图式 1.
硫氰化策略
Scheme 1.
Thiocyanation strategy

有机硫氰酸盐(RSCN)是一种重要的有机合成中间体, 通过简单的化学转换即可获得硫醚、硫醇、异硫氰、含硫含氮的杂环等多种类型的有机硫化物.此外, 这些硫氰酸酯类化合物具有抗微生物活性、抑制酶活性、杀虫活性等多种生物活性, 在医药、农药和材料等诸多领域中具有重要应用价值[1].优异的生物活性和广阔的应用前景, 极大地引起了有机合成化学家们的研究兴趣.
近年来, 有机化学家们对硫氰基化反应及应用开展了较为深入的研究和探索, 其构建策略主要包括过渡金属或非金属催化, 或化学计量氧化剂参与的传统硫氰基化策略(Scheme 1a), 其主要涉及亲核取代、亲电取代和自由基等反应类型[2]. 2016年, Gulea[3]和Pate[4]分别对硫氰酸酯和α-羰基硫氰酸酯类化合物的合成及应用做了很好的总结.最近, 金城安等[5]重点归纳总结了近三年来硫氰基化合物的合成进展, 对硫氰化研究领域的发展具有重要作用.
开发绿色、高效、高原子经济性的C—S键构筑策略是有机合成化学领域重要的研究内容之一[6].光催化是以光能作为直接能源, 电催化是以电能(电子传递)作为能源, 符合绿色化学要求.近年来, 有机合成工作者们利用光催化策略, 以空气作为绿色氧化剂已成功实现多种类型的硫氰基化反应.此外, 有机电化学合成作为一种新颖的、绿色的策略, 是利用连续不断的电子得失来替代传统的化学氧化还原试剂.有机合成工作者利用这一特性成功地实现了一系列无金属、无氧化剂参与的硫氰基化反应(Scheme 1b).在这篇综述中, 整理讨论了利用光催化、电催化的新型策略来实现硫氰基化反应, 以显示光催化、电催化硫氰基化反应的独特性和新型性, 以期为读者了解光、电催化硫氰化反应的研究动态提供帮助.
通过C—H键活化官能化反应构建C—S键是最直接、高效和绿色的途径, 具有原子经济性而被广泛关注[5].由于惰性的C(sp3)—H键的活化往往需要较高的活化能, 这增加了C(sp3)—H键活化官能化的难度和挑战.与传统热力学C(sp3)—H硫氰基化反应方法相比, 光催化氧化偶联C(sp3)—H硫氰基化反应是实现C(sp3)—H参与硫氰基化反应的有效策略.
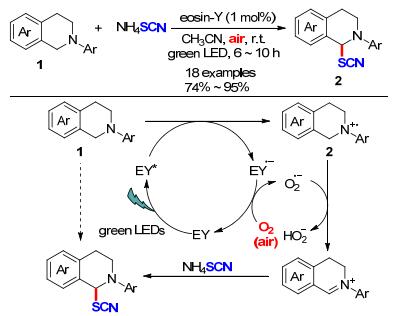
2015年, Yadav课题组[7]报道了一种以Eosin Y为光催化剂, 空气氛围中, 在绿光照射下实现叔胺α-位C(sp3)—H键的硫氰基化反应.该方法具有光催化剂用量低(Eosin Y 1 mol%)、反应温度低、底物适用范围宽等优点.该方法能够以74%~95%的产率获得一系列硫氰化四氢异喹啉衍生物2.根据研究, 作者提出了一种可能的机理:首先, EY被光激发到激发态EY*, 底物与激发态EY*经单电子转移得到中间体A和RB·-; 随后, 氧气氧化RB·-得到基态EY, 并生成O2·-, O2·-可攫取A的α位质子形成亚胺离子中间体B; 最后, SCN进攻B得到最终目标产物(Scheme 2).
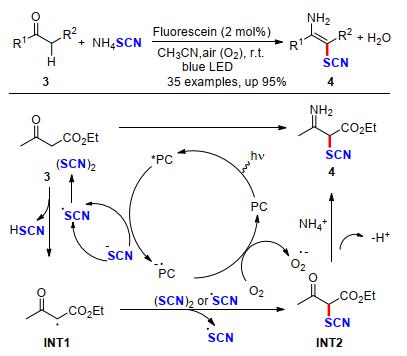
2018年, 刘强课题组[8]利用荧光素作为光催化剂, 在氧化条件下, 通过交叉偶联硫氰酸铵和活化羰基酮类化合物, 为氨化/硫氰化烯烃的构建发展了一种简单、高效、高原子经济的合成方法(Scheme 3).该方法以低毒、廉价的硫氰酸铵为胺源和硫氰基源, 可以方便、高效地获得一系列多取代烯烃, 副产物为水.作者结合机理实验和理论计算, 提出了一种可能的机理:反应开始于荧光素被可见光照射激发到激发态经单电子氧化硫氰酸铵生成•SCN自由基和PC•-, 氧气氧化PC•-生成超氧化物O2•-并再生光催化剂.然后, •SCN自由基从3中攫氢, 生成碳中心自由基INT1和硫氰酸, 获得的中间体INT1被其它•SCN捕获生成中间体INT2.由于INT2中羰基缺电子容易被NH3亲核进攻, 并消除水得到目标产物4.
光催化自由基硫氰基化反应是合成有机硫氰化物的一种重要途径. 1978年, Guy课题组[9]就发展了一种紫外光照射, 室温环境下, 通过自由基加成途径实现了烯烃的双硫氰基化反应, 该方法能够以24%~99%的收率获得一系列的双硫氰化烯烃(Scheme 4), 不足是该方法需要用高能量的紫外光作为光源.
2015年, Guan等[10]自由基加成/分子内环化串联反应, 以N-烷基-N-甲基丙烯基苯酰胺为原料实现了硫氰化异喹啉二酮类化合物的合成(Scheme 5).作者利用吖啶盐作为光催化剂、利用廉价易得的硫氰酸铵通过单电子氧化得到硫氰基自由基, 实现了21个具有高功能化药物样分子结构的硫氰化异喹啉二酮类化合物的绿色构建.作者根据一系列的机理实验以及相关文献基础上, 提出了一种可能的反应机理:首先, 在可见光照射下, Acr+-Mes被光激发为Acr+-Mes*, 激发态光催化剂Acr+-Mes*从硫氰酸盐中攫取一个电子得到•SCN, 同时生成Acr•-Mes.随后, Acr•-Mes被氧气氧化为Acr+-Mes完成光催化循环, 并产生超氧阴离子O2•-拿走NH4+质子得到HO2•.同时, 生成的硫氰基自由基与7发生自由基加成形成C—S键, 并得到烷基自由基中间体8, 8经自由基芳构化生成中间体9, HO2•氧化9得到目标产物10.

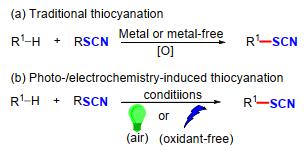
2018年, Bolm课题组[11]以N—Br亚砜亚胺与NH4SCN为原料, 以二氯甲烷(DCM)作为溶剂, 在常温下反应16 h制备了一种新型硫氰化试剂(11).这种新型的硫氰化试剂在蓝光照射下, 以Ir为催化剂, 可以很好地应用于烯烃的双官能团化反应, 能够以38%~81%的收率获得一系列新颖的硫氰化亚胺化产物13(Scheme 6).
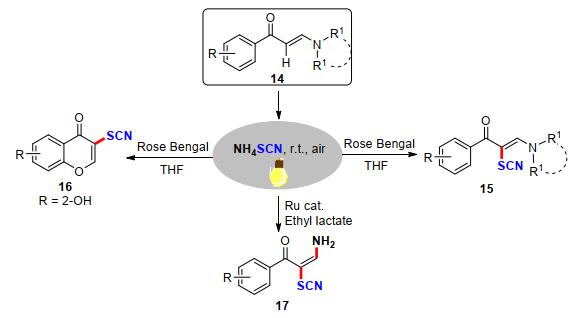
2019年, 万结平课题组[12]利用可见光诱导策略成功实现了叔烯胺酮α-C—H键活化硫氰化反应.该方法为构建硫氰化烯烃类衍生物提供了一种直接、简单、有效的方法.作者通过在底物14的邻位引入一个羟基官能团, 实现硫氰化色酮类衍生物16的合成.当将光催化剂由Rose Bengal改为Ru(bpy)3Cl2•6H2O时, 能够得到一系列具有氨基和硫氰基的双官能化烯烃17, 作者认为在该过程中Ru(bpy)3Cl2•6H2O的作用为路易斯酸(Scheme 7).
近几年来, 有机化学家们又发展了一系列高效节能的可见光催化硫氰化策略, 相对传统的紫外光催的硫氰化反应, 该类策略是直接以可见光为光源、空气作为氧化剂, 具有绿色环保、能耗低的优点.例如, 2014年, Li课题组[13]为吲哚β位引入硫氰基发展了一种绿色、高效的可见光催化策略(Scheme 8).以14 W的节能灯为光源, 廉价易得的Rose Bengal为光催化剂, 空气为氧化剂, 在四氢呋喃溶剂中照射24 h, 以73%~95%的产率获得15个不同种类的β-硫氰化吲哚化合物.作者认为该反应机理可能是:首先, 光照RB转变为激发态RB*, SCN被RB*经单电子氧化得到•SCN自由基A, 并产生RB•-自由基阴离子被氧气氧化还原为基态RB.随后, •SCN自由基A与底物18经亲电加成得到中间体B, B被进一步氧化产生阳离子中间体C.最后, C失去质子得到目标产物19.
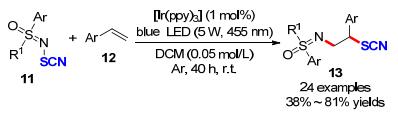
随后, He课题组[14]借用异相催化策略为吲哚β位引入硫氰基发展了一种无毒、易得、稳定的TiO2/MoS2纳米光催化剂.该方法对电中性的底物具有较好的催化效率, 并能获得优异的收率.然而, 该策略在底物的适用范围上还存在一定的局限性, 底物中有硝基或羧基等强吸电子时, 目标产物的产率较低, 甚至是N-Boc保护的底物仅能获得痕量的产物.但是, 催化剂的可重复利用性是该方法的一大优势(Scheme 9).
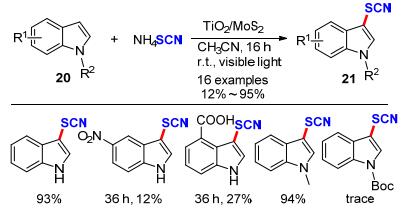
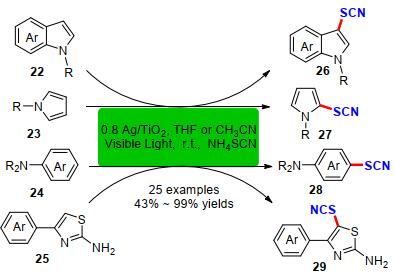
2018年, Sarvari课题组[15]通过简单的水热法合成了一种Ag/TiO2纳米管为光催化剂, 以空气为终端氧化剂, 通过可见光照射成功地实现了吲哚、苯胺、吡咯和2-氨基噻唑类化合物与硫氰酸铵的硫氰化反应(Scheme 10).该方法具有底物适用范围宽、光催化剂可循环利用、催化效率高等优点.其产物最高收率可达99%, 为一系列硫氰化的N-杂环化合物合成提供新思路.
2015年, 我们课题组[16]以KSCN为硫氰基源, 利用廉价易得的K2S2O8为氧化剂, 1, 2-二氯乙烷(DCE)为溶剂, 室温环境下, 成功实现了咪唑并[1, 2-a]吡啶类化合物C-3位硫氰基化反应(Scheme 11a).随后, Hajra课题组[17]利用蓝光照射Eosin Y的光催化策略也实现了咪唑并[1, 2-a]吡啶类化合物30的C-3位硫氰基化反应(Scheme 11b).通过对比两种策略不难发现, 两种方法都能够以高产率合成具有潜在生物活性的3-硫氰基咪唑并[1, 2-a]吡啶类化合物31, 从其底物扩展得知:无论是供电子基还是强吸电子基团的底物, 都能很好地适应两种催化体系.
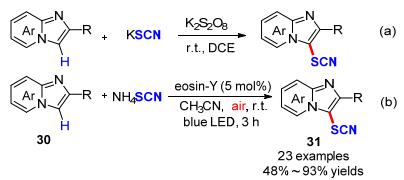
同年, Singh课题组[18]报道了利用光催化实现了异腈类衍生物32分子内环化硫氰化构建6-硫氰菲啶类化合物.该反应以Eosin Y为光催化剂, NH4SCN为硫氰基源, 乙腈反应溶剂, 在室温环境下, 绿光照射8~16 h即可高效合成17个带有不同官能团的6-硫氰菲啶衍生物33, 产物的收率最高可达94%, 这为6-硫氰菲啶化合物的有效合成提供了新思路(Scheme 12).
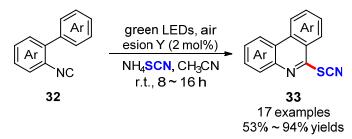
最近, Jain等[19]报道了以Eosin Y作为可见光催化剂, 空气作为末端氧化剂, 成功实现了2-芳基喹啉酮化合物34的C-3位硫氰基化反应(Scheme 13).该方法可方便快捷地获得多种2-芳基-3-硫氰基喹诺林-4-酮类化合物35.此外, 该方法还可用于选择性实现芳氨类衍生物36对位C—H键活化硫氰化构建硫氰基芳氨衍生物37, 且目标产物也有中等至良好的收率.
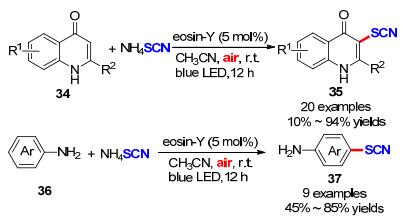
2018年, Kshirsagar课题组[20]开发了一种便捷、高效、无金属的3-硫氰基苯并噻吩的合成方法(Scheme 14).该方法以2-炔基硫代茴香醚化合物38作为底物, 利用Eosin Y作为光敏剂, 在蓝光照射介导下光催化氧化硫氰酸铵盐的阴离子为硫氰基自由基, 经自由基加成串联环化得到目标产物42.采用这种光催化策略成功实现了20种具有潜在生物活性的3-取代苯并噻吩衍生物的绿色合成.作者根据机理实验和以往文献报道, 提出了一种可能的机理:首先, SCN通过单电子转移过程被激发态光催化剂EY*氧化为硫氰基自由基, 同时生成EY·-, EY·-被氧气氧化为EY, 实现催化剂再生, 生成超氧自由基负离子.与此同时, 硫氰基自由基与底物38作用经自由基加成串联环化得到中间体41.随后, 在DMSO作为亲核试剂环境下, 中间体41经C—S断裂脱甲基过程得到目标产物42.
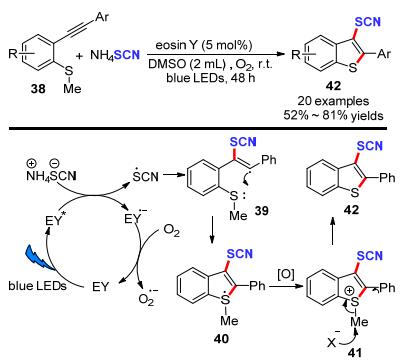
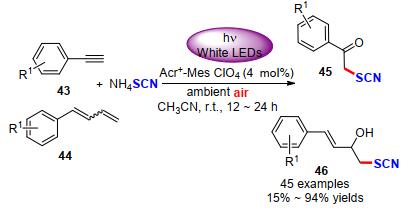
随后, Gullapalli等[21]采用光催化策略, 成功实现了末端炔烃(43)或联烯(44)类化合物选择性硫氰化反应构建α-硫氰基酮(45)或醇(46).该方法是以吖啶盐作为光敏剂, 空气为氧化剂, NH4SCN为硫氰基源, 经白光照射6~24 h, 以15%~94%的产率实现45个目标化合物的构筑(Scheme 15).
过去几十年以来, 氧化交叉偶联已被发展成为构建新化学键最直接的方法之一[22].选择廉价易得的底物作为起始原料, 具有无需预官能化、缩减了繁琐的合成步骤的优点.然而, 通常需要氧化剂促进氧化交叉偶联的进行, 造成了材料成本高, 降低了整体转化的原子经济性.此外, 化学计量的氧化剂的使用很有可能引起一些不必要的副反应或过氧化.因此, 非常有必要发展无化学氧化剂参与的氧化偶联反应策略.
近十年来, 电化学有机合成作为一种高效、绿色、环境友好的构筑平台受到了广大有机合成化学家们的持续关注[6, 22].电合成是利用电子作为氧化剂, 可避免化学计量的化学氧化剂的使用, 从而避免了一些化学废料的生成.尤其是近几年, 一些电化学氧化交叉偶联反应被用于C—H键活化硫氰化构建C—S键[22o].
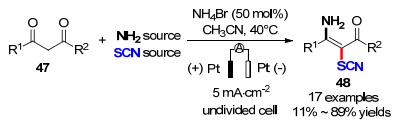
众所周知, 由于C(sp3)—H键的活化需要较高的活化能, 使得C(sp3)—H键活化要难于C(sp2)—H.因此, 该类型的反应受到有机化学家们广泛关注.例如, 2016年曾程初课题组[23]开发了一种成对电合成反应活化C—H键, 通过连续的C—S、C—N键的形成构建3-氨基-2-硫氰基-α, β-不饱和羰基衍生物(Scheme 16).作者利用50 mol%的NH4Br作为电催化媒介, 以铂片作为工作电极和对电极, 在无外加电解质的乙腈为溶剂中, 以电流密度为5 mA/cm2电解即可以11%~89%的收率获得17个不同类型的3-氨基-2-硫氰基-α, β-不饱和羰基衍生物(48).在该成对电解过程中, 氨基和硫氰酸酯分子的来源于单一试剂或乙酸铵和异氰酸钾的组合.
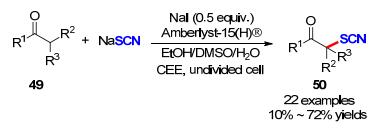
随后, 他们课题组[24]又发展了一种非均相固体盐Amberlyst-15(H) [A-15(H)]为质子催化剂, 以NaI作为电催化媒介, 采用恒电流电解的方法实现了酮羰基α-位C(sp3)—H键的硫氰基化反应构建硫氰化酮衍生物.与以往的合成方法相比, 这种方法无需外加电解质.值得一提的是, 这种新型的固体盐可多次重复使用, 且产率没有显著的降低(Scheme 17).
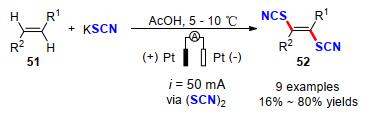
电催化烯烃硫氰基化反应的研究相对较早, 在1973年De Klein[25]利用电氧化硫氰酸盐生成硫氰与烯烃的加成反应即可获得双硫氰化产物52.他们总结了电氧化烯烃的硫氰化反应, 相对于传统化学合成路径具有以下优势: (1)双键不可能卤化(当双键与硫氰反应缓慢, 与Cl或Br反应快速时, 这特别有利); (2)通过控制氧化电位或电流, 从而减少副产物的生成.作者使用恒电流电解的方法, 在5~10 ℃的温度的乙酸环境下, 以16%~80%的收率获得一系列新颖的二硫氰酸盐产物, 为电催化硫氰化反应的研究奠定了理论基础(Scheme 18).
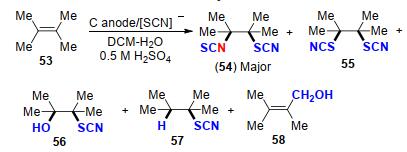
2015年, Becker课题组[26]采用“阳极一锅法”在水和二氯甲烷的两相酸性环境下实现了多取代烯烃硫氰基化和异硫氰基化反应(Scheme 19).作者以四甲基乙烯为模板底物, 优化考察了实验涉及到不同的酸、阳极、电流密度、电荷消耗和电解方法对反应的影响.研究发现在两相介质中, 以中等产率得到了两个主要产物54 (major)和55.其他烷基取代烯烃也产生了类似的加成产物, 但主要产物是两个SCN加成的产物.然而, 当苯乙烯和茚酮类底物在水-乙腈组成的均相介质中反应时, 产物则主要是SCN/OH加合物.
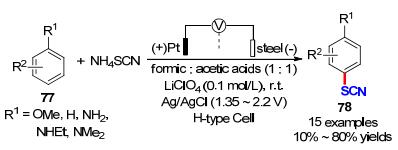
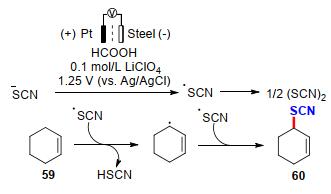
次年Becker课题组[27]还开发了一种两步阳极氧化策略, 实现了烯烃的硫氰化反应(Scheme 20).首先, 作者以铂片作为阳极, 钢片作为对电极, Ag/AgCl作为参比电极, 在以甲酸为溶剂, LiClO4为电解质的分离池中加入NH4SCN作为硫氰基源, 在电压为1.25 V的恒电压模式下经阳极氧化形成硫氰(SCN)2.随后, 加入不同种类的烯烃衍生物, 经自由基反应路径即可获得目标产物60.
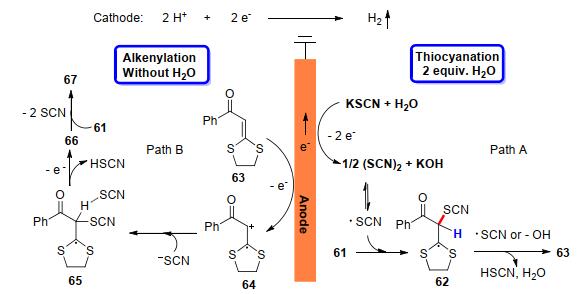
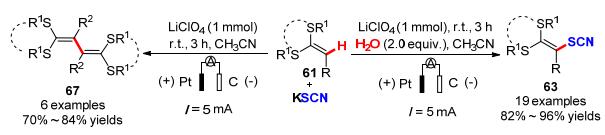
烯烃C—H键活化官能化一直是广大合成化学家关注的热点, 特别是内烯C—H键活化官能化反应途径成为多样取代烯烃最直接有效的构建方法之一.如何实现便捷、高效、可控性强、环境友好的内烯C—H键活化官能化呢? 2019年, 我们课题组[28]报道了一种电化学氧化条件下H2O控制内烯选择性硫氰化和烯基化反应.这是首例结合电化学-H2O控制来实现内烯选择性硫氰基化和烯基化的反应(Scheme 21).该反应以廉价易得的二硫代缩醛为原料, 以Pt作为阳极, 碳棒作为阴极, LiClO4为电解质, 乙腈为溶剂, 在恒电流常温反应条件下, 通过H2O控制反应的选择性, 实现了内烯的硫氰化和烯烃的自偶联反应.结果表明, 该体系具有底物适用范围广和官能团兼容性好等突出优点, 芳基、烷基以及杂环取代的二硫代缩醛类化合物均能在该反应条件下高选择性地制得相应的硫氰基化产物.为了深入探究反应机理, 课题组基于相关文献研究背景, 通过一系列控制实验以及结合循环伏安测试结果提出了涉及的反应机理途径.途径A:当水存在时, KSCN和水在阳极更容易被氧化生成(SCN)2, 通过(SCN)2均裂得到硫氰基自由基, 进而与底物发生自由基加成反应获得硫氰化产物63.途径B:无水条件下, 硫代缩醛底物在阳极失去一个电子被氧化生成自由基正离子64, 与碱性亲电试剂KSCN反应得到中间体65, 通过中间体65失去一个电子和质子生成氰基化物63, 最后, 硫氰根作为拟卤素基团离去获得自偶联产物67, 同时在阴极放出氢气(Scheme 22).
硫氰基衍生物是有机合成中很有价值的中间体, 特别是在杂环化合物的制备方面.在均相非水溶剂中电解的缺点是能耗高, 需要纯溶剂和昂贵的辅助电解质盐. 1992年, Gurjar课题组[29]采用一种两相电解硫氰酸铵生成(SCN)3-的策略, 成功实现了苯胺、苯酚类衍生物选择性硫氰化反应(Scheme 23).随后, 他们课题组还详细研究了电解温度、电解时间、非水溶剂性质、非水相体积分数等实验参数对电流效率的影响, 循环伏安测试研究表明(SCN)3-是产生于水相体系中后被带入非水相中参与反应[30].
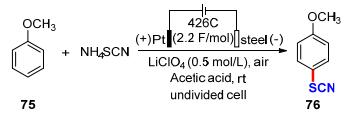
芳香硫氰酸盐是制备杂环化合物的重要中间体, 特别是用作农用化学品、染料和药品的前体.在各种已知的硫氰化方法中, 以硫氰(SCN)2作为硫氰化剂最为常见.然而, 由于(SCN)2对水、光和热的敏感性, 以及在非水介质中易于聚合, 其稳定性较差, 因此, 其取代产物的产率通常较低.基于此, Becker课题组[31]在2006年采用电化学方法成功实现了苯甲醚对位硫氰化反应(Scheme 24).作者以苯甲醚75为底物对比性地研究了恒电流、恒电压电解的区别, 考察了阳极材料、溶剂、耗电量、电流密度、电解质和浓度等因素对硫氰基化产物76形成的影响.
2010年, Becker课题组[32]又利用H-型的分离电解池在恒电压的酸性条件下, 选择性地实现了苯甲醚、甲苯和苯胺类衍生物对位硫氰基化反应(Scheme 25).作者认为该反应首先是硫氰酸根在阳极被氧化生成硫氰基自由基, 硫氰基自由基发生聚合得到硫氰(SCN)2.随后, (SCN)2被酸性溶剂极化, 并攻击底物的芳香核以提供相应的硫氰酸酯衍生物78.但遗憾的是, 该方法对芳香胺类底物收率较低, 仅有15%~42%的产率.
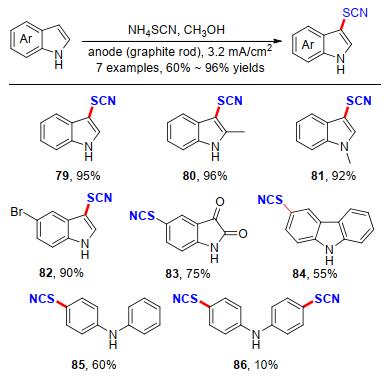
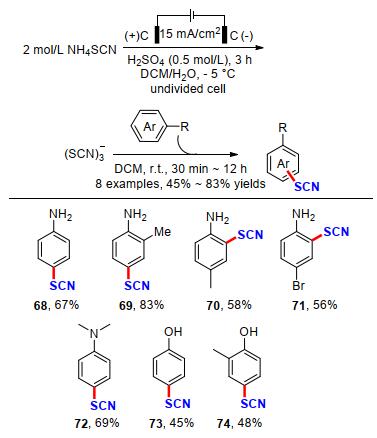
硫氰酸盐基团在化学和生物化学上具有广泛的用途, 已报道了多种芳族的硫氰化方法.例如I2/NH4SCN, Mn(OAc)3/NH4SCN, 酸性的蒙脱石(K-10), Zn(SCN)2/ Cl2, H5IO6/KSCN, FeCl3, 硝酸铈铵, 三氟异氰尿酸/SiO2等[33].然而, 这些方法中存在一些缺点, 包括产率低(特别是芳香胺的硫氰化)、强酸性或苛刻的氧化条件、反应时间长和/或高温以及使用昂贵和有毒的试剂.基于此, 2013年Nikoofar课题组[34]报道了一种含氮(杂)芳香族化合物在恒电流条件下, 以硫氰酸铵为原料, 在室温条件下进行阳极硫氰化反应.值得指出的是, 吲哚、咔唑、二苯胺类型的N-杂原子底物在该反应条件下都能以中等的收率得到目标产物79~85.此外, 利用该策略以二苯胺为原料还能得到双硫氰化的产物86 (Scheme 26).
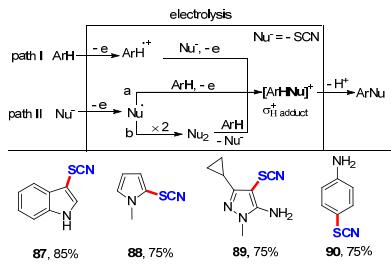
2014年Petrosyan课题组[35]在path I的基础上, 通过阳极氧化硫氰酸根生成硫氰, 与芳烃发生亲核取代反应(path II b), 成功实现了吲哚、N-甲基吡咯、吡唑和苯胺等N-杂环衍生物的硫氰化反应(Scheme 27).值得注意的是, 相比于Becker教授的工作, 该方法对于选择性实现苯胺对位硫氰化反应构建化合物90具有更高的收率(75%).
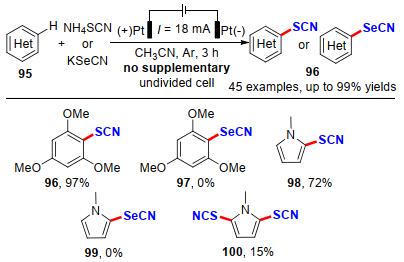
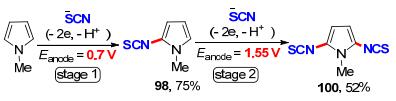
2018年, 江洪课题组[36]开发了一种低成本、高效的富电子芳烃硫代和硒代氰化策略(Scheme 28).在没有任何催化剂或氧化剂的温和条件下, 这种策略适用于各种情况(杂)芳香族化合物, 如吲哚、吡咯、苯胺和茴香醚等衍生物的硫氰基化和硒基化反应, 以52%~99%的产率实现45个不同取代基的硫代和硒代氰化芳杂环化合物的构建.遗憾的是, 该方法不能得到硒氰化的均三苯甲醚产物97和硒氰化的N-甲基吡咯产物99, 能够得到双硫氰化产物100, 但产率较低. 2019年, Petrosyan课题组[37]在此基础上, 提出了一种两步控制电压电解的策略, 成功实现了N-甲基吡咯的双C—H键活化硫氰化, 产率提升到了52% (Scheme 29).
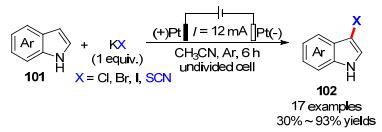
卤代吲哚作为生物活性的天然产物中必不可少的结构单元, 受到了合成化学家们极大关注.江洪课题组[38]同样在没有任何催化剂或氧化剂, 无额外电解质的温和条件的电化学策略, 成功实现吲哚类衍生物C-3位卤化(Cl, Br, I)或硫氰化反应, 各种含电子丰富的吲哚衍生物表现出了很大的转化效率(Scheme 30).
2016年, Kokorekin教课题组[39]采用电化学方法, 在室温条件下以Pt作为阳极, NH4SCN作为硫氰基源, NaClO4作为电解质, CH3CN作为溶剂, 实现了吡唑并[1, 5-a]嘧啶化合物103的3位选择性硫氰酸化反应, 产物104的收率为69%~87%.但是对于具有较高氧化电位的底物, 当作者在其反应体系中加入ZnCl2时, 其收率可从0%提高到70%.作者认为ZnCl2起着路易斯酸的作用.他们的研究揭示了电氧化吡唑并[1, 5-a]嘧啶硫氰酸化的反应的效率取决于底物的氧化电位, 路易斯酸的加入可以帮助调节底物的氧化电势(Scheme 31).
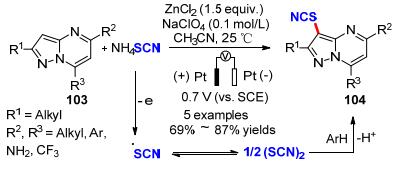
随后, 他们课题组[40]分别针对高活性和低活性的吡唑并[1, 5-a]嘧啶衍生物的电氧化硫氰化反应开发三种新的互补体系(32).第一种方法(电氧化-SCN生成(SCN)2)适用于高活性芳烃(Ep<1.70 V), 第二种方法(芳基阳离子生成法)和第三种方法[ZnCl2, via (SCN)2]适用于低反应性的芳烃(Ep>1.70 V).作者以硫氰酸盐(NH4SCN或KSCN)为硫氰基源, 在室温下电解硫氰酸根离子/吡唑并[1, 5-a]嘧啶105混合物, 成功地实现了11种具有抗真菌活性的3-硫氰酸基吡唑并[1, 5-a]嘧啶衍生物106的合成, 其目标产物的产率为60%~89% (Scheme 32).
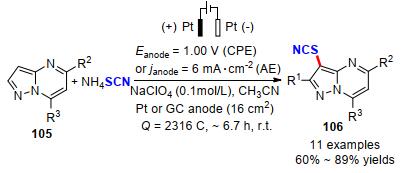
2019年, Gooßen等[41]提出了一种简单的、无过渡金属催化的异硫氰化芳基硼酸和三氟硼酸芳酯构建芳基硫氰化物的电化学方法.硫氰酸盐阴离子经阳极氧化原位生成SCN亲电试剂, 以高到100%的产率实现了22个不同类型的芳基硼酸衍生物的硫氰基化反应.遗憾的是, 带有强吸电子基团类芳基硼酸和烷基硼酸类底物在该反应条件下不能得到产物(Scheme 33).
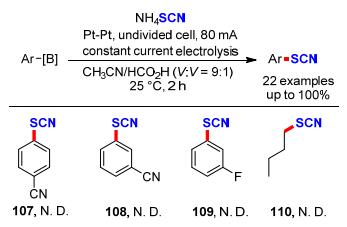
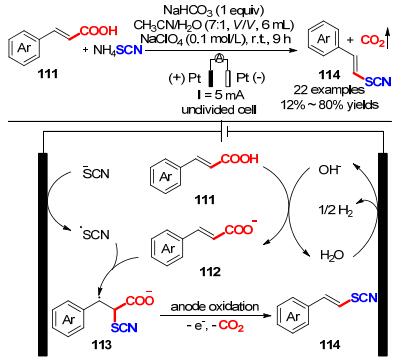
同年, Huang课题组[42]利用Kolbe反应, 报道了一种简单、绿色的电催化肉桂酸111与NH4SCN在水溶液中脱羧偶联制备芳基乙烯硫氰酸酯的电化学方法(Scheme 34).采用恒电流电解的方法, 以NH4SCN为硫氰基源, 乙腈和水作为溶剂, NaHCO3作为碱, 高氯酸钠作为电解质, 室温5 mA电解反应9 h, 以12%~80%产率合成22种芳基乙烯硫氰酸酯化合物.但不足的是, 带有强吸电子基团的烯烃作为底物时产率相对较低, 甚至烷基烯烃在该反应条件下不能够得到理想的目标产物.作者对该反应提出了一种可能的反应机理:首先, 硫氰酸根阴离子在阳极表面上经单电子氧化得到硫氰酸根自由基中间体.随后, 生成的硫氰酸根自由基与112发生自由基加成形成中间体113, 113经过脱羧得到目标产物乙烯基硫氰酸酯(114).但该反应也不能排除阳极氧化硫氰酸根阴离子生成硫氰(SCN)2参与反应的可能性.
近年来, 通过使用可见光、电催化介入的硫氰基化反应这两种新型、绿色的构筑策略, 可以实现一系列热力学上难以实现的硫氰基化反应.这为合成医药、功能材料等提供了新的方法, 且具有绿色、高效、安全等优势.尽管光、电催化介导的硫氰化反应策略已取得了一些进展, 但对于复杂分子和天然产物分子的硫氰化修饰仍然还处于研究初期.电化学有机合成是通过电子的得失来替代传统的氧化还原试剂, 可通过改变电流或电压及时调控反应活化能、控制反应的选择性, 是一种的绿色、经济的策略, 可实现一系列化学转化, 极具潜力.相信该类合成策略能够得到进一步的优化和拓展, 在更多的催化反应中应用, 适用于更多种类的底物, 为医药、功能材料领域中硫氰化物的制备提供绿色、高效、环境友好的平台.
(a) Capon, R. J.; Skene, C.; Liu, E. H.-T.; Lacey, E.; Gill, J. H.; Heiland, K.; Friedel, T. J. Org. Chem. 2001, 66, 7765.
(b) Dutta, S.; Abe, H.; Aoyagi, S.; Kibayashi, C.; Gates, K. S. J. Am. Chem. Soc. 2005, 127, 15004.
(c) Elhalem, E.; Bailey, B. N.; Docampo, R.; Ujváry, I.; Szajnman, S. H.; Rodriguez, J. B. J. Med. Chem. 2002, 45, 3984.
(d) Kokorekin, V.; Terent'ev, A.; Ramenskaya, G.; Grammatikova, N.; Rodionova, G.; Ilovaiskii, A. Pharm. Chem. J. 2013, 47, 422.
(e) Yasman, Y.; Edrada, R. A.; Wray, V.; Proksch, P. J. Nat. Prod. 2003, 66, 1512.
(a) Khalili, D. Chin. Chem. Lett. 2015, 26, 547.
(b) Zeng, Y.-F.; Tan, D.-H.; Chen, Y.; Lv, W.-X.; Liu, X.-G.; Li, Q.; Wang, H. Org. Chem. Front. 2015, 2, 1511.
(c) Guo, L.-N.; Gu, Y.-R.; Yang, H.; Hu, J. Org. Biomol. Chem. 2016, 14, 3098.
(d) Khalili, D. New J. Chem. 2016, 40, 2547.
(e) Chen, Q.; Lei, Y.; Wang, Y.; Wang, C.; Wang, Y.; Xu, Z.; Wang, H.; Wang, R. Org. Chem. Front. 2017, 4, 369.
(f) Ji, F.; Fan, Y.; Yang, R.; Yang, Y.; Yu, D.; Wang, M.; Li, Z. Asian J. Org. Chem. 2017, 6, 682.
(g) Jiang, G.; Zhu, C.; Li, J.; Wu, W.; Jiang, H. Adv. Synth. Catal. 2017, 359, 1208.
(h) Prieto, A.; Uzel, A.; Bouyssi, D.; Monteiro, N. Eur. J. Org. Chem. 2017, 2017, 4201.
(i) Chen, Y.; Wang, S.; Jiang, Q.; Cheng, C.; Xiao, X.; Zhu, G. J. Org. Chem. 2018, 83, 716.
(j) Khaikate, O.; Meesin, J.; Pohmakotr, M.; Reutrakul, V.; Leowanawat, P.; Soorukram, D.; Kuhakarn, C. Org. Biomol. Chem. 2018, 16, 8553.
(k) Qiu, J.; Wu, D.; Karmaker, P. G.; Yin, H.; Chen, F.-X. Org. Lett. 2018, 20, 1600.
(l) Wu, C.; Lu, L.-H.; Peng, A.-Z.; Jia, G.-K.; Peng, C.; Cao, Z.; Tang, Z.; He, W.-M.; Xu, X. Green Chem. 2018, 20, 3683.
(m) Dey, A.; Hajra, A. Adv. Synth. Catal. 2019, 361, 842.
(n) Noikham, M.; Yotphan, S. Eur. J. Org. Chem. 2019, 2019, 2759.
Castanheiro, T.; Suffert, J.; Donnard, M.; Gulea, M. Chem. Soc. Rev. 2016, 45, 494. doi: 10.1039/C5CS00532A
Vekariya, R. H.; Patel, H. D. Synth. Commun. 2017, 47, 87. doi: 10.1080/00397911.2016.1255973
Qin, X.; Zhang, L.-Y.; Feng, G.-F.; Jin, C.-A. Chin. J. Org. Chem. 2019, 39, 287(in Chinese).
(徐庆, 张连阳, 冯高峰, 金城安, 有机化学, 2019, 39, 287.)
(a) Hartwig, J. F. Nature 2008, 455, 314.
(b) Lu, Q.; Zhang, J.; Wei, F.; Qi, Y.; Wang, H.; Liu, Z.; Lei, A. Angew. Chem., Int. Ed. 2013, 52, 7156.
(c) Lu, Q.; Zhang, J.; Zhao, G.; Qi, Y.; Wang, H.; Lei, A. J. Am. Chem. Soc. 2013, 135, 11481.
(d) Xi, Y.; Dong, B.; McClain, E. J.; Wang, Q.; Gregg, T. L.; Akhmedov, N. G.; Petersen, J. L.; Shi, X. Angew. Chem., Int. Ed. 2014, 53, 4657.
(e) Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T. A.; Liu, X. Chem. Soc. Rev. 2015, 44, 291.
(f) Liu, Y.; Xiong, J.; Wei, L. Chin. J. Org. Chem. 2017, 37, 1667(in Chinese).
(刘云云, 熊进, 韦丽, 有机化学, 2017, 37, 1667.)
(g) Mei, H.; Yin, Z.; Liu, J.; Sun, H.; Han, J. Chin. J. Chem. 2019, 37, 292.
(h) Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338.
Yadav, A. K.; Yadav, L. D. S. Tetrahedron Lett. 2015, 56, 6696. doi: 10.1016/j.tetlet.2015.10.048
Yuan, P.-F.; Zhang, Q.-B.; Jin, X.-L.; Lei, W.-L.; Wu, L.-Z.; Liu, Q. Green Chem. 2018, 20, 5464. doi: 10.1039/C8GC02720J
Guy, R. G.; Thompson, J. J. Tetrahedron 1978, 34, 541. doi: 10.1016/0040-4020(78)80049-0
Chen, Y.-J.; He, Y.-H.; Guan, Z. Tetrahedron 2019, 75, 3053. doi: 10.1016/j.tet.2019.04.053
Zhang, D.; Wang, H.; Bolm, C. Chem. Commun. 2018, 54, 5772. doi: 10.1039/C8CC03178A
Gao, Y.; Liu, Y.; Wan, J.-P. J. Org. Chem. 2019, 84, 2243. doi: 10.1021/acs.joc.8b02981
Fan, W.; Yang, Q.; Xu, F.; Li, P. J. Org. Chem. 2014, 79, 10588. doi: 10.1021/jo5015799
Wang, L.; Wang, C.; Liu, W.; Chen, Q.; He, M. Tetrahedron Lett. 2016, 57, 1771. doi: 10.1016/j.tetlet.2016.03.028
Hosseini-Sarvari, M.; Hosseinpour, Z.; Koohgard, M. New J. Chem. 2018, 42, 19237. doi: 10.1039/C8NJ03128B
Yang, D.; Yan, K.; Wei, W.; Li, G.; Lu, S.; Zhao, C.; Tian, L.; Wang, H. J. Org. Chem. 2015, 80, 11073. doi: 10.1021/acs.joc.5b01637
Mitra, S.; Ghosh, M.; Mishra, S.; Hajra, A. J. Org. Chem. 2016, 47, 8275.
Singh, M.; Yadav, A. K.; Yadav, L. D. S.; Singh, R. Synlett 2018, 29, 176. doi: 10.1055/s-0036-1590921
Chauhan, P.; Ritu, R.; Preeti, P.; Kumar, S.; Jain, N. Eur. J. Org. Chem. 2019, 2019, 4334.
Tambe, S. D.; Jadhav, M. S.; Rohokale, R. S.; Kshirsagar, U. A. Eur. J. Org. Chem. 2018, 2018, 4867.
Gullapalli, K.; Vijaykumar, S. Org. Biomol. Chem. 2019, 17, 2232. doi: 10.1039/C9OB00054B
(a) Zhao, Y.; Wang, H.; Hou, X.; Hu, Y.; Lei, A.; Zhang, H.; Zhu, L. J. Am. Chem. Soc. 2006, 128, 15048.
(b) Li, C.-J. Acc. Chem. Res. 2009, 42, 335.
(c) Chen, M.; Zheng, X.; Li, W.; He, J.; Lei, A. J. Am. Chem. Soc. 2010, 132, 4101.
(d) Le Bras, J.; Muzart, J. Chem. Rev. 2011, 111, 1170.
(e) Liu, C.; Zhang, H.; Shi, W.; Lei, A. Chem. Rev. 2011, 111, 1780.
(f) Shi, W.; Liu, C.; Lei, A. Chem. Soc. Rev. 2011, 40, 2761.
(g) Yeung, C. S.; Dong, V. M. Chem. Rev. 2011, 111, 1215.
(h) He, C.; Guo, S.; Ke, J.; Hao, J.; Xu, H.; Chen, H.; Lei, A. J. Am. Chem. Soc. 2012, 134, 5766.
(i) Girard, S. A.; Knauber, T.; Li, C. J. Angew. Chem., Int. Ed. 2014, 53, 74.
(j) Liu, C.; Liu, D.; Lei, A. Acc. Chem. Res. 2014, 47, 3459.
(k) Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Chem. Rev. 2015, 115, 12138.
(l) Zhang, G.; Liu, C.; Yi, H.; Meng, Q.; Bian, C.; Chen, H.; Jian, J.-X.; Wu, L.-Z.; Lei, A. J. Am. Chem. Soc. 2015, 137, 9273.
(m) Song, C.; Yi, H.; Dou, B.; Li, Y.; Singh, A. K.; Lei, A. Chem. Commun. 2017, 53, 3689.
(n) Song, C.; Dong, X.; Yi, H.; Chiang, C.-W.; Lei, A. ACS Catal. 2018, 8, 2195.
(o) Song, C.; Liu, K.; Dong, X.; Chiang, C.-W.; Lei, A. Synlett 2019, 30, 1149.
Kang, L.-S.; Luo, M.-H.; Lam, C. M.; Hu, L.-M.; Little, R. D.; Zeng, C.-C. Green Chem. 2016, 18, 3767. doi: 10.1039/C6GC00666C
Liang, S.; Zeng, C. C.; Tian, H.; Sun, B.; Ren, F. Adv. Synth. Catal. 2018, 360, 1444. doi: 10.1002/adsc.201701401
De Klein, W. J. Electrochim. Acta 1973, 18, 413. doi: 10.1016/0013-4686(73)80044-1
Levy, A.; Becker, J. Y. Electrochim. Acta 2015, 178, 294. doi: 10.1016/j.electacta.2015.07.127
Gitkis, A.; Becker, J. Y. Electroanalysis 2016, 28, 2802. doi: 10.1002/elan.201600197
Wen, J.; Zhang, L.; Yang, X.; Niu, C.; Wang, S.; Wei, W.; Sun, X.; Yang, J.; Wang, H. Green Chem. 2019, 21, 3597. doi: 10.1039/C9GC01351B
Krishnan, P.; Gurjar, V. G. Synth. Commun. 1992, 22, 2741. doi: 10.1080/00397919208021538
Krishnan P., Gurjar V. G.. J.[J]. Appl. Electrochem., 1995, 25(792): .
Gitkis, A.; Becker, J. Y. J. Electroanal. Chem. 2006, 593, 29. https://www.sciencedirect.com/science/article/abs/pii/S0022072805006261
Gitkis, A.; Becker, J. Y. Electrochim. Acta 2010, 55, 5854. doi: 10.1016/j.electacta.2010.05.035
(a) Nair, V.; George, T. G.; Nair, L. G.; Panicker, S. B. Tetrahedron Lett. 1999, 40, 1195.
(b) Chakrabarty, M.; Sarkar, S. Tetrahedron Lett. 2003, 44, 8131.
(c) Yadav, J.; Reddy, B.; Shubashree, S.; Sadashiv, K. Tetrahedron Lett. 2004, 45, 2951.
(d) Yadav, J.; Reddy, B.; Krishna, A.; Reddy, C. S.; Narsaiah, A. Synthesis 2005, 2005, 961.
(e) Pan, X.-Q.; Lei, M.-Y.; Zou, J.-P.; Zhang, W. Tetrahedron Lett. 2009, 50, 347.
(f) Akhlaghinia, B.; Pourali, A.-R.; Rahmani, M. Synth. Commun. 2012, 42, 1184.
(g) Khazaei, A.; Zolfigol, M. A.; Mokhlesi, M.; Panah, F. D.; Sajjadifar, S. Helv. Chim. Acta 2012, 95, 106.
Fotouhi, L.; Nikoofar, K. Tetrahedron Lett. 2013, 54, 2903. doi: 10.1016/j.tetlet.2013.02.106
Kokorekin, V. A.; Sigacheva, V. L.; Petrosyan, V. A. Tetrahedron Lett. 2014, 55, 4306. doi: 10.1016/j.tetlet.2014.06.028
Zhang, X.; Wang, C.; Jiang, H.; Sun, L. RSC Adv. 2018, 8, 22042. doi: 10.1039/C8RA04407D
Yaubasarova, R. R.; Kokorekin, V. A.; Ramenskaya, G. V.; Petrosyan, V. A. Mendeleev Commun. 2019, 29, 334. doi: 10.1016/j.mencom.2019.05.032
Sun, L.; Zhang, X.; Li, Z.; Ma, J.; Zeng, Z.; Jiang, H. Eur. J. Org. Chem. 2018, 2018, 4949. doi: 10.1002/ejoc.201800267
Kokorekin, V. A.; Yaubasarova, R. R.; Neverov, S. V.; Petrosyan, V. A. Mendeleev Commun. 2016, 5, 413.
Kokorekin, V. A.; Yaubasarova, R. R.; Neverov, S. V.; Petrosyan, V. A. Eur. J. Org. Chem. 2019, 2019, 4233. doi: 10.1002/ejoc.201900390
Dyga, M.; Hayrapetyan, D.; Rit, R. K.; Gooßen, L. J. Adv. Synth. Catal. 2019, 361, 3548. doi: 10.1002/adsc.201900156
Yang, S.-M.; He, T.-J.; Lin, D.-Z.; Huang, J.-M. Org. Lett. 2019, 21, 1958. doi: 10.1021/acs.orglett.8b04136

图式 5 光催化活化的烯酰胺衍生物的硫氰化反应
Scheme 5 Photocatalytic thiocyanation of activated enamide derivatives

图式 6 光催化烯烃的硫氰化和磺酰胺化反应
Scheme 6 Photocatalytic thiocyanation and sulfonamidation of olefins

图式 9 可见光促进的TiO2/MoS2纳米复合物催化吲哚的硫氰化反应
Scheme 9 Visible-light-promoted TiO2/MoS2 nanocomposite catalyzed thiocyanation of indoles

图式 10 可见光促进Ag/TiO2纳米管催化N-杂环化合物的硫氰化反应
Scheme 10 Visible-ligh-promoted thiocyanation of N-heterocy- clic compounds catalyzed by Ag/TiO2 nanotubes

图式 11 咪唑并[1, 2-a]吡啶类化合物C-3位硫氰基化反应
Scheme 11 C-3 thiocyanation of imidazo[1, 2-a]pyridines

图式 12 光催化合成6-硫氰菲啶衍生物
Scheme 12 Photocatalytic synthesis of 6-thiophenanthridine derivatives

图式 13 可见光促进的2-芳基喹啉-4-酮的C-3硫氰化反应
Scheme 13 Visible-light-promoted C-3 thiocyanation of 2-aryl- quinolin-4-ones

图式 14 可见光催化合成3-硫氰基苯并噻吩
Scheme 14 Visible light-catalyzed synthesis of 3-thiocyano- benothiophene

图式 15 可见光诱导合成α-硫氰基酮/硫氰基醇
Scheme 15 Visible light induced synthesis of aryl α-thiocyano ketones/thiocyano alcohols

图式 22 烯烃的硫氰化和烯基化反应的机理
Scheme 22 Mechanism of thiocyanation and alkenylation of alkenes

图式 25 取代茴香醚、甲苯和苯胺衍生物的电化学硫氰化反应
Scheme 25 Electrochemical thiocyanation of substituted anisole, toluene and aniline derivatives

图式 26 含氮杂环的电化学硫氰化反应
Scheme 26 Electrochemical thiocyanation of nitrogen-contain- ing heterocycles

图式 27 N-杂芳烃衍生物的电化学硫氰化反应
Scheme 27 Electrochemical thiocyanation of N-heteroaromatic derivatives

图式 28 富电子芳烃的硫代和硒代氰化策略
Scheme 28 Thio- and seleno-cyanation strategies for electron-rich arenes

图式 29 N-甲基吡咯的阳极双C—H硫氰化反应
Scheme 29 Double anodic C—H thiocyanation of N-methyl- pyrrole

图式 33 芳基硼酸和三氟硼酸芳基的电化学硫氰化反应
Scheme 33 Electrochemical thiocyanation of arylboronic acids and aryl trifluoroborates

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:

















 下载:
下载: