

图式 1.
基于邻炔基苯胺的多取代吲哚的合成
Scheme 1.
Synthesis of muti-substituted indoles based on 2-alkynyl aniline

炔烃是一类非常重要的有机合成子, 常用于合成具有生物活性的杂环化合物骨架[1].炔烃的环化反应是构建碳(杂)环化合物重要方法之一, 已受合成化学家们广泛关注[2].这类碳(杂)环化合物广泛存在天然产物或药物中, 是构建具有生理活性的化合物和药物的重要组成; 同时也是合成某些染料、功能材料的重要原料[3].
利用炔烃的环化来构建碳(杂)环化合物的研究大多采用过渡金属催化, 比如钯、铂、铑和金等贵金属, 然而贵金属的使用存在价格昂贵、金属残留等缺点[4].如何更加高效和经济地实现炔烃的选择性环化反应, 是目前研究的热点之一[5].利用邻位基团参与, 实现炔烃的高选择性环化反应是近年来发展起来的一种有效策略.此类方法能有效避免过渡金属的使用, 并且反应底物大都具有简单易得、条件温和、原子经济性高等优点, 在有机合成中具有重要研究意义.本文将基于邻位基团参与的无过渡金属催化体系下, 就不同邻位基团参与的炔烃环化反应研究进展进行简要综述.
吲哚是一类非常重要的氮杂环化合物[6], 寻找更加经济和简便的方法来合成此类化合物具有重要意义. 2003年, Knochel等[7]报道了在强碱条件下, 避免过渡金属使用, 利用邻位氨基参与, 实现炔烃分子内五元环化反应, 合成一系列吲哚类化合物, 提供了氮杂环化合物的简便合成方法(Eq. 1).
|
|
(1) |
随后, Fiandanese等[8]报道以邻二乙炔基苯胺为底物, 同样加入氢化钾(KH), 在N-甲基吡咯烷酮(NMP)中室温下反应, 邻位氨基参与二乙炔关环反应.值得关注的是, 远离苯胺的另一炔键得到完整保存, 并顺利得到2-炔基吲哚类化合物(Eq. 2).
|
|
(2) |
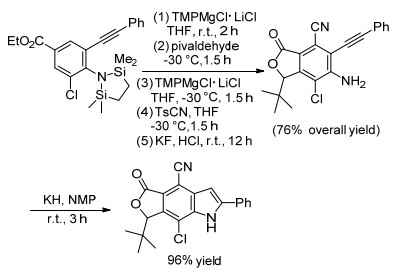
Knochel等[9]在前期研究吲哚骨架构建的基础上, 报道了一种通过区域选择性金属化合物保护的高取代的邻炔基苯胺合成多取代吲哚的新方法.选择性金属化合物保护的邻炔基苯胺在TMPMgCl•LiCl和KH/NMP条件下一锅法制备多取代吲哚化合物, 该反应过程中得到去保护化的多取代邻炔基苯胺中间产物, 最后邻位氨基参与炔烃的五元关环, 合成高取代的吲哚化合物(Scheme 1).
Arcadi等[10]发展了一种避免在金属催化剂或有机酸、碱条件下合成吲哚的方法, 采用温和绿色电化学方法. 0 ℃恒电流控制在25 mA•cm-2下, 在电解质体系为CH3CN/0.1 mol/L四乙基四氟硼酸铵(TEATFB)的铂电极中进行电解, 电解产生的-CH2CN促使邻位氨基参与炔烃的关环, 实现吲哚化合物的合成(Eq. 3).另外文中还提到在电解结束时再加入邻炔基苯胺, 其目的是避免底物中敏感官能团的还原.
|
|
(3) |
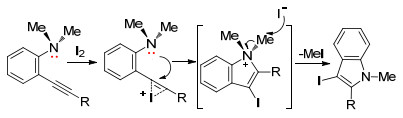
2004年, Lorack等[11]采用碘单质代替过渡金属催化剂, 实现炔烃的五元环化反应.在I2/CH2Cl2体系中, 以邻位叔胺为参与基团制备N-烷基-3-碘吲哚系列化合物(Eq. 4).当胺基上的氮连接两个不同的烷基, 该反应呈现出有趣的选择性, 其中氮上带有甲基和正丁基的化合物进行亲电环化反应, 以72:28(质量比)生成N-正丁基-3-碘吲哚和N-甲基-3-碘吲哚混合化合物.
|
|
(4) |
另外, 该课题组对此反应提出可能反应机理:在亲电试剂I2作用下, 活化炔烃生成碘鎓离子中间体, 随后N, N-二烷基胺基上的氮进攻鎓离子产生碘吲哚正离子中间体, 然后甲基以碘代甲烷的方式离去生成吲哚类化合物(Scheme 2).
Erker等[12]报道通过受阻路易斯酸碱对(FLPs)实现了无金属催化下胺参与炔烃的分子内环化反应.在B(C6F5)3路易斯酸条件下, 利用邻位胺进攻分子内炔烃生成五元杂环硼酸铵衍生物(Eq. 5).该发现证实了FLPs策略在杂环反应中应用的实用性.
|
|
(5) |
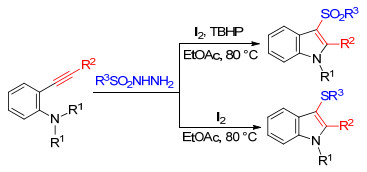
Kuhakarn等[13]发展了一种经济且环境友好的I2- TBHP(过氧化叔丁醇)和I2反应体系, 以N, N-二烷基邻炔基苯胺和磺酰肼为底物, 选择性地合成了具有潜在生物活性的N-烷基-3-磺酰基吲哚和N-烷基-3-硫基吲哚类化合物.该反应具有良好的官能团耐受性, 产率适中至良好(Scheme 3).
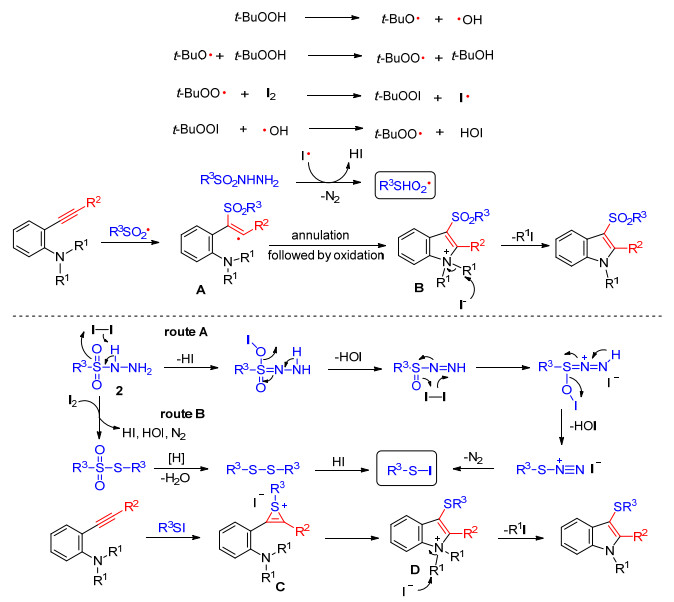
该课题组对反应机理进行了研究, 在无碘单质或对甲苯磺酰碘代替磺酰肼条件下, 均无目标产物生成; 而在I2-TBHP反应体系中加入自由基抑制剂四甲基哌啶氧化物(TEMPO), 该反应被抑制.根据机理研究实验和前人研究的结果, 对在I2-TBHP体系下生成磺酰基吲哚的反应提出了磺酰基自由基机理, 其可能反应机理为: TBHP受热不稳定, 分解成叔丁氧自由基和叔丁过氧自由基, 这两个自由基与碘和磺酰肼反应生成磺酰基自由基, 同时释放N2, 紧接着磺酰基自由基进攻炔烃生成乙烯基自由基中间体A, 随后炔烃邻位的氮原子参与氧化环化生成吲哚中间体B, 最后在碘离子作用下脱烷基, 最终生成3-磺酰基吲哚化合物.对I2反应体系生成的硫基吲哚提出了R3SI对炔烃亲电加成环化的反应机理, 其可能机理为:磺酰肼与碘反应生成R3SI, 然后R3SI进攻炔烃发生亲电加成生成烯基正离子中间体C, 随后炔烃邻位的氮原子亲核进攻环化生成吲哚中间体D, 在碘离子作用下脱烷基, 生成3-硫基吲哚化合物(Scheme 4).
为了合成多取代吲哚类化合物, 范仁华等[14]发展了无过渡金属催化下N-Ts邻炔基苯胺与对甲基苯硫酚的一锅煮反应(Eq. 6).为研究反应的可能途径, 进行了密度泛函理论(DFT)计算.其机理研究表明, 该反应过程涉及二乙酰氧基碘苯(PIDA)介导的氧化脱芳构化和Brønsted酸促进邻位氮原子参与的炔烃环化.
|
|
(6) |
此外, 王磊等[15]采用可见光诱导2-炔基苯胺与二硫化物(二硒醚)的氧化环化反应, 合成了3-硫基吲哚和3-硒基吲哚类化合物(Eq. 7).该反应避免过渡金属和光催化剂的使用, 室温下反应, 产物收率最高可达97%.
|
|
(7) |
韩丙等[16]以邻炔基芳基叠氮化合物为研究对象, 实现了邻位叠氮基参与炔烃的自由基五元环化反应.在叔丁基过氧化氢(TBHP)条件下, 通过2-炔基芳基叠氮化物与亚磺酸的反应, 开发了一种简便、有效的构建3-磺酰基吲哚化合物的方法, 该方法具有良好的选择性且无副产物(Eq. 8).通过自由基抑制实验, 试验表明该反应是自由基历程, 提出可能反应机理为:亚磺酸在加热条件下与TBHP反应生成磺酰基自由基A, 随后磺酰基自由基进攻炔烃生成烯基自由基中间体B, 烯基自由基立即与邻位叠氮基发生分子内环化生成吲哚N-自由基中间体C, 同时释放N2, 最后中间体C从亚磺酸或周围环境获得氢原子, 生成3-磺酰基吲哚化合物(Scheme 5).
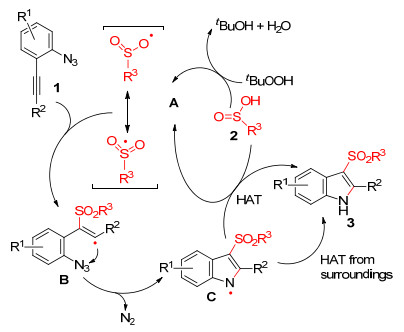
与韩丙课题组不同的是, 朱成建等[17]采用2-乙炔酮基芳基叠氮化合物代替2-炔基芳基叠氮化合物, 在TBHP条件下与芳基亚磺酸反应, 实现2-乙炔酮基芳基叠氮化合物的六元环化反应, 同时为构建3-磺酰基喹诺酮化合物提供了一条高效途径(Eq. 9).反应机理表明, 该反应也是自由基历程, 在TBHP下产生的磺酰基自由基引发的六元环化反应.
Yamamoto等[18]报道了无金属催化下邻丙炔基芳基叠氮化合物亲电环化合成喹啉类化合物.室温条件下, 在CH3NO2溶剂中, 邻丙炔基芳基叠氮化合物在亲电试剂(I2, IC1, NIS, Br2, NBS和HNTf2)作用下发生分子内六元环化反应, 实现喹啉类化合物的合成(Eq. 10).该反应历程是亲电试剂进攻活化炔烃, 随后邻位叠氮基参与炔烃的关环, 最后脱去N2和H+得到喹啉类化合物.
|
|
(10) |
此外, Yamamoto等[19]还报道了在碘介导作用下, 2-炔基苄基叠氮化合物利用邻位叠氮基参与炔烃环化反应合成1, 3, 4-三取代异喹啉化合物(Eq. 11).根据底物结构, 碘、Barluenga试剂(Py2IBF4/HBF4)或N-碘代琥珀酰亚胺在该反应中可被选用I+源.
|
|
(11) |
异吲哚啉-1-酮衍生物是一类重要的生物活性分子, 化学家们已陆续寻找绿色方法来合成该衍生物. Arcadi等[20]利用之前采用的电化学方法实现了2-炔基苯胺合成吲哚杂环化合物的方法, 发现2-炔基苯甲酰胺在同样的电解质体系(CH3CN/0.1 mol/L TEATFB)条件下, 也能实现五元环化反应, 有效地合成3-乙烯基-异吲哚啉酮类化合物(Eq. 12).
|
|
(12) |
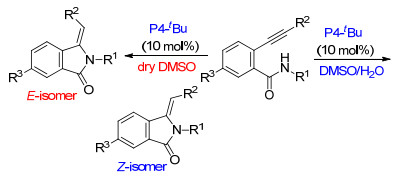
Terada等[21]发展了一种在有机超强碱作用下, 利用有水和无水条件可以有效控制异吲哚啉-1-酮产物的E/Z构型.在10 mol%的有机超强碱P4-tBu存在下, 邻炔基苯甲酰胺在二甲基亚砜(DMSO)/H2O共溶剂体系中Z-选择性环化, 在无水DMSO溶剂中E-选择性环化(Scheme 6).
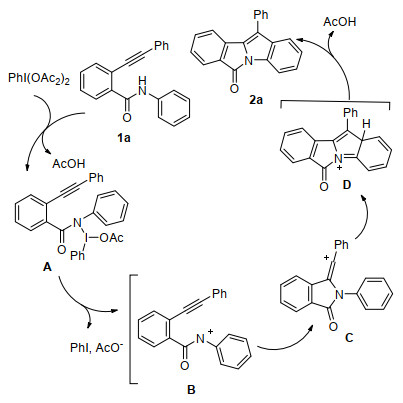
Maurya等[22]开发了一种无过渡金属催化、高价碘(Ⅲ)条件下合成吲哚并异吲哚啉酮四环稠合化合物的有效方法.以PIDA为高价碘, 2-(1-芳基乙炔基)苯甲酰胺在室温下区域选择性地合成11-芳基-6H-异吲哚[2, 1-a]吲哚-6-酮化合物, 产率中等至良好(Eq. 13).该课题组提出PIDA介导的串联氧化反应机理:首先PIDA与2-(1-芳基乙炔基)苯甲酰胺底物反应生成N-碘酰胺中间体A, 中间体A分解生成酰胺氮正离子中间体B, 随后中间体B发生分子内氮正离子进攻炔烃环化生成碳正离子中间体C, 然后发生亲电芳基取代反应生成中间体D, 原位生成的乙酸阴离子夺取氢后生成目标产物(Scheme 7).
Mehta等[23]报道了在n-BuLi-I2/ICl条件下, 邻炔基苯甲酰胺中的酰胺氮参与炔烃的碘代N-环化反应, 区域选择性地合成异吲哚啉-1-酮衍生物, 并且产物的碳碳双键为Z-立体化构型(Eq. 14).底物酰胺为伯酰胺时, 也能生成异吲哚啉-1-酮化合物, 产率为38%~94%.该反应机理也为离子机理, 反应过程可能涉及异吲哚啉酮乙烯基阴离子中间体或离子对中间体产生.
|
|
(14) |
另外陈知远等[24]报道了碱促进区域选择性和立体选择性地合成(Z)-3-亚烷基异吲哚啉-1-酮衍生物.前面Terada[21]和Mehta[23]课题组分别采用在超强碱P4-tBu和强极性碱n-BuLi来促进邻炔基苯甲酰胺的环异构化反应, 而陈知远等采用碱盐Na2CO3和超过化学计量的水实现邻炔基苯甲酰胺中的酰胺氮参与炔烃的N-环化反应.该反应中过量的水是必要的, 对反应的转化有促进作用, 且反应绿色温和, 产率适中至良好(Eq. 15).
|
|
(15) |
为寻找利用更加温和、更加绿色化的方法合成具有生物活性的O-杂环化合物, 近年来利用邻位含氧原子参与的炔烃分子内环化反应已引起化学家们的广泛关注, 在构建O-杂环化合物方面也取得了较大的进展. 2009年, Larock等[25]报道了2-炔基苄醇在I2/NaHCO3条件下, 邻位羟基参与的炔烃分子内关环反应, 生成碘取代的异色烯和二氢异苯并呋喃类化合物(Eq. 16).该反应的区域选择性取决于底物结构, 特别是当底物带有叔醇结构, 更易发生五元外环化反应, 生成二氢异苯并呋喃类化合物; 而带有伯或仲醇基的底物, 更倾向于六元内环化反应, 生成异色烯类化合物.
|
|
(16) |
杜纪元等[26]发展了一种无金属催化邻羟炔丙基苯酚和膦氧化物的一锅煮反应.邻羟炔丙基苯酚原位生成o-QMs, 通过磷氧化物的膦-迈克尔加成以及邻位氧原子参与关环, 有效地合成了3-膦酰基苯并呋喃衍生物(Eq. 17).在C(sp2)—P键构建的基础上, 利用光学纯的RP-(—)-甲基苯基膦氧化物, 探讨不对称合成含有手性P-立体中心的3-酰基苯并呋喃衍生物.
|
|
(17) |
Kim等[27]报道了1-甲氧基-2-乙炔基苯在I2/NaHCO3条件下, 利用邻位甲氧基参与炔烃关环反应, 合成碘代苯并呋喃类化合物(Eq. 18).其中碘代苯并呋喃产物通过Suzuki偶联反应可以合成一类基于苯并呋喃骨架的天然寡萜类化合物.
|
|
(18) |
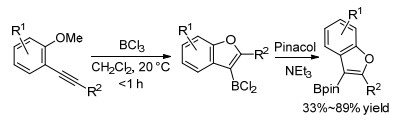
同时Ingleson等[28]报道了在BCl3催化下, 利用邻位甲氧基参与的炔烃环化反应, 简便有效地合成了C3-硼化苯并呋喃衍生物.另外二氯硼代苯并呋喃产物在频哪醇条件下可以合成广泛存在的频哪醇硼酸酯类化合物(Scheme 8).在该反应体系下, 炔烃邻位除了甲氧基可以参与炔烃环化, 甲硫基同样适用, 可以合成C3-硼化苯并噻吩衍生物.
Verma等[29]发展了在碘介导下邻炔基芳甲醛的亲电碘代环化反应.在I2/K2CO3体系条件下, 邻炔基芳甲醛在二氯甲烷溶剂中, 与适当的亲核试剂反应, 通过醛基参与炔烃的环化反应, 形成吡喃并[4, 3-b]喹啉衍生物(Eq. 19).另外, 如果使用醇既作为亲核试剂又作为溶剂, 不会发生环化反应, 而是氧化生成邻炔基芳基酯衍生物.
|
|
(19) |
紧接着, Youn等[30]开发了一种1, 3-双(2, 4, 6-三甲苯基)氯化咪唑鎓(NHC)催化邻炔基苯甲醛的分子内亲电环化反应, 区域选择性地生成苯酞和异香豆素类化合物(Eq. 20).该反应利用空气中氧气作为氧原子来源, 将醛氧化成相应的苯甲酸.在NHC有机催化体系下, 通过高效的双重激活, 该反应以原子经济型实现两个连续C—O键的构建.
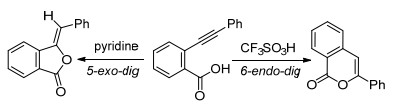
近些年来研究报道显示邻位羧基也能参与炔烃的分子内环化反应. 2006年, Uchiyama等[31]报道了2-炔基苯甲酸中的邻位羧酸参与炔烃的环化反应, 有意思的是利用酸碱条件调控分子内环化反应的区域选择性.在强酸CF3SO3H下, 2-炔基苯甲酸区域选择性地发生六元环化反应生成异香豆素类化合物; 而在弱碱吡啶下发生五元环化反应生成苯酞类化合物(Scheme 9).
|
|
(20) |
2007年, Terada等[32]报道在有机碱1, 5-二氮杂二环[5.4.0]十一烯-5 (DBU)催化下, 邻炔基苯甲酸中羧基参与炔烃的五元环化反应, 区域选择性地合成了苯酞类化合物(Eq. 21).产率中等至良好, 为合成具有生物活性的苯酞类化合物提供了有效方法.
|
|
(21) |
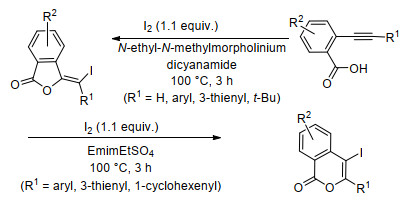
2017年, Mancuso等[33]报道了在没有外部碱情况下, 在非常规溶剂和以分子碘为碘源的离子液体(ILs)中, 2-炔基苯甲酸发生邻位羧酸参与炔烃的环化反应, 该环化反应的区域选择性强烈依赖于ILs介质(Scheme 10).另外, 从与二乙醚的反应混合物中提取产物后, ILs溶剂可以回收.
2018年, 我们课题组[34]以溴代四丁基铵(TBAB)为溴盐, Oxone为氧化剂, 在二氯乙烷和水共溶剂下实现了邻炔基苯甲酸的五元溴环化反应, 区域选择性地合成了溴取代的苯酞类化合物(Eq. 22).反应条件温和, 且后处理操作简便; 对于产物的应用, 可以通过交叉偶联反应进行产物的衍生化, 另外使其产物与水合肼在四氢呋喃和水共溶剂下60 ℃反应, 可以专一性地合成具有核心结构的酞嗪-1(2H)-酮化合物.
|
|
(22) |
紧接着, 我们课题组发现将1.5 equiv. TBAB改成催化量的TBAB, 同样以Oxone为氧化剂, 邻炔基苯甲酸在水溶剂下区域选择性地实现了分子内六元环化反应, 合成了异香豆素类化合物(Eq. 23).
|
|
(23) |
邻位酰胺基作为参与基团时, 除了氮参与炔烃环化反应, 邻位酰胺基氧也能参与炔烃的专一环化反应. 2012年, Opatz等[35]报道了在I2/NaHCO3条件下, 邻炔基苯甲酰胺发生碘环化反应.利用邻位酰胺基氧参与的炔烃环化反应合成异苯并呋喃-1-亚胺和异色烯-1-亚胺, 取代了内酰胺的合成(Eq. 24).并且通过DFT计算, 反应倾向C—O键形成途径.
|
|
(24) |
同时, Larock等[36]报道指出2-炔基苯甲酰胺在亲电试剂作用下环化生成异苯并呋喃-1-亚胺和异色烯-1-亚胺类化合物, 并非是邻位酰胺氮参与的炔烃环化反应生成多取代异喹啉-1-酮和3-乙烯基异吲哚酮类化合物(Eq. 25).利用X射线晶体学和光谱技术对产物进行了表征, 纠正该课题组先前发表产物结构的错误.
|
|
(25) |
前面报道的2-炔基苯甲酰胺在发生氧环化反应时, 同时生成五元和六元环化产物.为提高反应区域选择性, 我们课题组[37]报道了在TBAB/Oxone体系下, 2-炔基苯甲酰胺在碳酸钾下能够专一选择性地发生五元溴环化反应, 合成异苯并呋喃-1-亚胺类化合物.在反应完全生成异苯并呋喃-1-亚胺化合物后, 加入酸能够完全转化成异苯并呋喃酮类化合物(Eq. 26).
|
|
(26) |
针对该反应, 我们课题组提出两种可能反应机理.第一条反应途径可能是经过自由基溴化氧环化反应:在此过程中, 溴化物被氧化成溴自由基, 然后溴自由基进攻炔烃生成烯基自由基中间体A, 中间体A被氧化成烯基正离子中间体B, 随后被分子内酰胺氧捕获发生关环得到异苯并呋喃-1-亚胺.第二条反应途径可能是:溴被氧化成溴正离子, 然后溴正离子活化炔烃生成中间体C, 从而促使邻位酰胺氧参与的五元亲电环化反应.然而根据Larock等[36]研究, 2-炔基苯甲酰胺的亲电环化不具有区域选择性.若反应是通过亲电环化反应进行, 将产生五元环化产物和六元环化产物混合物.根据上述结果, 该反应优先以自由基途径区域选择性地合成目标产物(Scheme 11).
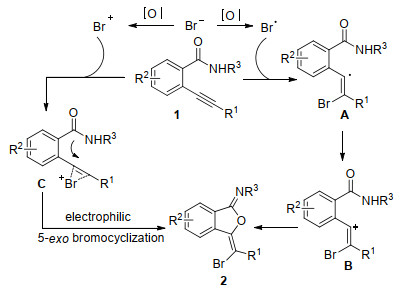
接着, 我们课题组[37]发现将TBAB量降低到催化量(0.1 equiv.)时, 在TBAB/Oxone反应体系下, 2-炔基苯甲酰胺发生氧环化反应, 区域选择性地生成异色烯-1-亚胺类化合物.机理研究表明, 3-溴甲基异苯并呋喃-1-亚胺是一个关键的中间体, 经过C—O键迁移和脱溴反应生成最终的异色烯-1-亚胺类化合物(Eq. 27).
|
|
(27) |
除邻位氮、氧原子外, 无过渡金属催化下邻位硫原子也能参与炔烃的环化反应, 为实现构建结构多样的杂环化合物提供了重要途径. 2008年, Larock等[38]报道了在亲电试剂I2下, 实现了无过渡金属催化邻位硫原子参与的炔烃五元环化反应.对于邻位多取代的二芳基炔烃, 炔烃邻位分别有甲硫基和甲氧基(或酯基), 能选择性地发生硫参与的炔烃环化反应(Eq. 28).
|
|
(28) |
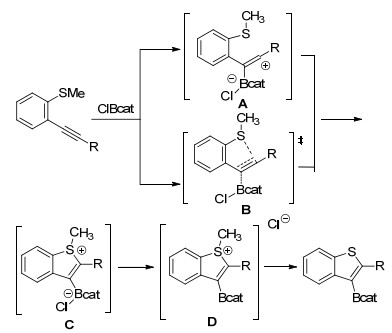
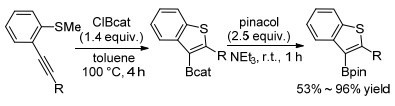
2016年, Blum等[39]首次报道无过渡金属催化下炔烃的硫硼化反应.以B-氯代邻苯二酚硼烷为(ClBcat)亲电硼源, 邻位甲硫基参与炔烃环化反应生成硼化苯并噻吩类化合物(Scheme 12).该化合物可以参与多种原位衍生反应, 在频哪醇作用下反应生成更加稳定的频哪醇硼酸酯代苯并噻吩类化合物.另外该课题组专门在一篇文章通过动力学、计算等研究对反应机理进行详细论述, 提出可能反应机理为:利用路易斯酸硼诱导活化2-炔基硫代苯甲醚中碳碳叁键生成两性离子中间体C, 随后中间体C通过氯离子脱甲基或经过离子对中间体D生成3-硼化苯并噻吩化合物(Scheme 13).该反应途径表明ClBcat具有关键双功能作用, 首先是作为亲电活化试剂, 其次是亲核氯化物的来源.
2017年, 宋秋玲等[40]报道了2-炔基硫代苯甲醚和芳基亚磺酸在TBHP条件下甲硫基参与炔烃的自由基环化反应, 合成3-芳基磺酰基苯并噻吩(Eq. 29).该反应条件温和、操作简单并且反应时间短, 产率中等至良好.
|
|
(29) |
利用无过渡金属催化邻位苯基参与炔烃的环化反应, 是合成稠环芳烃的一种经济有效方法. 1994年, Swager等[41]在三氟乙酸(TFA)条件下设计了含邻苯基乙炔基-1, 1'-联苯核心对称结构底物, 利用邻位苯基参与炔烃的环化反应, 定向专一地合成了具有特殊结构的稠环芳烃(Eq. 30).另外该课题组还合成了全碳梯形共轭聚合物或石墨带, 证实了该反应的实用性.
|
|
(30) |
2004年, Larock等[42]报道了在亲电试剂如ICl作用下, 利用2-(芳基乙炔基)联苯的邻位苯基参与炔烃的环化反应, 合成了碘取代的稠环芳烃化合物.芳烃取代基可以是供电子基团或吸电子基团, 具有良好的官能团耐受性, 并且该方法已经广泛应用于合成多环或杂环化合物中(Eq. 31).
|
|
(31) |
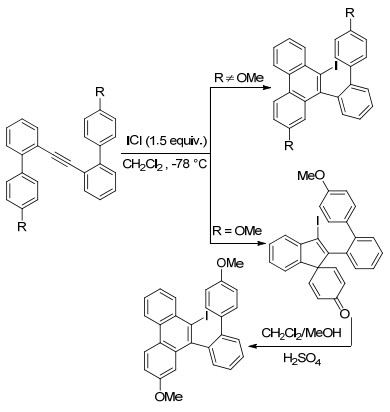
2007年, 刘瑞雄等[43]也报道了在亲电试剂ICl作用下, 双(二芳基)炔烃发生分子内环化合成碘菲类化合物, 该产物通过Mizoroki-Heck偶联反应可以合成二苯并[g, p]联苯.另外该课题组发现以4'-甲氧基-2-乙炔基联苯为底物, 在ICl作用下发生ipso环化得到双环螺环己二烯酮.在MeOH/H2SO4条件下, 这些螺酮产物通过选择性的1, 2-烯基转移进行重排, 得到碘菲类化合物(Scheme 14).
为实现七元环化反应, Chen等[44]设计了1-([1, 1'-联苯]-2-基)炔酮底物代替1-([1, 1'-联苯]-2-基)炔烃化合物, 在氯化碘条件下, 利用邻位苯基参与的炔烃环化反应合成了碘代二苯并环庚-5-酮.通过改变炔酮底物上的取代基, 可能发生分子内6元环化反应生成螺环共轭化合物(Eq. 32).
|
|
(32) |
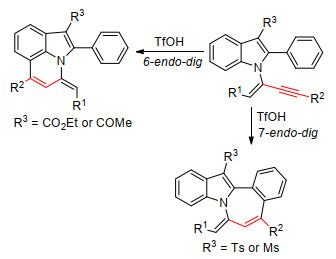
Park等[45]设计了吲哚炔胺类底物, 邻位苯基选择性地参与炔烃的环化反应.在三氟甲磺酸(TfOH)介导下, 通过吲哚C3上的吸电子官能团来控制环异构化, 含羰基的吲哚类底物(如CO2Et和COMe)发生六元环化, 生成吡咯并[3, 2, 1-ij]喹啉化合物; 而含有磺酰基的吲哚类底物(如Ts和Ms)发生七元环化, 生成苯并[3, 4]氮杂环庚烷[1, 2-a]吲哚化合物, 产率中等至以上(Scheme 15).
综上所述, 基于无过渡金属催化下邻位基团参与的炔烃环化反应近些年已取得很大进展, 为氮、氧、硫杂环化合物以及多元并环结构化合物的合成提供了一条绿色、原子经济型途径.该类方法具有以下优点: (1)利用亲电试剂等代替过渡金属的催化, 节约成本; (2)底物具有很高的原子经济型, 并且反应操作简单, 反应区域选择性高; (3)构建了结构多样、价值有用的杂环化合物骨架以及具有潜在价值的杂环化合物新骨架, 给天然产物和药物的合成提供了新的选择.然而, 该类发应尚具有较大发展和应用空间:首先, 目前研究较多的是邻位氮、氧、硫和苯基参与炔烃的环化, 还需发展新的邻位基团参与炔烃的(多元)环化反应; 其次, 很多药物分子具有手性结构, 邻位基团参与的炔烃对映选择性环化报道还较少; 此外, 多元并环结构的合成机理尚不明确, 值得进一步探究.
(a) Boyarskiy, V. P.; Ryabukhin, D. S.; Bokach, N. A.; Vasilyev, A. V. Chem. Rev. 2016, 116, 5894.
(b) Zheng, Z. T.; Wang, Z. X.; Wang, Y. L.; Zhang, L. M. Chem. Soc. Rev. 2016, 45, 4448.
(c) Yuan, S. T.; Zhou, H. W.; Gao, L.; Liu, J. B.; Qiu, G. Y. S. Org. Lett. 2018, 20, 562.
(d) Wang, Y. C.; Wang, R. X.; Qiu, G. Y. S.; Zhou, H. W.; Xie, W. L.; Liu, J. B. Org. Chem. Front. 2019, 6, 2471.
(a) Huang, S. Y.; Li, X. X.; Lin, C. L.; Tang, W. P. Chem. Commun. 2012, 48, 2204.
(b) Chen, D. P.; Xing, G. D.; Zhou, H. W. Org. Chem. Front. 2015, 2, 947.
(c) Zhou, P.; Qiu, H. H.; Shi, J. C. Chin. J. Org. Chem. 2016, 36, 425(in Chinese).
(周鹏, 邱会华, 施继成, 有机化学, 2016, 36, 425.)
(d) Yu, J. J.; Yang, S.; Wu, Z.; Zhu, C. Chin. J. Org. Chem. 2019, 39, 223(in Chinese). (余佳佳, 杨珊, 吴镇, 朱晨, 有机化学, 2019, 39, 223.)
(e) Mei, H. B.; Yin, Z. Z.; Liu, J.; Sun, H. L.; Han, J. L. Chin. J. Chem. 2019, 37, 292.
(a) Kirsch, S. F. Synthesis 2008, 3183.
(b) Jung, H. H.; Floreancig, P. E. J. Org. Chem. 2007, 72, 7359.
(c) Zhang, J.; Xiao, G. M. Petrochem. Technol. 2011, 40, 579(in Chinese).
(张进, 肖国民, 石油化工, 2011, 40, 579.)
(d) Wang, Y.-C.; Wang, R.-X.; Qiu, G.; Zhou, H.; Xie, W.; Liu, J.-B. Org. Chem. Front. 2019, 6, 2471.
(e) Wang, Y. D.; Li, J. H.; Gao, Y.; Yang, Y.; Gao, Y.; Xu, Z. F. Hydrometallurgy 2020, 191, 105220.
(f) Wang, Y. C.; Liu, J. B.; Zhou, H. W.; Xie, W. L.; Rojsitthisak, P.; Qiu, G. Y. S. J. Org. Chem. 2020, 85, 1906.
(a) Wang, Z.; Yang, L.; Liu, H. L.; Tan, Y. Z.; Bao, W. H.; Wang, M.; Tang, Z. W.; He, W. M. Chin. J. Org. Chem. 2018, 38, 2639(in Chinese). (王峥, 杨柳, 刘慧兰, 谭英芝, 包文虎, 汪明, 唐子龙, 何卫民, 有机化学, 2018, 38, 2639.)
(b) Lu, L. H.; Wang, Z.; Xia, W.; Cheng, P.; Zhang, B.; Cao, Z.; He, W. M. Chin. Chem. Lett. 2019, 30, 1237.
(c) Cao, Z.; Zhu, Q.; Lin, Y. W.; He, W. M. Chin. Chem. Lett. 2019, 30, 2132.
(d) Wu, C.; Wang, Z.; Hu, Z.; Zeng, F.; Zhang, X. Y.; Cao, Z.; Tang, Z.; He, W. M. Org. Biomol. Chem. 2018, 16, 3177.
(e) Wu, C.; Xiao, H. J.; Wang, S. W.; Tang, Z. L.; Xia, W.; Li, W. F.; Cao, Z.; He, W. M. ACS Sustainable Chem. Eng. 2019, 7, 2169.
(f) Yuan, S.; Wang, Y.; Qiu, G.; Liu, J.-B. Chin. J. Org. Chem. 2017, 37, 566(in Chinese).
(袁斯甜, 王艳华, 邱观音生, 刘晋彪, 有机化学, 2017, 37, 566.)
(a) Wen, B.; Petersen, J. L.; Wang, K. K. Org. Lett. 2010, 13, 168.
(b) Zhu, H. T.; Ji, K. D.; Yang, F.; Wang, L. J.; Zhao, S. C.; Ali, S.; Liu, X. Y.; Liang, Y. M. Org. Lett. 2011, 13, 684.
(c) Yang, X. B.; Hu, F. Z.; Di, H. J.; Cheng, X. X.; Li, D.; Kan, X. L.; Zou, X. M.; Zhang, Q. C. Org. Biomol. Chem. 2014, 12, 8947.
(d) Luo, P. S.; Tang, R. Y.; Zhong, P.; Li, J. H. Chin. J. Org. Chem. 2009, 29, 1924(in Chinese).
(罗培松, 汤日元, 钟平, 李金恒, 有机化学, 2009, 29, 1924.)
Somei, M.; Yamada, F. Nat. Prod. Rep. 2005, 22, 73. doi: 10.1039/b316241a
Koradin, C.; Dohle, W.; Rodriguez, A. L.; Schmid, B.; Knochel, P. Tetrahedron 2003, 59, 1571. doi: 10.1016/S0040-4020(03)00073-5
Fiandanese, V.; Bottalico, D.; Marchese, G.; Punzi, A. Tetrahedron 2008, 64, 7301. doi: 10.1016/j.tet.2008.05.059
Stoll, A. H.; Knochel, P. Org. Lett. 2008, 10, 113. doi: 10.1021/ol7025872
Arcadi, A.; Bianchi, G.; Inesi, A.; Marinelli, F.; Rossi, L. Eur. J. Org. Chem. 2008, 2008, 783. doi: 10.1002/ejoc.200701011
(a) Yue, D.; Larock, R. C. Org. Lett. 2004, 6, 1037.
(b)Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2006, 71, 62.
Voss, T.; Chen, C.; Kehr, G.; Nauha, E.; Erker, G.; Stephan, D. W. Chem.-Eur. J. 2010, 16, 3005. doi: 10.1002/chem.200903483
Meesin, J.; Pohmakotr, M.; Reutrakul, V.; Soorukram, D.; Leowanawat, P.; Kuhakarn, C. Org. Biomol. Chem. 2017, 15, 3662. doi: 10.1039/C7OB00366H
Han, D. D.; Li, Z. M.; Fan, R. H. Org. Lett. 2014, 16, 6508. doi: 10.1021/ol5034139
Shi, Q.; Li, P. H.; Zhang, Y.; Wang, L. Org. Chem. Front. 2017, 4, 1322. doi: 10.1039/C7QO00152E
Chen, F.; Meng, Q.; Han, S. Q.; Han, B. Org. Lett. 2016, 18, 3330. doi: 10.1021/acs.orglett.6b01427
Zhou, N. N.; Yan, Z. F.; Zhang, H. L.; Wu, Z. K.; Zhu, C. J. Org. Chem. 2016, 81, 12181. doi: 10.1021/acs.joc.6b01847
Huo, Z. B.; Gridnev, I. D.; Yamamoto, Y. J. Org. Chem. 2010, 75, 1266. doi: 10.1021/jo902603v
(a) Fischer, D.; Tomeba, H.; Pahadi, N. K.; Patil, N. T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2007, 46, 4764.
(b) Fischer, D.; Tomeba, H.; Pahadi, N. K.; Patil, N. T.; Huo, Z. B. Yamamoto, Y. J. Am. Chem. Soc. 2008, 130, 15720.
Bianchi, G.; Chiarini, M.; Marinelli, F.; Rossi, L.; Arcadi, A. Adv. Synth. Catal. 2010, 352, 136. doi: 10.1002/adsc.200900668
Kanazawa, C.; Terada, M. Chem.-Asian J. 2009, 4, 1668. doi: 10.1002/asia.200900342
Dev, K.; Maurya, R. RSC Adv. 2015, 5, 13102. doi: 10.1039/C4RA10452H
Brahmchari, D.; Verma, A. K.; Mehta, S. J. Org. Chem. 2018, 83, 3339. doi: 10.1021/acs.joc.7b02903
Tan, J.; Tong, Y.; Chen, Z. Y. ChemistrySelect 2018, 3, 3886. doi: 10.1002/slct.201800666
Mancuso, R.; Mehta, S.; Gabriele, B.; Salerno, G.; Jenks, W. S.; Larock, R. C. J. Org. Chem. 2009, 75, 897.
Du, J. Y.; Ma, Y. H.; Yuan, R. Q.; Nie, S. Z.; Ma, C. L.; Li, C. Z.; Zhao, C. Q. Org. Lett. 2018, 20, 477. doi: 10.1021/acs.orglett.7b03863
Jung, Y.; Singh, D. K.; Kim, I. Beilstein. J. Org. Chem. 2016, 12, 2689. doi: 10.3762/bjoc.12.266
Warner, A. J.; Churn, A.; McGough, J. S.; Ingleson, M. J. Angew. Chem., Int. Ed. 2017, 56, 354. doi: 10.1002/anie.201610014
Verma, A. K.; Rustagi, V.; Aggarwal, T.; Singh, A. P. J. Org. Chem. 2010, 75, 7691. doi: 10.1021/jo101526b
Park, J. H.; Bhilare, S. V.; Youn, S. W. Org. Lett. 2011, 13, 2228. doi: 10.1021/ol200481u
Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K. Org. Lett. 2006, 8, 5517. doi: 10.1021/ol062190+
Kanazawa, C.; Terada, M. Tetrahedron Lett. 2007, 48, 933. doi: 10.1016/j.tetlet.2006.12.015
Mancuso, R.; Pomelli, C. C.; Malafronte, F.; Maner, A.; Marino, N.; Chiappe, C.; Gabriele, B. Org. Biomol. Chem. 2017, 15, 4831. doi: 10.1039/C7OB00962C
(a) Zheng, Y. N.; Liu, M. L.; Qiu, G. Y. S.; Xie, W. L.; Wu, J. Tetrahedron 2019, 75, 1663.
(b) Wang, Y. H.; Qiu, G. Y. S.; Zhou, H. W.; Xie, W. L.; Liu, J. B. Tetrahedron 2019, 75, 3850.
Schlemmer, C.; Andernach, L.; Schollmeyer, D.; Straub, B. F.; Opatz, T. J. Org. Chem. 2012, 77, 10118. doi: 10.1021/jo3017378
Mehta, S.; Yao, T.; Larock, R. C. J. Org. Chem. 2012, 77, 10938. doi: 10.1021/jo301958q
(a) Wang, R. X.; Yuan, S. T.; Liu, J. B.; Wu, J.; Qiu, G. Y. S. Org. Biomol. Chem. 2018, 16, 4501.
(b) Wang, Y. H.; Ouyang, B.; Qiu, G. Y. S.; Zhou, H. W.; Liu, J. B. Org. Biomol. Chem. 2019, 17, 4335.
Mehta, S.; Waldo, J. P.; Larock, R. C. J. Org. Chem. 2008, 74, 1141.
(a) Faizi, D. J.; Davis, A. J.; Meany, F. B.; Blum, S. A. Angew. Chem., Int. Ed. 2016, 55, 14286.
(b)Issaian, A.; Faizi, D. J.; Bailey, J. O.; Mayer, P.; Berionni, G.; Singleton, D. A. Blum, S. A. J. Org. Chem. 2017, 82, 8165.
Xu, J.; Yu, X. X.; Yan, J. X.; Song, Q. L. Org. Lett. 2017, 19, 6292. doi: 10.1021/acs.orglett.7b02971
(a) Goldfinger, M. B.; Swager, T. M. J. Am. Chem. Soc. 1994, 116, 7895.
(b) Goldfinger, M. B.; Crawford, K. B.; Swager, T. M. J. Am. Chem. Soc. 1997, 119, 4578.
(a) Yao, T.; Campo, M. A.; Larock, R. C. Org. Lett. 2004, 6, 2677.
(b) Yao, T.; Campo, M. A.; Larock, R. C. J. Org. Chem. 2005, 70, 3511.
Li, C. W.; Wang, C. I.; Liao, H. Y.; Chaudhuri, R.; Liu, R. S. J. Org. Chem. 2007, 72, 9203. doi: 10.1021/jo701504m
Chen, Y.; Huang, C. L.; Liu, X. C.; Perl, E.; Chen, Z. W.; Namgung, J.; Zhang, G.; Hersh, W. H. J. Org. Chem. 2014, 79, 3452. doi: 10.1021/jo5001803
Alam, K.; Kim, J. G.; Kang, D. Y.; Park, J. K. Adv. Synth. Catal. 2019, 361, 1683. doi: 10.1002/adsc.201801506

图式 1 基于邻炔基苯胺的多取代吲哚的合成
Scheme 1 Synthesis of muti-substituted indoles based on 2-alkynyl aniline

图式 2 N, N-二烷基邻炔基苯胺的环化反应机理
Scheme 2 Mechanism for cyclization reaction of N, N-dialkyl- 2-(1-alkynyl)anilines

图式 3 3-磺酰基吲哚和3-硫基吲哚的选择性合成
Scheme 3 Selective synthesis of 3-sulfonylindoles and 3-sulfanylindoles

图式 4 N-烷基-3-磺酰基吲哚和N-烷基-3-硫基吲哚类化合物合成反应机理
Scheme 4 Mechanism for the synthesis of N-alkyl-3-sulfonylindoles and N-alkyl-3-sulfanylindoles

图式 5 2-炔基芳基叠氮化物与亚磺酸的环化反应机理
Scheme 5 Mechanism for cyclization reaction of 2-alkynyl-arylazides and sulfinic acids

图式 7 2-(1-芳基乙炔基)苯甲酰胺的环化反应机理
Scheme 7 Mechanism for cyclization reaction of 2-(1-arylethy- nyl)benzamides

图式 9 区域调控的2-炔基苯甲酸的分子内环化反应
Scheme 9 Regiocontrolled intramolecular cyclizations of 2-alkynylbenzoic acid

图式 10 2-炔基苯甲酸的区域选择性碘代内酯化反应
Scheme 10 Regioselective iodolactonization of 2-alkynylben- zoic acids

图式 11 2-炔基苯甲酰胺的环化反应机理
Scheme 11 Mechanism for cyclization reaction of 2-alkynyl- benzamide

图式 13 2-炔基硫代苯甲醚的硫硼化反应机理
Scheme 13 Mechanism for thioboration reaction of 2-alkynyl- thioanisoles

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



























 下载:
下载:





 下载:
下载: