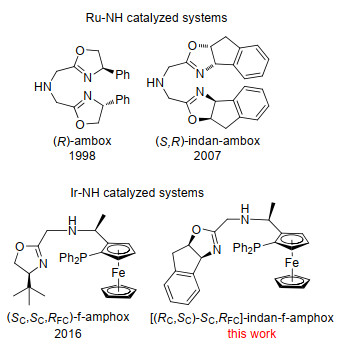
Scheme 1.
Evolution of NH-tridentate ligands in Zhang's group
Indan-f-amphox: An Efficient PNN Ligand for Iridium-Catalyzed Asymmetric Hydrogenation of β-Aryl β-Ketoesters
Guoxian Gu , Yang Hu , Shaodong Liu , Xiu-Qin Dong , Xumu Zhang

Enantiopure compounds are important building blocks in the fields of pharmaceutical and agrochemical industries due to their distinct biological activities.[1] In principle, the preparation of enantiopure compounds through catalytic asymmetric hydrogenation (AH) of readily available unsaturated compounds such as olefins, ketones and imines is one of the most straightforward and operationally simple approaches.[2] Although many chiral catalysts have been developed in this field, there is no universal catalyst suitable for many substrates.[3] Development of readily available new ligand systems is the key factor for asymmetric catalysis.
In many catalysts, the M-NH system has proved to be a key moiety for achieving high catalytic activities and excellent enantioselectivities in the hydrogenation of ketones and imines.[4] In 1995, Noyori and co-workers[5] reported ruthenium bisphosphine/diamine system, which showed high catalytic activity for hydrogenation of simple ketones. In this system, a proton from the NH2 ligand and a hydride from the Ru center interact with the C=O bond of ketone via a six-membered pericyclic Ru-NH transition state, and this "NH effect" has resulted in the high catalytic efficiency of simple aromatic ketones.[6] In 2011, Zhou and co- workers[7] have made a breakthrough using catalysts with Ir-NH species for hydrogenation of simple ketones. Introducing a coordination pyridine group to the SpiroAP ligand can inhibit dimerization, which is crucial important for the exceptionally high stability and activity in hydrogenation reaction. The resulting modified ligand SpiroPAP afforded chiral alcohols products in the hydrogenation of ketones with up to 99.9% ee and TONs as high as 4550000. In 1998, Zhang and co-workers[8] developed a new tridentate phosphine free ligand, bis(oxazo- linylmethyl)amine (ambox), and successfully applied the in situ generated Ru(Ⅱ)-ambox complex in the transfer hydrogenation of simple ketones. M-NH was demonstrated to be an important element for the hydrogenation of aromatic ketones. In 2007, indan-ambox was successfully prepared and applied in direct AH of simple ketones achieving high enantioselectivities and decent activity.[9] Based on our success with chrial ambox systems, more electron donating ligands were designed and synthesized to achieve higher activity. Lately, (SC, SC, RFC)-f-amphox was reported by our group and it was also successfully applied in AH of simple and functionalized ketones (Scheme 1).[10]
In Ru-NH catalyzed hydrogenation of β-alkyl β-keto- esters, Ru(Ⅱ)-BINAP/diamine system offered excellent ee and turnovers under an acidic condition. However, for hydrogenation of β-aryl β-ketoesters, an important reaction for synthesizing key drug intermediates such as duloxetine and atomoxetine, much lower ee's and turnovers were observed.[2c, 11] In 2012, Zhou et al.[12] reported an Ir-NH species for AH of β-aryl β-ketoesters in the Ir-SpiroPAP system with excellent ee's. A slight drawback is that the ligand synthesis is tedious. Considering the success of these Ir-NH catalyzed systems, we envisioned that Ir/f-amphox catalyst could also promote the AH of β-ketoesters.
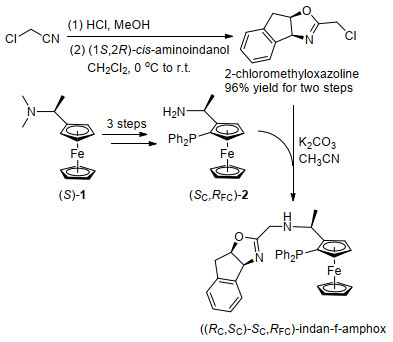
Here we report our achievements in the synthesis of another steric hindered and electron-rich tridentate ligand, [(RC, SC)-SC, RFC]-indan-f-amphox, which has a deeper chiral concave pocket around the metal center than its sister ligand of (SC, SC, RFC)-f-amphox. As shown in Scheme 2, the synthesis of indan-f-amphox started from functionalization of (S)-Ugi's amine 1 to obtain (SC, RFC)-2 based on previous literature.[13] Compound (SC, RFC)-2 reacted with 2-chloromethyloxazoline, which was obtained from chiral cis-amino indanol and 2-chloroacetoneitrile, to afford indan-f-amphox with an overall yield of 23%. This short synthetic route with four steps allows easy modification and both enantiomers are readily to be prepared.
Our initial study began with ethyl 3-oxo-3-phenyl- propanoate 3a as the model substrate. Under 2 MPa of hydrogen with 0.1 mol% Ir-ligand complex generated in situ, a brief screening of ligands in i-PrOH was conducted and the results is showed in Table 1. Preliminary results displayed that indan-f-amphox (L1) provided moderate ee value (Entry 1) but with only 15% conversion. Then a screening of different solvents with Ir/indan-f-amphox was carried out and methanol was turned out to be the best solvent for this reaction, affording 96% ee and > 99% conversion (Entry 5). It is interesting that the iPrOH, which had been proved to be beneficial for Ir/f-amphox in the AH of other functionalized ketones, [10] was proved to be negative for Ir/indan-f-amphox in this reaction (Entry 1). Non-polar or polar aprotic solvents gave low reactivities or low enantioselectivities (Entries 7~9). If the oxazoline arm is replaced by pyridine (L3), which is planar, [14] ee value dropped dramatically to 33% (Entry 9). It is inexplicable that (SC, SC, RFC)-f-amphox (L2), the sister ligand of [(RC, SC)-SC, RFC]-indan-f-amphox, did not work in either iPrOH or MeOH in this reaction (Entries 2 and 10) with 0.1 mol% catalyst loading. The other Ir-NH ligand, f-ampha, which works well in AH of α-keto- esters[15] performed poorly in this reaction (Entry 4). When the catalyst loading decreased from 0.1 to 0.01 mol%, the Ir/indan-f-amphox also provided 99% conversion and 96% ee (Entry 11, S/C=10000).
 下载:
导出CSV
下载:
导出CSV
 | ||||
| Entry | Ligand | Solvent | Conv.b/% | eec/% |
| 1 | L1 | i-PrOH | 15 | 70 |
| 2 | L2 | i-PrOH | NR | NA |
| 3 | L3 | i-PrOH | > 99 | 33 |
| 4 | L4 | i-PrOH | < 5% | NA |
| 5 | L1 | MeOH | > 99 | 96 |
| 6 | L1 | EtOH | > 99 | 64 |
| 7 | L1 | CH2Cl2 | > 99 | 10 |
| 8 | L1 | Toluene | NR | NA |
| 9 | L1 | THF | 5 | 60 |
| 10 | L2 | MeOH | NR | NA |
| 11d | L1 | MeOH | > 99 | 96 |
| a Reaction conditions: 0.2 mmol 3a, 0.05 mol% [Ir(COD)Cl]2, 0.1 mol% ligand, 1 mol% tBuOK, 2.0 mL solvent, reaction time 16 h. b Determined by 1H NMR analysis. c ee was determined by high performance liquid chromatography (HPLC) analysis; the absolute configuration was assigned by the comparison of the optical rotation with reported standard.[16] d S/C=10000, 24 h. | ||||
With the optimized condition in hand, a variety of methyl 3-oxo-3-arylpropanoates (3a~3o) have been reduced to provide the corresponding chiral secondary alcohols (Table 2) with Ir/indan-f-amphox at S/C=1000. High yields (95%~99%) and excellent enantioselectivities (88%~99% ee) were obtained. Change of substituents on substrates has little effect on the reactivities (> 99% conv.). Substrates with electron-donating groups at meta- and para- or ortho-position, provided the hydrogenated products with excellent ee's (4b, 4c, 4d, 4k and 4l). In contrast, substrates with a electron-withdrawing substituent on para-position (4f and 4g) gave product in diminished ee's. The substrates with naphthyl (4m and 4n) were hydrogenated smoothly with both reactivity (both > 99% conv.) and enantioselectivities (96% ee for 4m and 94% ee for 4n). Particularly, the heterocyclic substrate (4o) also proceed smoothly affording the desired hydrogenation product with 90% ee. Interestingly, transesterification occurs under this reaction condition, giving methyl ester products exclusively (3a' and 3m').
下载:
导出CSV
 | ||||||
| Entry | 3 | Ar | R | Product | Yieldb/% | eec/% |
| 1 | 3a | C6H5 | Me | 4a | 99 | 96 |
| 2 | 3a' | C6H5 | Et | 4a | 98 | 96 |
| 3 | 3b | 4-MeC6H4 | Me | 4b | 99 | 99 |
| 4 | 3c | 4-EtC6H4 | Me | 4c | 99 | 96 |
| 5 | 3d | 4-MeOC6H4 | Me | 4d | 99 | 95 |
| 6 | 3e | 4-FC6H4 | Me | 4e | 96 | 96 |
| 7 | 3f | 4-F3CC6H4 | Me | 4f | 96 | 88 |
| 8 | 3g | 4-NO2C6H4 | Me | 4g | 97 | 90 |
| 9 | 3h | 4-ClC6H4 | Me | 4h | 96 | 93 |
| 10 | 3i | 2-ClC6H4 | Me | 4i | 99 | 88 |
| 11 | 3j | 3-ClC6H4 | Me | 4j | 95 | 91 |
| 12 | 3k | 2-MeC6H4 | Me | 4k | 98 | 95 |
| 13 | 3l | 3-MeC6H4 | Me | 4l | 99 | 96 |
| 14 | 3m | 1-Naphthyl | Me | 4m | 97 | 96 |
| 15 | 3m' | 1-Naphthyl | Et | 4m | 99 | 96 |
| 16 | 3n | 2-Naphthyl | Me | 4n | 97 | 94 |
| 17 | 3o | 2-Thienyl | Me | 4o | 96 | 90 |
| aReaction conditions: 0.2 mmol of substrate 3, 0.05 mol% [Ir(COD)Cl]2, 0.1 mol% ligand, 2.0 mL of MeOH, 0.1 mol% tBuOK, 20 h. bIsolated yield. cee was determined by chiral HPLC analysis. | ||||||
A new chiral tridentate ligand, indan-f-amphox, was synthesized. In contrast to its sister ligand f-amphox, indan-f-amphox showed highly reactivity and enantioselectivity in iridium catalyzed AH of β-aryl β-ketoesters. By using an Ir/indan-f-amphox complex, a variety of β-keto- esters were reduced efficiently to chiral alcohols with high optical purities (up to 96% ee at S/C=10000).
1H NMR (400 MHz), 13C NMR (101 MHz) and 31P NMR (162 MHz) spectra were recorded on a Bruker ADVANCE Ⅲ spectrometer with CDCl3 as solvent and tetramethylsilane (TMS) as internal standard. Chemical shifts (δ) were reported up field to TMS for 1H NMR and relative to CDCl3 (δ 77.0) for 13C NMR. HRMS was recorded on APEXII and ZAB-HS spectrometer. HPLC analyses were performed using an Agilent 1260 Infinity Ⅱ instrument. Column chromatography was performed with silica gel Merck 60 (300~400 mesh). All reactions and manipulations, which are sensitive to moisture or air, were performed in an argon-filled glove box or using standard schlenk techniques. Aromatic ketones were purchased from commercial suppliers and purified by simple distillation or flash column chromatography prior to use. Anhydrous MeOH, EtOH, DCM, iPrOH, THF and toluene were purchased from Sigma-Adrich. [Ir(COD)Cl]2 was prepared according to the literature.[17]
2-Chloromethyloxazoline was synthesized following the procedure reported by Ref. [18] and (S, RFC)-2 was synthesized by the method reported in Ref. [11].
(S, R)-3 (500 mg, 1.20 mmol), K2CO3 (1.66 g, 12.0 mmol) and oxazoline (274 mg, 1.33 mmol) were placed in a round bottom flask, and purged by nitrogen twice. 15 mL of anhydrous acetonitrile was added by using syringe. Then the reaction was refluxed overnight. The solvent was removed under vacuum. Crude product was purified by column chromatography with DCM/MeOH (V:V=99:1) as eluent to give 230 mg of pure product as yellow solid (33% yield). m.p. 210.7~212.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.40 (d, J=6.6 Hz, 3H), 3.07~2.99 (m, 2H), 3.18 (d, J=17.9 Hz, 1H), 3.32 (dd, J=17.9, 6.9 Hz, 1H), 3.74 (d, J=10.0 Hz, 1H), 4.02~3.90 (m, 7H), 4.26 (t, J=2.4 Hz, 1H), 4.43 (s, 1H), 5.10~5.06 (m, 1H), 5.36 (d, J=7.9 Hz, 1H), 7.58~7.05 (m, 14H); 13C NMR (101 MHz, CDCl3) δ: 19.15, 39.62, 43.17, 50.65, 50.74, 69.02, 69.66, 69.72, 71.24, 75.20, 75.28, 76.22, 83.00, 96.87, 97.11, 125.18, 125.50, 127.31, 128.07, 128.15, 128.20, 128.28, 128.37, 129.04, 132.58, 132.77, 134.76, 134.97, 137.10, 139.77, 142.07, 166.02; 31P NMR (162 MHz, CDCl3) δ: -25.07; HRMS calcd for C35H34N2OPFe 585.1753, found 585.1750.
In an argon-filled glove box, a 4.0 mL vial was charged with the metal precursor [Ir(COD)Cl]2 (3.4 mg, 5.0×10-3 mmol), indan-f-amphox (6.1 mg, 10.5×10-3 mmol) and anhydrous MeOH (1.0 mL). The mixture was stirred for 1.0 h at 25 ℃, giving an orange red solution. The resulting solution (20 μL) and solution of tBuOK in MeOH (20 μL, c=0.1 mol/L) were transferred by a syringe into a 5.0 mL vial charged with β-keto esters (0.2 mmol) in 1.0 mL of anhydrous MeOH. The vial was transferred to an autoclave, which was then charged with 2 MPa of H2 and stirred at room temperature for 16 h. The hydrogen gas was released slowly in a well-ventilated hood and the solution was passed through a short column of silica gel to remove the metal complex. The crude product was analyzed by 1H NMR for conversion and chiral HPLC for ee values.
Methyl (R)-3-hydroxy-3-phenylpropanoate (4a):[19] Colorless oil, 36 mg, > 99% conv., 99% yield, 96% ee.
Methyl (R)-3-hydroxy-3-(p-tolyl)propanoate (4b):[20] Colorless oil, 39 mg, > 99% conv., 99% yield, 99% ee.
Methyl (R)-3-(4-ethylphenyl)-3-hydroxypropanoate (4c): Colorless oil, 41 mg, > 99% conv., 98% yield, 96% ee.
Methyl (R)-3-hydroxy-3-(4-methoxyphenyl)propanoate (4d): Colorless oil, 42 mg, > 99% conv., 99% yield, 96% ee.
Methyl (R)-3-(4-fluorophenyl)-3-hydroxypropanoate (4e): Colorless oil, 38 mg, > 99% conv., 96% yield, 96% ee.
Methyl (R)-3-hydroxy-3-(4-(trifluoromethyl)phenyl)pro- panoate (4f): White solid, 48 mg, > 99% conv., 97% yield, 88% ee. m.p. 42.3~44.2 ℃ (lit.[20] 42.2~43.7 ℃);
Methyl (R)-3-hydroxy-3-(4-nitrophenyl)propanoate (4g):[21] Colorless oil, 44 mg, > 99% conv., 97% yield, 88% ee.
Methyl (R)-3-(4-chlorophenyl)-3-hydroxypropanoate (4h):[20] Colorless oil, 41 mg, > 99% conv., 96% yield, 93% ee.
Methyl (R)-3-(2-chlorophenyl)-3-hydroxypropanoate (4i):[22] Colorless oil, 42 mg, > 99% conv., 99% yield, 88% ee.
Methyl (R)-3-(3-chlorophenyl)-3-hydroxypropanoate (4j):[22] Colorless oil, 41 mg, > 99% conv., 95% yield, 91% ee.
Methyl (R)-3-hydroxy-3-(o-tolyl)propanoate (4k):[23]Colorless oil, 38 mg, > 99% conv., 98% yield, 95% ee.
Methyl (R)-3-hydroxy-3-(m-tolyl)propanoate (4l): Colorless oil, 38 mg, > 99% conv., 98% yield, 96% ee.
Methyl (R)-3-hydroxy-3-(naphthalen-1-yl)propanoate (4m): Colorless oil, 41 mg, > 99% conv., 95% yield, 91% ee.
Methyl (R)-3-hydroxy-3-(naphthalen-2-yl)propanoate (4n):[20] Colorless oil, 45 mg, > 99% conv., 97% yield, 94% ee.
Methyl (R)-3-hydroxy-3-(thiophen-2-yl)propanoate (4o):[20] Yellow oil, 36 mg, > 99% conv., 96% yield, 90% ee.
In an argon-filled glove box, a 4.0 mL vial was charged with the metal precursor [Ir(COD)Cl]2 (3.4 mg, 5.0×10-3 mmol), indan-f-amphox (6.1 mg, 10.5×10-3 mmol) and anhydrous MeOH (1.0 mL). The mixture was stirred for 1.0 h at 25 ℃, giving an orange red solution. The resulting solution (2 μL) and t-BuOK (1 mg) were transferred by a syringe into a 5.0 mL vial charged with 3a (0.2 mmol) in 1.0 mL of anhydrous MeOH. The vial was transferred to an autoclave, which was then charged with 1 MPa of H2 and stirred at room temperature for 24 h. The work-up was identical to that described for the asymmetric hydrogenation at S/C=1000. (R)-3-Hydroxy-3-phenylpropanoate (4a): > 99% conversion, 96% ee.
Supporting Information 1H NMR and 13C NMR spectra of all compounds, and chromatography of 4a~4o. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
Johnson, R. A.; Sharpless, K. B. In Catalytic Asymmetric Synthesis, Ed.: Ojima, I., Wiley-VCH, New York, 2000, p. 13.
(a) Noyori R.; Kitamura M. In Modern Synthetic Methods, Ed.: Scheffold, R., Springer, Berlin, 1989, Vol. 5, p. 115.
(b) Takaya, H.; Ohta, T.; Noyori, R. In Catalytic Asymmetric Synthesis, Ed.: Ojima, I., VCH, New York, 1993, p. 1.
(c) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029.
(a) Chen, S.; Yang, W.; Yao, Y.; Yang, X.; Deng, Y.; Yang, D. Chin. J. Org. Chem. 2018, 38, 2534(in Chinese).
(陈姝琪, 杨文, 姚永祺, 杨新, 邓颖颍, 杨定乔, 有机化学, 2018, 38, 2534.)
(b) Shan, H.; Ling, L.; Hu, J.; Zhang, H. Chin. J. Org. Chem. 2019, 39, 1548(in Chinese).
(单贺, 伶俐, 胡剑锋, 张浩, 有机化学, 2019, 39, 1548.)
(c) Yuan, Q.; Zhang, W. Chin. J. Org. Chem. 2016, 36, 274(in Chinese).
(袁乾家, 张万斌, 有机化学, 2016, 36, 274.
For M-NH catalysis see review: (a) Zhao, B.; Han, Z.; Ding, K. Angew. Chem., Int. Ed. 2013, 52, 4744.
The recent advances in M-NH ligands see: (b) Li, Y.; Yu, S.; Wu, X.; Xiao, J.; Shen, W.; Dong, Z., Gao, J. J. Am. Chem. Soc. 2014, 136, 4031.
(c) Bao, D.-H.; Wu, H.-L.; Liu, C.-L.; Xie, J.-H.; Zhou, Q.-L. Angew. Chem., Int. Ed. 2015, 54, 8791.
Ohkuma, T.; Ooka, H.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 2675. doi: 10.1021/ja00114a043
(a) Hashigushi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 7562.
(b) Gao, J.; Ikariya, T.; Noyori, R. Organometallics 1996, 15, 1087.
(c) Gamez, P.; Fache, F.; Lemaire, M. Tetrahedron: Asymmetry 1995, 6, 705.
Xie, J.-H.; Liu, X.-Y.; Xie, J.-B.; Wang, L.-X.; Zhou, Q.-L. Angew. Chem., Int. Ed. 2011, 50, 7329. doi: 10.1002/anie.201102710
Jiang, Y.; Jiang, Q.; Zhang, X. J. Am. Chem. Soc. 1998, 120, 3817. doi: 10.1021/ja974095p
Li, W.; Hou, G.; Wang, C.; Jiang, Y.; Zhang, X. Chem. Commun. 2010, 46, 3979. doi: 10.1039/b927028k
(a) Wu, W.; Liu, S.; Duan, M.; Tan, X.; Chen, C.; Xie, Y.; Lan, Y.; Dong, X.-Q.; Zhang, X. Org. Lett. 2016, 18, 2938.
(b) Wu, W.; You, C.; Yin, C.; Liu, Y.; Dong, X.-Q.; Zhang, X. Org. Chem. Front. 2017, 4, 1499.
(c) Wu, W.; You, C.; Yin, C.; Liu, Y.; Dong, X.-Q.; Zhang, X. Org. Lett. 2017, 19, 2548.
(d) Wu, W.; Xie, Y.; Li, P.; Li, X.; Liu, Y.; Dong, X.-Q.; Zhang, X. Org. Chem. Front. 2017, 4, 555.
(e) Gu, G.; Yang, T.; Yu, O.; Qian, H.; Wang, J.; Dang, L.; Zhang, X. Org. Lett. 2017, 19, 5920.
Noyori, R. Angew. Chem., Int. Ed. 2002, 41, 2008. doi: 10.1002/1521-3773(20020617)41:12<2008::AID-ANIE2008>3.0.CO;2-4
Xie, J.-H.; Liu, X.-Y.; Yang, X.-H.; Xie, J.-B.; Wang, L.-X.; Zhou, Q.-L. Angew. Chem., Int. Ed. 2012, 51, 201. doi: 10.1002/anie.201105780
Hayashi, T.; Mise, T.; Fukushima, M.; Nagashima, N.; Hamada, Y.; Matsumoto, A.; Kawakami, S.; Konishi, M.; Yamamoto, K. Bull. Chem. Soc. Jpn. 1980, 53, 1138. doi: 10.1246/bcsj.53.1138
Nie, H.; Zhou, G.; Wang, Q.; Chen, W.; Zhang, S. Tetrahedron:Asymmetry 2013, 24, 1567. doi: 10.1016/j.tetasy.2013.10.012
Gu, G.; Yang, T.; Lu, J.; Wen, J.; Dang, L.; Zhang, X. Org. Chem. Front. 2018, 5, 1209. doi: 10.1039/C8QO00047F
Jiang, W.; Zhao, Q.; Tan, W. Chin. J. Chem. 2018, 36, 153. doi: 10.1002/cjoc.201700645
(a) Schenck, T. G.; Downes, J. M.; C. Milne, R. C.; Mackenzie, P. B.; Boucher, H.; Whelan, J.; Bosnich, B. Inorg. Chem. 1985, 24, 2334.
(b) Pohl, M.; Lyon, D. K.; Mizuno, N.; Nomiya, K.; Finke, R. G. Inorg. Chem. 1995, 34, 1413.
Ye, M.-C.; Li, B.; Zhou, J.; Sun, X.-L.; Tang, Y. J. Org. Chem. 2005, 70, 6108. doi: 10.1021/jo050595m
Jiang, W.; Zhao, Q.; Tang, W. Chin. J. Chem. 2018, 36, 153. doi: 10.1002/cjoc.201700645
Zhang, Y.; Yu, D.; Huang, Z.; Xu, J.; Wu, X.; Wang, Y.; Wang, M.; Yang, S.; Webster, R. D.; Chi, Y. R. J. Am. Chem. Soc. 2015, 137, 2416. doi: 10.1021/ja511371a
White, N. A.; Rovis, T. J. Am. Chem. Soc. 2014, 136, 14674. doi: 10.1021/ja5080739
Chen, X.-S.; Hou, C.-J.; Qin, C.; Liu, H.; Liu, Y.-J.; Huang, D.-Z.; Hu, X.-P. RSC Adv. 2017, 7, 12871. doi: 10.1039/C7RA00472A
Ohtani, I. I.; Hotta, K.; Ichikawa, Y.; Isobe, M. Chem. Lett. 1995, 7, 513. doi: 10.1246/cl.1995.513
Andrea, K.; Michel, S.; Barbare, M. Org. Lett. 2013, 15, 1614. doi: 10.1021/ol400393j
Table 1. AH of ethyl 3-oxo-3-phyneylpropanoatea
| ||||
| Entry | Ligand | Solvent | Conv.b/% | eec/% |
| 1 | L1 | i-PrOH | 15 | 70 |
| 2 | L2 | i-PrOH | NR | NA |
| 3 | L3 | i-PrOH | > 99 | 33 |
| 4 | L4 | i-PrOH | < 5% | NA |
| 5 | L1 | MeOH | > 99 | 96 |
| 6 | L1 | EtOH | > 99 | 64 |
| 7 | L1 | CH2Cl2 | > 99 | 10 |
| 8 | L1 | Toluene | NR | NA |
| 9 | L1 | THF | 5 | 60 |
| 10 | L2 | MeOH | NR | NA |
| 11d | L1 | MeOH | > 99 | 96 |
| a Reaction conditions: 0.2 mmol 3a, 0.05 mol% [Ir(COD)Cl]2, 0.1 mol% ligand, 1 mol% tBuOK, 2.0 mL solvent, reaction time 16 h. b Determined by 1H NMR analysis. c ee was determined by high performance liquid chromatography (HPLC) analysis; the absolute configuration was assigned by the comparison of the optical rotation with reported standard.[16] d S/C=10000, 24 h. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Iridium-catalyzed AH of β-ketoestersa
| ||||||
| Entry | 3 | Ar | R | Product | Yieldb/% | eec/% |
| 1 | 3a | C6H5 | Me | 4a | 99 | 96 |
| 2 | 3a' | C6H5 | Et | 4a | 98 | 96 |
| 3 | 3b | 4-MeC6H4 | Me | 4b | 99 | 99 |
| 4 | 3c | 4-EtC6H4 | Me | 4c | 99 | 96 |
| 5 | 3d | 4-MeOC6H4 | Me | 4d | 99 | 95 |
| 6 | 3e | 4-FC6H4 | Me | 4e | 96 | 96 |
| 7 | 3f | 4-F3CC6H4 | Me | 4f | 96 | 88 |
| 8 | 3g | 4-NO2C6H4 | Me | 4g | 97 | 90 |
| 9 | 3h | 4-ClC6H4 | Me | 4h | 96 | 93 |
| 10 | 3i | 2-ClC6H4 | Me | 4i | 99 | 88 |
| 11 | 3j | 3-ClC6H4 | Me | 4j | 95 | 91 |
| 12 | 3k | 2-MeC6H4 | Me | 4k | 98 | 95 |
| 13 | 3l | 3-MeC6H4 | Me | 4l | 99 | 96 |
| 14 | 3m | 1-Naphthyl | Me | 4m | 97 | 96 |
| 15 | 3m' | 1-Naphthyl | Et | 4m | 99 | 96 |
| 16 | 3n | 2-Naphthyl | Me | 4n | 97 | 94 |
| 17 | 3o | 2-Thienyl | Me | 4o | 96 | 90 |
| aReaction conditions: 0.2 mmol of substrate 3, 0.05 mol% [Ir(COD)Cl]2, 0.1 mol% ligand, 2.0 mL of MeOH, 0.1 mol% tBuOK, 20 h. bIsolated yield. cee was determined by chiral HPLC analysis. | ||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: