甲酰化产物; 当CO2压力较小或减少CO2用量时, 中间体主要以二聚体CH2(OSiHPh2)为主, 与甲基苯胺反应后得到二胺产物, 该产物经过甜菜碱的进一步催化, 可进一步转化为氮甲基化的产物.该方法不仅实现了无过渡金属催化的氮甲基化反应, 而且通过改变实验条件, 可以选择性地生成三种目标产物, 极大地推进了此类反应的发展和应用.
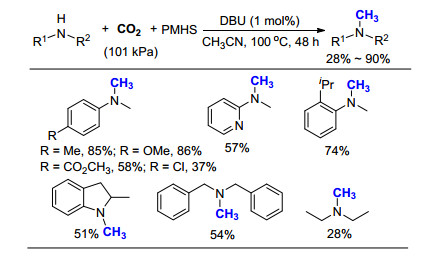
2018年, 夏纪宝课题组[32]发现单一的有机碱二环[4.3.0]-1, 5-二氮-5-十一烯(DBU), 仅需1 mol%催化剂用量, 101 kPa CO2下即可高效催化二级胺的氮甲基化反应(Scheme 20).该方法最大的特色是未使用苯硅烷作为还原剂, 而是使用价格相对低廉的聚甲基硅烷(PMHS)作为还原剂, 大大降低了成本, 是目前使用CO2作为C1来源时最为简便的方法, 但是相对其反应活性仍需进一步提高, 尤其是脂肪胺的产率仍然偏低.
甲酸在自然界中来源十分广泛, 如蚂蚁的分泌物, 植物的叶和根以及水果, 被大量应用于各类工业产品的生产中.同时, 甲酸也可以经由甲酸甲酯水解或CO2氢化反应, 非常简便地制得.甲酸为无毒的液体, 可以被用作保存食品的防腐剂.在有机合成中, 甲酸可用于氢转移反应, 也被证明是一个潜在的储氢材料.尽管利用CO2作为C1来源的氮甲基化反应已有报道, 但是其反应条件通常需要高温和加压, 不利于实验室大规模合成.而如果能够使用无毒、生物降解性好的甲酸作为C1来源, 不仅简化了实验操作, 也进一步优化了氮甲基化的反应条件, 扩大了反应的适用范围.基于这一目标, 利用甲酸作为C1来源的均相和非均相催化条件也相继被报道, 其所参与的氮甲基化反应也表现出优良的反应活性和化学选择性.
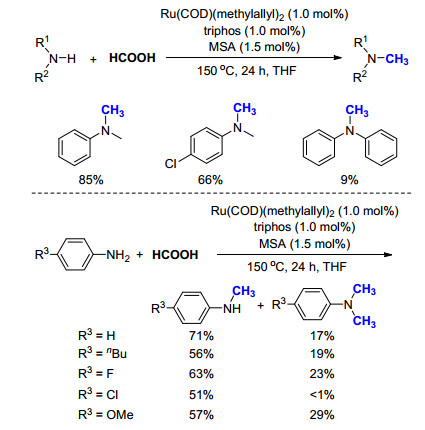
2014年, Beller课题组[33]报道了一例利用甲酸作为C1来源的氮甲基化反应(Scheme 21).作者首先对一系列金属催化剂进行筛选, 发现金属铂的化合物效果最好, 其中商品化的Karstedt催化剂在不使用任何配体的情况下, 仅需0.5 mol%即可有效实现甲基苯胺的氮甲基化反应(>99% GC yield).增加苯硅烷的用量, 在相同条件下对一级胺PhNH2进行考察, 同样以高产率(90% GC yield)得到N, N-二甲基胺PhN(CH3)2.进一步通过对商品化配体的筛选, 发现在使用1, 3-双(二苯基膦基)丙烷(dppp)时, 一级胺的收率可以进一步提高(>99% GC yield).由此可见, 在使用甲酸作为C1来源时, Pt/dppp表现出相当优异的催化性能.
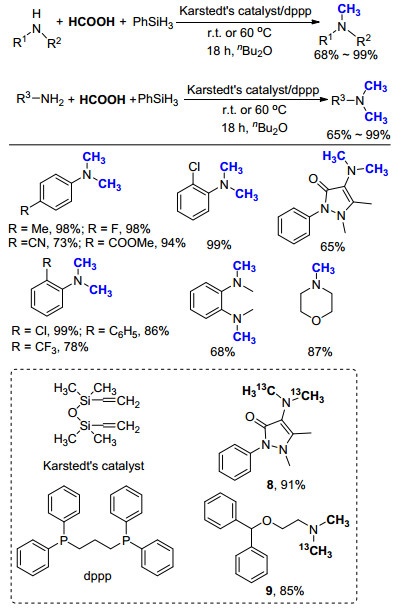
这一结果在底物拓展中再次得到了证实, 无论使用一级胺或二级胺、脂肪胺或芳香胺、含吸电子基团或推电子基团、是否有空间位阻, 均可以非常高的产率得到目标产物, 而且反应条件十分温和(Scheme 21).对于活性底物, 在室温下即可反应, 对于相对困难底物, 只需要将温度提高至60 ℃便可得到不错的产率.而后, 作者使用价格相对低廉的H13COOH作为碳源, 成功在4-氨基安替吡啉和2-(二苯基甲氧基)-N-甲基乙胺两个药物分子中引入13CH3, 以91%和85%产率得到13C标记的化合物8和9, 其具有潜在的医用价值.这项工作不仅证明HCOOH同样可以作为氮甲基化反应的C1来源, 而且为后续的研究开辟了道路.接下来的工作主要集中于降低催化剂成本、研究反应活性与选择性、扩大生产规模以及工业化应用等方面.
2014年, Cantat等[34]利用甲酸作为碳源, 报道了钌催化的氮甲基化反应, 但是该反应存在着较大的局限性.首先, 反应的活性不高, 需要使用高达6 equiv. HCOOH, 反应温度需要150 ℃.当底物为一级胺时, 反应的选择性不够, 会同时得到甲基胺和N, N-二甲胺, 限制了该方法的应用范围(Scheme 22).其次, 底物拓展结果表明, 该反应的催化活性严重依赖于底物的官能团, 例如同样为一级胺, 以甲基苯胺为原料时产率为85%, 以4-氯甲基苯胺为原料时产率为66%, 而当使用二苯胺时产率仅为9% (Scheme 22).作者通过理论计算对反应机理进行了研究, 认为在反应过程中, HCOOH不仅充当了C1的来源, 而且HCOOH也可以在Ru催化剂作用下脱氢生成CO2, 所生成的CO2也同样作为C1来源参与了甲基化过程.
2015年, 傅尧课题组[35]以甲酸为碳源, 报道了无需金属催化剂, 而是采用B(C6F5)3为催化剂的氮甲基化反应(Scheme 23).值得一提的是, 该反应的还原剂从传统使用的苯硅烷变为价格相对低廉的聚甲基硅烷(PMHS), 大大降低了反应成本, 提高了该方法的应用价值.利用所发展的方法, 作者从4-叔丁基苄胺出发, 经过两步反应, 以91%的总收率制备得到已商品化的药物布替萘芬(butenafine, 3).该工作最大的亮点在于, 如果将甲酸换为其他的羧酸, 氮甲基化反应可以转变为氮烷基化反应.作者利用这一策略, 成功制备出商品化药物分子吡贝地尔(piribedil, 10)和西那卡塞(cinacalcet, 11), 且都具有较好的产率.
非均相催化剂相较于均相催化剂而言, 往往具有分离简单、操作方便、可回收利用等优点[36-37]. 2016年, 我们课题组[38]报道了商品化Pt/C非均相催化的胺和亚胺的氮甲基化反应(Scheme 24).通过对反应条件的优化, 发现当使用甲苯作为溶剂时, 效果最佳, 可以将PhNH- CH3全部转化为PhN(CH3)2, 产率为97%, 而且几乎没有副产物PhN(CH3)CHO的生成, 选择性非常好.通过过滤实验及ICP残留金属检测, 证实该反应所经历的确为非均相催化的过程.该反应体系对于吸电子或推电子基团取代的一级胺或二级胺都有不错的效果, 并且能够顺利地催化空间位阻相对较大的各类亚胺底物, 进一步扩大了反应的适用范围.当利用H13COOH为碳源时, 成功地在目标分子中引入13CH3, 为发展同位素标记药物提供了新的方法.这项工作中, 整个反应为非均相, 仅需要使用很低的催化剂用量, 且无需额外添加配体, 即可实现反应物较高的转化数, TON为1700, 而且催化剂在反应结束后可以很方便地由过滤除去.同时, 该方法使用商品化催化剂, 反应条件温和, 简便易操作, 有利于实际生产中的应用.
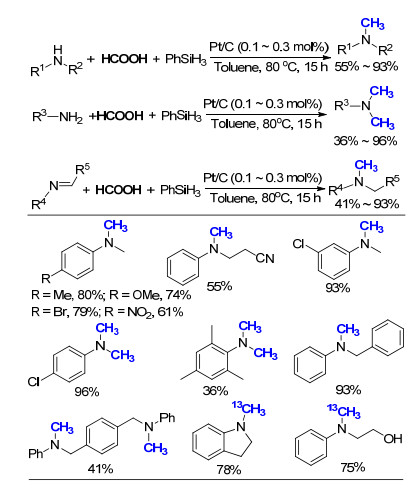
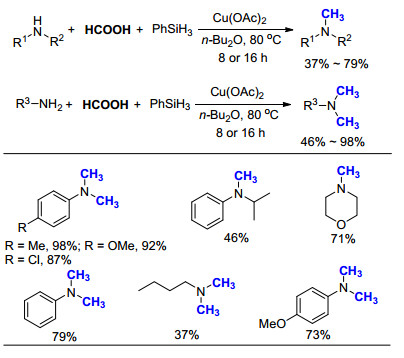
已有报道中, 以甲酸为C1来源, 过渡金属催化的氮甲基化反应主要集中使用Pt[33]、Ru[34]、Ir[39]等贵金属, 以及我们所发展的非均相Pt/C[38]催化剂.在2017年, 何良年课题组[40]取得了重大突破, 使用价格相对低廉的Cu(OAc)2作为催化剂, 成功实现了胺的氮甲基化反应(Scheme 25).以PhNHCH3为模板底物, 在使用Cu(OTf)2, CuF2和Cu(OAc)2为催化剂时, 目标产物N, N-二甲基苯胺的GC产率均为99%, 最终作者选择价格较低的Cu(OAc)2进行底物范围研究.除硝基甲基苯胺外, 芳香胺和脂肪胺均可以顺利地完成转化, 相比而言脂肪胺产率稍低.该体系最大的特色在于, 使用Cu(OAc)2作为催化剂, 不仅大大降低了反应成本, 而且对环境友好, 符合绿色化学的需求.
甲醇作为一种来源丰富的常用溶剂, 被广泛应用于药物合成和材料制备中.在有机合成中, 甲醇作为重要的C1来源, 可参与各种类型的反应, 如碳甲基化反应、碳甲氧基化反应、氮甲酰化反应、甲氧羰基化反应、氧化甲酯化反应和氮甲基化反应, 用于构建碳碳键、碳氧键和碳氮键[41-43].其中, CH3OH在胺和亚胺氮甲基化反应中的应用, 近年来取得了很大的发展.
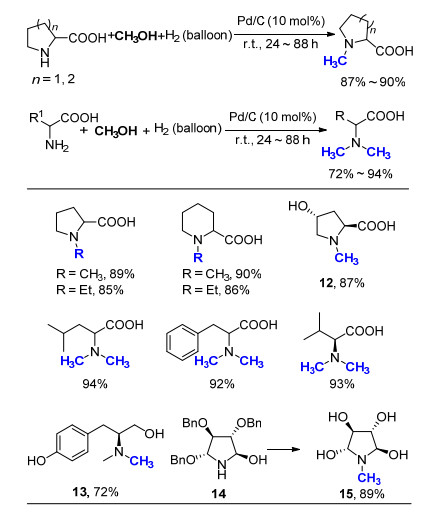
2010年, 黄培强课题组[44]以CH3OH为C1来源, 氢气为还原剂, 报道了一例Pd/C催化氨基酸的氮甲基化反应(Scheme 26).该反应在室温下即可进行, 当氨基酸反应底物为一级胺时, 可选择性地生成N, N-二甲基化产物, 即使底物中含有羟基, 该反应依然能够顺利进行, 如产物12和13.将CH3OH更换为EtOH, 同样的底物也可发生氮乙基化反应, 取得不错的产率.当反应底物含有Bn保护基, 如四氢吡咯衍生物14, 在此反应条件下, 可一步完成脱除苄基和氮甲基化的过程, 以89%产率得到含有四个羟基的氮甲基化产物15. 2017年, Feringa和Barta等[45]利用金属钌与茂环的络合物为催化剂, 着重研究了氨基酸的氮烷基化反应.他们使用了近二十余种不同类型的脂肪醇, 均取得了令人满意的结果, 为氨基酸的官能化提供了一种新方法.
通常, 胺和亚胺的氮甲基化反应都需要在加热的条件下进行, 2015年, 石峰课题组[46]扩大了CH3OH碳源的适用范围, 以一级胺和二级胺为反应底物, 报道了一例室温下光催化的氮甲基化反应(Scheme 27).作者使用Pd/TiO2为光催化剂, CH3OH为C1来源, 无需额外的还原剂, 即可很好地催化一级胺和二级胺的氮甲基化反应.这是因为甲醇在光引发的作用下, 首先生成HCHO和Pd-H活性中间体, 之后HCHO与胺生成亚胺, 亚胺再经Pd-H还原最终得到甲基化产物.如果以硝基苯为起始原料, 则无需添加额外试剂, 在相同的条件下可一步得到PhN(CH3)2.虽然该反应历经光催化的过程, 但是含有卤素的苯胺(4-ClC6H4NH2、4-BrC6H4NH2)依然可以顺利地转化为相应的甲基化产物.
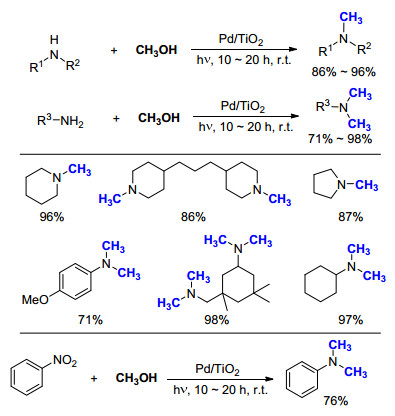
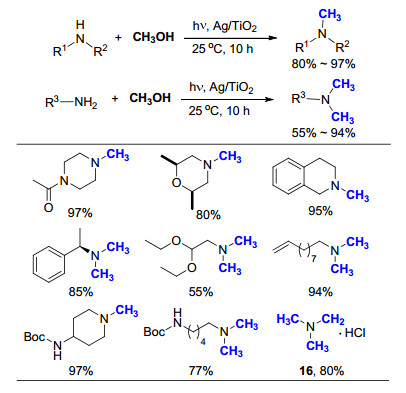
2015年, Saito等[47]报道了室温下以甲醇为C1来源, Ag/TiO2光催化胺的氮甲基化反应(Scheme 28).发现催化剂Ag/TiO2十分稳定, 可以在室温、空气下保存三个月以上, 而且底物适用范围很广.例如, 在最优化条件下, 含有N-Boc基团的一级胺和二级胺均可以高效地完成转化, 保护基不会受到影响; 当使用氨水为底物时, 也顺利地得到了相应的三甲基化产物, 经盐酸酸化后得到产物16.与之相较, Au/TiO2, Pd/TiO2或Pt/TiO2在类似的条件下会发生诸如双键被还原, N-Allyl基团脱除等副反应.
2015年, Seayad小组[48]报道了在以[RuCp*Cl2]2和dpePhos原位形成的配合物为催化剂、CH3OH为C1来源、5 mol% LiOtBu为添加的条件下, 芳香和脂肪一级胺以及磺酰胺均可以高效发生氮甲基化反应(Scheme 29).值得注意的是, 同样以甲醇为溶剂和碳源, 芳香类一级胺和磺酰胺选择性地生成了甲基胺产物, 而脂肪一级胺的产物则以N, N-二甲基胺为主.作者成功运用这一方法, 制备得到了天然产物(-)-N-methylephedrine (17)和药物分子萘替芬(Naftifine, 18).在研究中还发现, 如果将甲醇换为乙醇, 氮乙基化反应同样可以发生, 但是产率非常低.在催化循环中, 碱LiOtBu的添加是至关重要的, 能够将CH3OH活化为CH3OLi, 与Ru形成Ru-OCH3中间体后, 再脱H生成HCHO.事实上, 在很多以CH3OH为碳源的催化体系中[43], 反应都会首先生成HCHO, 与胺形成亚胺中间体后, 再经过还原剂或M-H还原, 最终得到氮甲基化产物.

2015年, Crabtree小组[49]报道了一例Ir-NHC催化, 以甲醇为溶剂, 微波作用下芳香胺的氮甲基化反应(Scheme 30).该方法表现出十分优秀的选择性, 对于一系列不同取代的苯胺均高选择地生成单一氮甲基化的产物, 副产物N, N-二甲基产物含量低于5%.底物适应性研究表明, 吸电子基团如NO2、CF3影响苯胺的亲核性, 从而降低反应的活性.
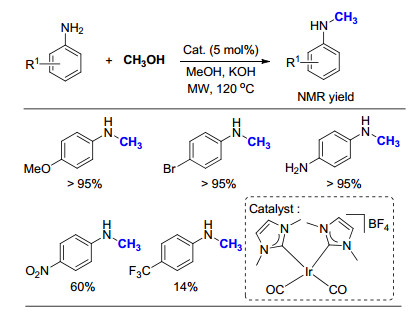
2016年, Beller小组[50]制备了一系列相比于贵金属价格相对低廉的锰配合物, 在研究苯胺与苄醇的氮烷基化反应时, 发现P的取代基对反应活性影响很大, 其中异丙基效果最佳(Scheme 31).进而将该催化剂应用于取代苯胺的氮甲基化反应时, 发现具有十分良好的底物适用范围, 各种取代的苯胺均可以选择性地生成相应的甲基苯胺产物.但是, 该反应需要添加100 mol%的t-BuOK, 以CH3OH为溶剂才能够顺利进行.
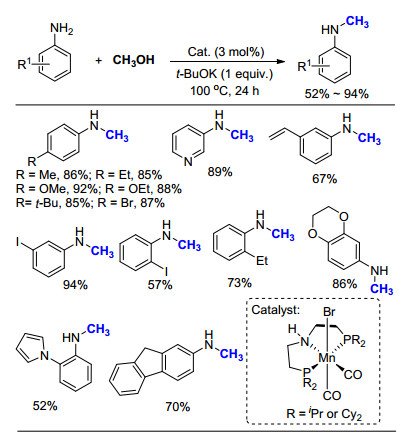
2017年, Sortais等[51]报道了Mn和Pincer的络合物(Scheme 32), 在添加20 mol% KOtBu的条件下, 以CH3OH为C1来源, 可以选择性地将一级胺转化为甲基胺产物.该方法是在先前Beller工作基础上[50]进行了改进, 通过改变Mn络合物的配体结构, 引入吡啶单元, 成功将t-BuOK的用量从100 mol%降低至20 mol%.
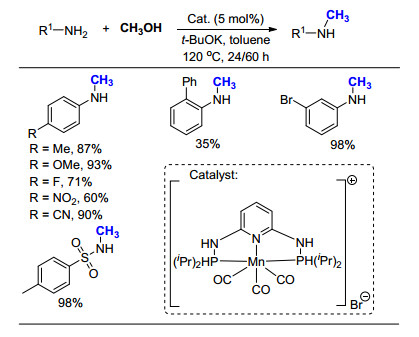
2017年, 刘志敏课题组[52]以甲醇为甲基化试剂, 报道了基于Co(acac)2的催化体系(Scheme 33).为了提高反应产率, 需要向反应体系中添加K3PO4, 并且使用P(CH2CH2PPh2)3配体.当以二级胺为原料时, 芳香胺和脂肪胺均可以高产率地实现氮甲基化; 当以脂肪一级胺为原料时, 因为CH3OH作为溶剂和甲基化试剂大量存在, 得到的产物主要以N, N-二甲基胺为主; 但是当以芳香一级胺为原料时, 如苯胺及其取代物, 即使增加K3PO4的用量, 产物仍然以甲基苯胺产物为主.作者经过实验发现, 这是由于苯胺的单一氮甲基化产物PhNHMe在此条件下, 无法进一步被甲基化所导致.这一结果不仅说明芳香和脂肪一级胺在此条件下反应活性是不同的, 而且说明本方法仅适用于脂肪族二级胺.
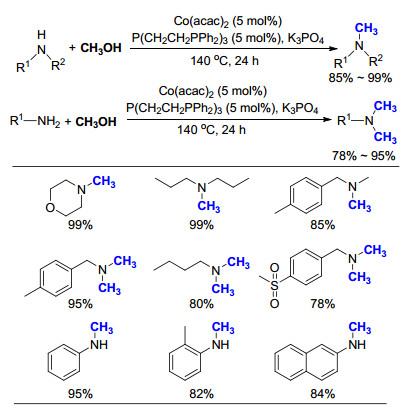
2019年, Natte等[53]报道了利用商业化Pd/C催化的氮甲基化反应, 通过添加2倍的tBuOK, 在130 ℃反应12 h, 可实现胺的氮甲基化反应(Scheme 34).在反应过程中, 无需添加额外的还原剂, CH3OH同时作为C1和H2的来源, 参与到整个催化循环中.该方法可适用于芳香胺和非芳香胺, 但是对于一级胺的选择性不是很高:环己胺的产物为N-甲基产物, 而对羟基苯乙胺的产物为N, N-二甲基产物.
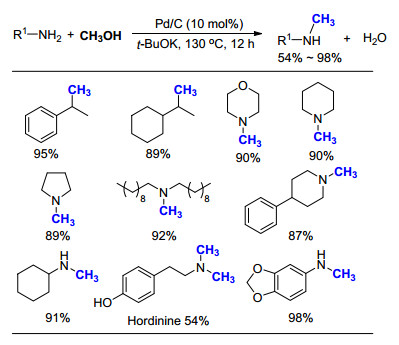
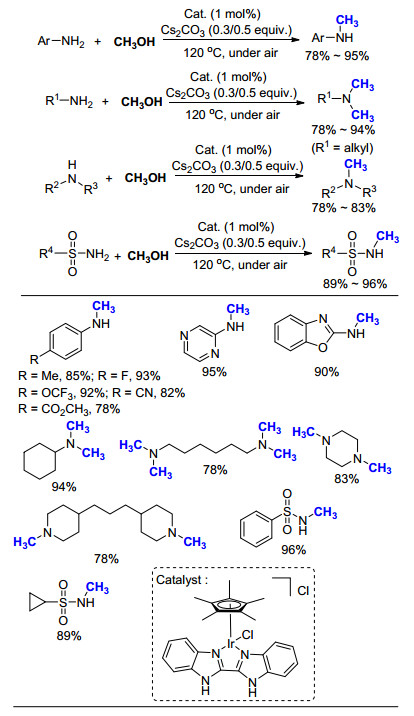
同样以CH3OH为C1来源, 李峰课题组[54]将催化剂种类拓展至Cp*Ir的络合物(Scheme 35).先前我们课题组也报道了Ir配合物在光催化反应中的应用[55].通过添加一定量的碱Cs2CO3, 无需惰性气体保护, 在空气下即可以实现胺类化合物的氮甲基化反应.与Co的催化体系相同[50], 芳香一级胺的主要产物为甲基胺, 而脂肪一级胺的主要产物为N, N-二甲基胺.当使用磺酰胺作为底物时, 该方法同样取得了不错的效果.可以看出, 以CH3OH为碳源所发展的方法中, 除了光催化过程外, 其他反应都需要在较高温度(>100 ℃)下进行, 催化剂的活性有待进一步提高.
随着保护社会环境意识的增强, CO2的转化和利用成为热门的研究领域, 因此近年来胺和亚胺氮甲基化反应的发展主要集中在使用CO2和CO2的加氢产物HCOOH, 以及甲醇作为C1来源.除此之外, 其他碳源, 如二甲基亚砜(DMSO)、多聚甲醛等也可以作为氮甲基化反应的C1来源, 相应的催化体系在文献中被陆续报道.
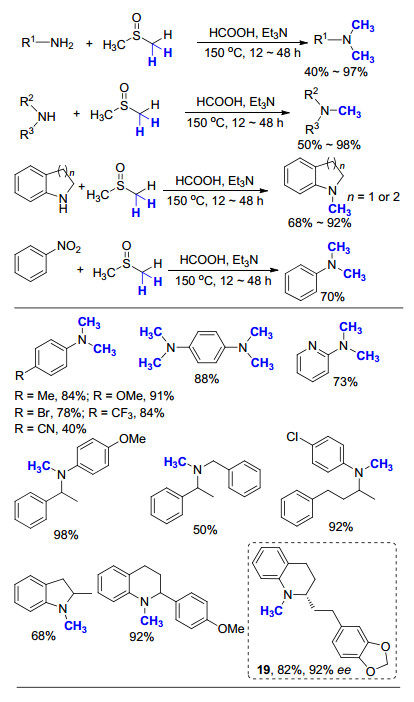
2014年, 肖建良课题组[56]报道了一例Et3N/ DMSO/HCOOH的反应体系, 在一级胺和二级胺的氮甲基化反应中均有不错的效果(Scheme 36).通过控制实验, 推测产物中的甲基主要来自于DMSO, DMSO既作为溶剂又作为甲基化试剂, 而所引入的CH3中有两个H来自DMSO, 一个H来源于HCOOH.在这项工作中, 作者通过在氮甲基化反应中添加FeCl2•7H2O, 成功实现了硝基苯的一锅法转化, 首先将硝基苯还原为苯胺, 之后再进行苯胺的氮甲基化, 两步转化的总体产率最高, 可达89%.这一策略不同于之前讲述的过渡金属的催化过程, 因此反应需要在较高温度下(150 ℃)进行, 并且需要使用大大过量的HCOOH和Et3N, 转化的原子经济性不高.此外, 作者将此方法应用于Galipinine (19)的全合成中, 发现此一级胺的手性中心在氮甲基化反应前后基本保持不变(95%→92% ee).
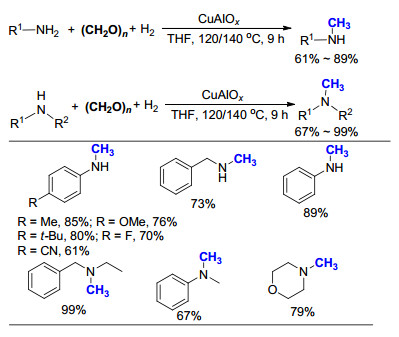
2013年, 石峰课题组[57]利用多聚甲醛为C1来源, 利用CuAlOx为催化剂, H2为还原剂, 报道了一系列胺的氮甲基化反应(Scheme 37).该反应可以很好地适用于一级胺和二级胺, 其特色在于当使用一级胺为底物时, 反应可以选择性地得到甲基胺产物.而该反应的局限性在于, 此体系无法适用于长烷基链的一级胺, 催化活性偏低.
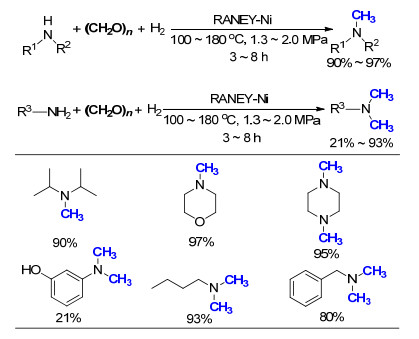
2014年, 钱超课题组[58]报道了RANEY-Ni催化的氮甲基化反应, 以多聚甲醛为C1来源, H2为还原剂, 反应的副产物仅仅为水(Scheme 38).值得注意的是, 这一催化体系对于脂肪胺具有非常好的活性, 这是其他催化体系较难达到的.作者通过理论计算认为, 反应的机理是多聚甲醛首先与胺作用生成亚胺中间体, 之后通过H2还原得到甲基化产物.该反应的一大特色在于, 催化剂RANEY-Ni在反应结束后, 可以通过简单过滤分离, 甲醇冲洗回收再利用, 循环使用四次后, 催化剂依然保持活性, 产率仅略微降低.
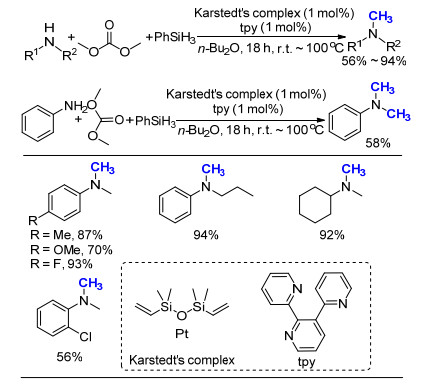
先前, Beller课题组[59]利用商品化的Karstedt催化剂, 成功发展出以HCOOH为C1来源的氮甲基化反应[33].在此基础上, 他们发现利用相同的催化剂, 碳酸二甲酯也可以作为甲基化试剂参与一级胺的氮甲基化反应(Scheme 39).通过对反应条件的优化, 作者发现通过添加2, 2':6', 2''-terpyridine (tpy)作为配体后, 反应活性得到大幅提升, 在室温下即可有效完成转化.在这项工作中, 作者主要考察了二级胺底物, 产率在56%~94%之间, 仅给出一例一级胺即苯胺, 产率为58%.总体比较而言, 即使在添加额外配体tpy的情况下, 碳酸二甲酯的活性还要低于HCOOH, 但是这项工作最重要的意义是, 证实了碳酸二甲酯同样可以作为甲基化试剂.
胺和亚胺的氮甲基化反应为天然产物和药物分子的制备及修饰提供了重要的方法, 也为发展同位素标记药物建立了有效途径, 逐渐成为近年来研究热点之一.总体而言, 胺和亚胺氮甲基化反应的发展是伴随着所使用的甲基化试剂种类而改变的, 从多聚甲醛、甲醇到CO2、HCOOH, 根据不同的C1来源, 一系列使用不同催化剂、不同反应条件的方法陆续被报道出来, 其反应的活性和选择性也略有不同.该反应的发展过程基本沿着降低成本, 更加符合绿色环保需求的方向进行, 例如在催化剂的选取中, 从贵金属Pt、Ru、Rh、Ir等过渡到Cu, 从均相催化剂过渡到可回收利用的非均相催化剂; 在C1来源上, CO2无疑是最佳选择, 但是较高的反应压力又不利于大规模合成, 使得人们又发展出HCOOH作为替代物.在将来的发展中, 胺和亚胺氮甲基化反应的反应活性和选择性仍有待进一步提高, 尤其对于脂肪胺类底物.此外, 非均相催化体系仍需继续发展, 力求实现催化剂的循环和回收再利用, 降低反应成本, 简化操作步骤, 为实际生产和工业化应用奠定基础.
Bissember, A. C.; Lundgren, R. J.; Creutz, S. E.; Peters, J. C.; Fu, J. C. Angew. Chem., Int. Ed. 2013, 52, 5129. doi: 10.1002/anie.201301202
Cho, S. H.; Kim, J. Y.; Lee, S. Y.; Chang, S. Angew. Chem., Int. Ed. 2009, 48, 9127. doi: 10.1002/anie.200903957
Tanaka, R.; Yamashita, M.; Nozaki, K. J. Am. Chem. Soc. 2009, 131, 14168. doi: 10.1021/ja903574e
Jacquet, O., Gomes, C. D. N., Ephritikhine, M.; Cantat, T. J. Am. Chem. Soc. 2012, 134, 2934. doi: 10.1021/ja211527q
Arakara, H.; Aresta, M.; Armor, J. N.; Barteau, M. A.; Beckman, E. J.; Bell, A. T.; Bercaw, J. E.; Creutz, C.; Dinjus, E.; Dixon, D. A.; Domen, K.; DuBois, D. L.; Eckert, J.; Fujita, E.; Gibson, D. H.; Goddard, W. A.; Goodman, D. W.; Keller, J.; Kubas, G. J.; Kung, H. H.; Lyons, J. E.; Manzer·, L. E.; Marks, T. J.; Morokuma, K.; Nicholas, K. M.; Periana, R.; Que, L.; Rostrup-Nielson, J.; Sachtler, W. M. H.; Schmidt, L. D.; Sen, A.; Somorjai, G. A.; Stair, P. C.; Stults, B. R.; Tumas, W. Chem. Rev. 2001, 101, 953. doi: 10.1021/cr000018s
Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365. doi: 10.1021/cr068357u
Wang, W.; Wang, S.-P.; Ma, X.-B.; Gong, J.-L. Chem. Soc. Rev. 2011, 40, 3703. doi: 10.1039/c1cs15008a
Fernández-Alvarez, F. J.; Aitani, A. M.; Oro, L. A. Catal. Sci. Technol. 2014, 4, 611. doi: 10.1039/C3CY00948C
Gomes, C. D. N.; Jacquet, O.; Villiers, C.; Thuéry, P.; Ephritikhine, M.; Cantat, T. Angew. Chem., Int. Ed. 2012, 51, 187. doi: 10.1002/anie.201105516
Khandelwal, M.; Wehmschulte, R. J. Angew. Chem., Int. Ed. 2012, 51, 7323. doi: 10.1002/anie.201201282
Jacquet, O.; Gomes, C. D. N.; Ephritikhine, M.; Cantat, T. ChemCatChem 2013, 5, 117. doi: 10.1002/cctc.201200732
Li, Y.; Fang, X.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 9568. doi: 10.1002/anie.201301349
Tlili, A.; Frogneux, X.; Blondiaux, E.; Cantat, T. Angew. Chem., Int. Ed. 2014, 53, 2543. doi: 10.1002/anie.201310337
Li, Y.; Sorribes, I.; Yan, T.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 12156. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM24115562
Jacquet, O.; Frognuex, X.; Gomes, C. D. N.; Cantat, T. Chem. Sci. 2013, 4, 2127. doi: 10.1039/c3sc22240c
Blondiaux, E.; Pouessel, J.; Cantat, T. Angew. Chem., Int. Ed. 2014, 53, 12186. doi: 10.1002/anie.201407357
Beydoun, K.; Stein, T. v.; Klankermayer, J.; Lertner, W. Angew. Chem., Int. Ed. 2013, 52, 9554. doi: 10.1002/ange.201304656
Beydoun, K.; Ghattas, G.; Thenert, K.; Klankermayer, J.; Leitner, W. Angew. Chem., Int. Ed. 2014, 53, 11010. doi: 10.1002/anie.201403711
Cui, X.; Dai, X.; Zhang, Y.; Deng, Y.; Shi, F. Chem. Sci. 2014, 5, 649. doi: 10.1039/C3SC52676C
Cui, X.; Zhang, Y.; Deng, Y.; Shi, F. Chem. Commun. 2014, 50, 13521. doi: 10.1039/C4CC05119J
Kon, K.; Siddiki, S. M. A. H.; Onodera, W.; Shimizu, K. Chem.- Eur. J. 2014, 20, 6264. doi: 10.1002/chem.201400332
Yang, Z.; Yu, B.; Zhang, H.; Zhao, Y.; Ji, G.; Ma, Z.; Gao, X.; Liu, Z. Green Chem. 2015, 17, 4189. doi: 10.1039/C5GC01386K
Yang, Z.; Yu, B.; Zhang, H.; Zhao, Y.; Ji, G.; Liu, Z. RSC Adv. 2015, 5, 19613. doi: 10.1039/C5RA00380F
Yang, Z.; Yu, B.; Zhang, H.; Zhao, Y.; Yu, C.; Ma, Z.; Ji, G.; Gao, X.; Han, B.; Liu, Z. ACS Catal. 2016, 6, 1268. doi: 10.1021/acscatal.5b02583
Liger, F.; Eijsbout, T.; Cadarossanesaib, F.; Tourvieille, C.; Bars, D. L.; Billard, T. Eur. J. Org. Chem. 2015, 6434. doi: 10.1002/ejoc.201500924
Du, X.-L.; Tang, G.; Bao, H.-L.; Jiang, Z.; Zhong, X.-H.; Su, D. S.; Wang, J.-Q. ChemSusChem 2015, 8, 3489. doi: 10.1002/cssc.201500486
Lucero, G.-S; Marcos, F.-A; Juventino, J. G. Organometallics 2015, 34, 763. doi: 10.1021/om501176u
Santoro, O.; Lazreg, F.; Minenkov, Y.; Cavallo, L.; Cazin, C. S. J. Dalton. Trans. 2015, 44, 18138. doi: 10.1039/C5DT03506F
Chen, W.-C.; Shen, J.-S.; Jurca, T.; Peng, C.-J.; Lin, Y.-H.; Wang, Y.-P.; Shih, W.-C.; Yap, G. P. A.; Ong, T.-G. Angew. Chem., Int. Ed. 2015, 54, 15422. http://www.ncbi.nlm.nih.gov/pubmed/26489967
Nguyen, T. V. Q.; Yoo, W.-J.; Kobayashi, S. Adv. Synth. Catal. 2016, 358, 452. doi: 10.1002/adsc.201500875
Liu, X.-F.; Li, X.-Y.; Qiao, C.; Fu, H.-C.; He, L.-N. Angew. Chem., Int. Ed. 2017, 56, 7425. doi: 10.1002/anie.201702734
Li, G.; Chen, J.; Zhu, D.-Y.; Chen, Y.; Xia, J.-B. Adv. Synth. Catal. 2018, 360, 2364. doi: 10.1002/adsc.201800140
Sorribes, I.; Junge, K.; Beller, M. Chem.-Eur. J. 2014, 20, 7878. doi: 10.1002/chem.201402124
Savourey, S.; Lefèvre, G.; Berthet, J.-C.; Cantat, T. Chem. Commun. 2014, 50, 14033. doi: 10.1039/C4CC05908E
Fu, M.-S.; Shang, R.; Cheng, W.-M.; Fu, Y. Angew. Chem., Int. Ed. 2015, 54, 9042. doi: 10.1002/anie.201503879
Zhu, L.; Li, B.-J.; Wang, S.; Wang, W.; Wang, L.-S.; Ding, L.; Qin, C.-Q. Polymers 2018, 10, 385. doi: 10.3390/polym10040385
Wen, W.; Han, B.; Yan, F.; Ding, L.; Li, B.-J.; Wang, L.-S.; Zhu, L. Nanomaterials 2018, 8, 326. doi: 10.3390/nano8050326
Zhu, L.; Wang, L.-S.; Li, B.-J.; Li. W.; Fu. B.-Q. Catal. Sci. Technol. 2016, 6, 6172. doi: 10.1039/C6CY00674D
Andrew, K. G.; Summers, D. M.; Donnelly, L. J.; Denton, R. M. Chem. Commun. 2016, 52, 1855. doi: 10.1039/C5CC08881J
Qiao, C.; Liu, X.-F.; Liu, X.; He, L.-N. Org. Lett. 2017, 19, 1490. doi: 10.1021/acs.orglett.7b00551
Natte, K.; Neumann, H.; Beller, M.; Jagadeesh, R. V. Angew. Chem., Int. Ed. 2017, 56, 6384. doi: 10.1002/anie.201612520
Chen, Y. Chem.-Eur. J. 2019, 25, 3405. doi: 10.1002/chem.201803642
Dominguez-Huerta A.; Dai X.-J.; Zhou, F.; Querard, P.; Qiu, Z.; Ung, S.; Liu, W.; Li, J.-B.; Li, C.-J. Can. J. Chem. 2019, 97, 67. doi: 10.1139/cjc-2018-0357?src=recsys
Xu, C.-P.; Xiao, Z.-H.; Zhuo, B.-Q.; Wang, Y.-H.; Huang, P.-Q. Chem. Commun. 2010, 46, 7834. doi: 10.1039/c0cc01487g
Yan, T.; Feringa, B. L.; Barta, K. Sci. Adv. 2017, 3, eaao6494. doi: 10.1126/sciadv.aao6494
Zhang, L.; Zhang, Y.; Deng, Y.; Shi, F. RSC. Adv. 2015, 5, 14514. doi: 10.1039/C4RA13848A
Tsarev, V. N.; Morioka, Y.; Caner, J.; Wang, Q.; Ushimaru, R.; Kudo, A.; Naka, H.; Saito, S. Org. Lett. 2015, 17, 2530. doi: 10.1021/acs.orglett.5b01063
Dang, T. T.; Ramalingam, B.; Seayad, A. M. ACS Catal. 2015, 5, 4082. doi: 10.1021/acscatal.5b00606
Campos, J.; Sharninghausen, L. S.; Manas, M. G.; Crabtree, R. H. Inorg. Chem. 2015, 54, 5079. doi: 10.1021/ic502521c
Elangovan, S.; Neumann, J.; Sortais, J.-B.; Junge, K.; Darcel, C.; Beller, M. Nat. Commun. 2016, 7, 12641. doi: 10.1038/ncomms12641
Bruneau-Voisine, A.; Wang, D.; Dorcet, V.; Roisnel, T.; Darcel, C.; Sortais, J.-B. J. Catal. 2017, 347, 57. doi: 10.1016/j.jcat.2017.01.004
Liu, Z.; Yang, Z.; Yu, X.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. Adv. Synth. Catal. 2017, 359, 4278. doi: 10.1002/adsc.201701044
Goyal, V.; Gahtori, J.; Narani, A.; Gupta, P.; Bordoloi, A.; Natte, K. J. Org. Chem. 2019, 84, 15389. doi: 10.1021/acs.joc.9b02141
Liang, R.; Li, S.; Wang, R.; Lu, L.; Li, F. Org. Lett. 2017, 19, 5790. doi: 10.1021/acs.orglett.7b02723
Zhu, L.; Wang, L.-S.; Li, B.-J.; Fu, B.-Q.; Zhang, C.-Q.; Li, W. Chem. Commun. 2016, 52, 6371. doi: 10.1039/C6CC01944G
Jiang, X.; Wang, C.; Wei, Y.; Xue, D.; Liu, Z.; Xiao, J. Chem.-Eur. J. 2014, 20, 58. doi: 10.1002/chem.201303802
Wang, H.; Huang, Y.; Dai, X.; Shi, F. Chem. Commun. 2017, 53, 5542. doi: 10.1039/C7CC02314F
Ge, X.; Luo, C.; Qian, C.; Yu, Z.; Chen, X. RSC Adv. 2014, 4, 43195. doi: 10.1039/C4RA04414B
Li, Y.; Sorribes, I.; Vicent, C.; Junge, K.; Beller, M. Chem.-Eur. J. 2015, 21, 16759. doi: 10.1002/chem.201502917

图式 1 以苯硅烷为还原剂钌催化的氮甲基化反应
Scheme 1 Ru-catalyzed N-methylation of amines with phen- ylsilane

图式 3 以苯硅烷为还原剂锌催化的氮甲基化反应
Scheme 3 Zn-catalyzed N-methylation of amines with phen- ylsilane

图式 4 以9-BBN为还原剂VBMe催化的氮甲基化反应
Scheme 4 VBMe catalyzed N-methylation of amines with 9-BBN

图式 7 CuAlOx催化胺、硝基苯和芳香腈的氮甲基化反应
Scheme 7 CuAlOx catalyzed N-methylation of amines, nitrobenzenes and aromatic nitriles

图式 8 Pd/CuZrOx催化胺和硝基苯的氮甲基化反应
Scheme 8 Pd/CuZrOx catalyzed N-methylation of amines and nitrobenzenes

图式 9 Pt-MoOx/TiO2催化二级胺的甲基化反应
Scheme 9 Pt-MoOx/TiO2 catalyzed N-methylation secondary amines

图式 11 F-PNHC-Zn催化胺的氮甲酰化和氮甲基化反应
Scheme 11 F-PNHC-Zn catalyzed N-formylation and N-meth- ylation of amines

图式 12 钌-氮杂三苯基膦聚合物催化芳香二级胺的氮甲基化反应
Scheme 12 Poly(PPh3)-azo-Ru catalyzed N-methylation of aro- matic secondary amines

图式 13 11CO2参与锌-NHC络合物催化芳香胺的氮甲基化反应
Scheme 13 IPrZnCl2 catalyzed N-methylation of aromatic amines with 11CO2

图式 14 氧化铝负载纳米金催化胺的氮甲基化反应
Scheme 14 Al2O3 supported AuNPs catalyzed N-methylation of amines

图式 17 硼烷、CO2参与含碳二卡宾催化胺的氮甲基化反应
Scheme 17 Carbodicarbenes catalyzed N-methylation of amines with borane and CO2

图式 18 含氮杂环卡宾-铑催化胺的氮甲基化反应
Scheme 18 Bis(tzNHC)-Rh complex catalyzed N-methylation of amines

图式 21 苯硅烷、甲酸参与铂催化胺的氮甲基化反应
Scheme 21 Pt catalyzed N-methylation of amines with PhSiH3 and HCOOH

图式 24 铂碳非均相催化芳香胺以及亚胺的氮甲基化反应
Scheme 24 Heterogeneous Pt/C catalyzed N-methylation of aromatic amines and imines

图式 27 钯/二氧化钛光催化胺与硝基化合物的氮甲基化反应
Scheme 27 Pd/TiO2-hν catalyzed N-methylation of amines and nitro-compound

图式 29 钌催化一级芳香、脂肪胺以及磺酰胺的氮甲基化反应
Scheme 29 Ru catalyzed N-methylation of primary aromatic and aliphatic amines and sulfonamide

图式 30 甲醇参与铱卡宾催化取代苯胺的氮甲基化反应
Scheme 30 Ir-NHC catalyzed N-methylation of anilines with methanol

图式 31 甲醇参与锰催化取代苯胺的氮甲基化反应
Scheme 31 Mn catalyzed N-methylation of anilines with methanol

图式 32 甲醇、氢气参与锰催化胺的氮甲基化反应
Scheme 32 Mn-complex catalyzed N-methylation of amines with CH3OH and H2

图式 33 甲醇参与基于钴的催化体系催化胺的氮甲基化反应
Scheme 33 Co-complex catalyzed N-methylation of amines with methanol

图式 35 甲醇参与铱催化胺的氮甲基化反应
Scheme 35 Cp*Ir complex catalyzed N-methylation of amines with CH3OH

图式 36 二甲基亚砜、三乙胺、甲酸参与胺的氮甲基化反应
Scheme 36 N-Methylation of amines with Et3N, HCOOH and DMSO

图式 37 多聚甲醛、氢气参与CuAlOx催化胺的氮甲基化反应
Scheme 37 CuAlOx catalyzed N-methylation of amines with paraformaldehyde and H2

图式 38 多聚甲醛、氢气参与RANEY-Ni催化胺的氮甲基化反应
Scheme 38 RANEY-Ni catalyzed N-methylation of amines with paraformaldehyde and H2

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:















 下载:
下载: