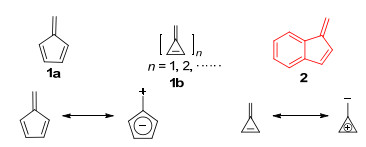
图 1.
富烯及其共振结构
Figure 1.
Fulvene and its resonance structure

富烯[1]是一类具有环外碳-碳双键的环戊二烯骨架的分子(Figure 1).早期, 其特指5-亚甲基-1, 3-环戊二烯(1a).随着对非苯芳香现象的认识的深入, 大量结构类似于富烯的分子被发现, 富烯的概念得到了进一步的拓展, 泛指含有奇数个原子的共轭环与一个环外双键形成的具有交叉共轭(cross-conjugation)结构的分子(1b).由于共振的存在, 环外双键的电子与环上共轭体系的π电子发生部分转移以满足分子中环状结构单元的芳香性.与此同时, 由于环外双键π电子部分转移至环上或环上的π电子部分转移至外双键上(图 1), 使得分子内存在一个较大的偶极矩[2], 表现出独特的物理和化学性质.
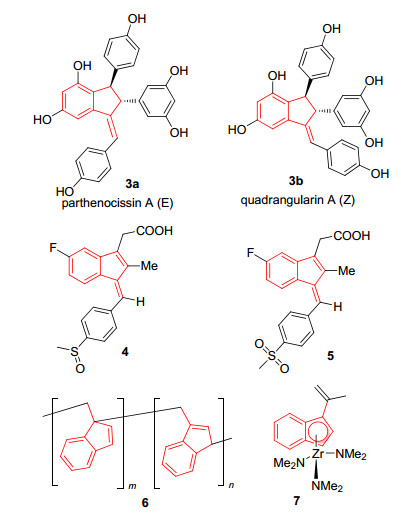
苯并富烯2及其衍生物则是富烯结构中非常重要的一类化合物.苯并富烯及其衍生物含有一个茚骨架, 又称为1-亚甲基茚.茚是许多药物分子和天然产物的基本骨架[3], 也是众多材料分子的重要前体[4].与茚骨架相比, 苯并富烯骨架含有一个环外的双键, 容易被官能团化.苯并富烯不仅是一些活性生物分子[5]和天然产物的核心结构骨架, 同时也是一类重要的合成中间体[6](图 2). 2009年, Sarpong课题组[6b]报道了一例利用苯并富烯骨架中间体合成二聚白黎芦醇的方法, 由甲氧基取代苯基苯并富烯经过一系列反应可得到两种具有活性的天然产物quadrangularin A和parthenocissin A.而非固醇类抗炎药物舒林酸(4)和舒林酸砜(5)的核心骨架也是含有氟取代的苯并富烯结构[5b].苯并富烯也被应用于高分子功能材料研究中, 如具有此类新颖结构的高聚物6, 可以用于光电材料并且前景广阔[7].通过改变官能团可以改变聚合物的性质.苯并富烯及其衍生物含有一个环戊二烯的骨架, 可用于制备后茂金属催化剂[8](Post- metallocene catalysts), 如金属锆络合物7, 应用于不对称的烯烃聚合反应[9]、Diels-Alder反应等[10].
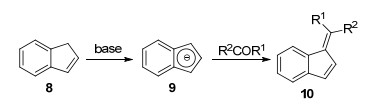
茚与醛或酮在碱催化作用下的缩合反应是合成苯并富烯的传统方法.首先, 茚8在碱的作用下, 失去质子转化为茚负离子9, 然后进攻醛/酮的羰基, 脱水形成苯并富烯10 (Scheme 1).这一方法官能团兼容性存在局限, 难以应用于不同取代基结构的苯并富烯合成[11].
苯并富烯及其衍生物的应用广泛, 其合成方法也得到了迅速的发展.按照关键的环化反应的不同引发方式, 构建苯并富烯骨架的主要方法可以大致分为五大类:光催化或者热引发的联烯-烯炔或者烯二炔环化反应、过渡金属催化的串联环化反应、亲核或亲电试剂进攻引发的环化反应、自由基引发的环化反应, 以及路易斯酸促进的环化反应.本文将按照上述反应分类, 对苯并富烯类化合物的合成方法进行总结.
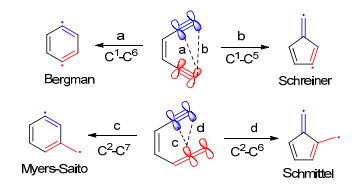
Bergman环芳香化及其相关反应是有机化学、药物化学和材料化学领域里的一类非常重要的反应[12].这类反应以烯二炔类和烯炔-联烯类化合物为底物, 常常会经历一个双自由基中间体[12]. Bergman环芳香化反应也是烯二炔类抗生素具有高效抗肿瘤活性的决定性因素之一.烯二炔类抗生素结构中含有双烯双炔共轭大环, 通过Bergman反应形成苯基双自由基活性中间体, 直接破坏肿瘤细胞DNA链, 最终引发肿瘤细胞凋亡.
Bergman环化反应发生在烯二炔的C1-C6位, 而Myers-Saito环化反应[13]发生在烯炔-联烯的C2-C7位, 这两类反应都是经历双自由基的中间体, 最终形成苯环(Scheme 2).从反应机理上来看, 这两种底物形成富烯的可能性同样也是存在的. 2001年Peter等[14]对于此类环化反应进行了细致的理论计算.结果表明上述的C1-C5环化反应相比C1-C6和C2-C7环化反应确实需要更高的反应活化能, 因而更容易生成苯环.但是芳环体系的存在, 以及大位阻末端取代基的应用, 可以促进C1-C5环化反应.如果烯二炔末端含有吸电子取代基, 则可以使环化反应所需要的能量进一步降低.
1995年, Schmittel等[15]研究苯并烯炔-联烯类底物11的C2-C6环化反应时, 发现含有特殊取代基的烯炔-联烯类底物12在50 ℃时发生C2-C7的常规Myers-Saito环化形成苯环, 当温度升高至84 ℃时, 可以发生C2-C6的环化反应, 以中等的收率得到了苯并富烯骨架的产物(Scheme 3a).虽然原料12可由烯炔11经过简单的转化而得, 但是利用这种方法进行合成, 其底物适用性仍然比较局限[16].
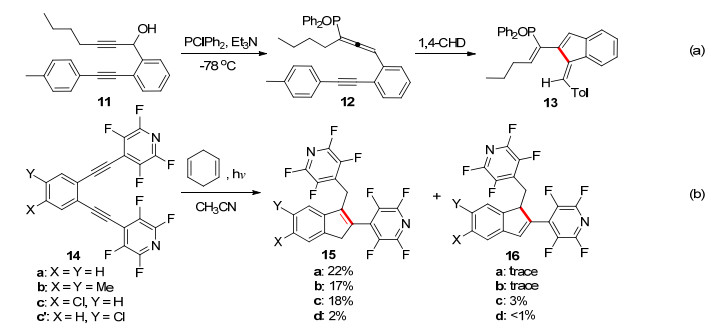
2002年, Serguei课题组[17]报道了一例光催化的1, 2-二乙炔基苯14的C1-C5的环化反应(Scheme 3b).由于Peter预测末端吸电子基团取代的环化反应的能垒较低, 并且实验进行了验证.所以作者合成了炔基末端是强吸电子基团的底物14, 尝试在光照的条件下合成苯并富烯, 但是未能成功, 以较低收率得到了C1-C5环化的茚的产物15, 16.实验证明, 仅利用光照, 由1, 2-二乙炔基苯合成苯并富烯比较困难.
2006年Schmittel课题组[18]报道了一例苯并烯炔-联烯17, 通过热引发生成苯并富烯衍生物的方法.他们在分子结构上引入自由基时钟模块(Radical clock)环丙烷, 得到了环丙烷开环产物, 证明了这个反应的自由基历程.这例反应伴有副产物20和21的产生, 反应产率比较低(Scheme 4a).

同年, Pascal课题组[19]报道了一例热引发1, 2-二苯基乙炔构建苯并富烯的反应(Scheme 4b), 这个反应与传统的利用这类底物构建苯并富烯的方法类似, 会发生C1-C6, C1-C5两种环化反应(Scheme 2).他们课题组也尝试反应.由于会有副产物24的生成, 导致产率较低, 分离收率仅为19%.这种方法需要260 ℃的高温, 不能广范应用于苯并富烯底物的合成.该研究也没有对底物进行更广泛的考察, 但是证明了苯并烯二炔类的底物可以通过热引发的方式, 发生C1-C5的环化反应构建苯并富烯.
2008年, Schmittel课题组[20]报道了一例光催化烯炔-联烯的25的C2-C6环化反应.根据经验他们合成了联烯基末端羰基取代, 炔基末端TIPS取代的底物25, 通过筛选不同溶剂与照射波长, 由苯并烯炔-联烯25在波长为300 nm的光源照射下得到苯并富烯的三种产物, 这例反应的底物类型对该反应的影响比较大, 特定的几个底物能得到苯并富烯的产物, 最高分离收率可达51% (Scheme 5). Schmittel课题组发展的这例光催化构建苯并富烯的方法, 可以以简单的方式获得了苯并富烯.
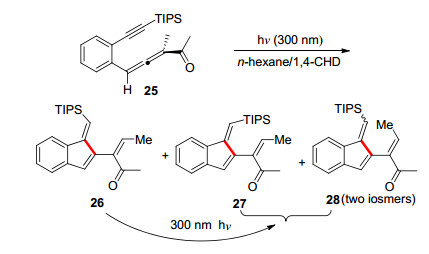
光或者热引发烯二炔, 烯炔-联烯构建苯并富烯的方法简单方便, 一般来说联烯-烯炔类底物的环化反应条件比二炔类底物的相对温和一些.不过这些方法都不可避免地会产生Bergman环化产物(C1-C6)或Myers- Saito环化产物(C2-C7)等副产物.此外, 已报道的相关方法在底物的适用性以及环外双键构建等方面还有待进一步的研究.
近年来, 过渡金属在有机化学中的应用获得空前发展, 使得高效地构建碳-碳以及碳-杂原子键日趋成熟.这一发展也为苯并富烯骨架的构建提供了很多新途径.在过渡金属催化下, 发生串联的环化反应为构建苯并富烯类化合物提供了高效的方法.
碳氢键活化是近些年过渡金属催化的一个研究热点.应用Rh, Ru, Ir, Pd, Co等催化的C—H键活化引发反应, 进而通过炔基插入和环化是构建苯并富烯的有效方法.
2008年, Shibata等[21]报道了一例羰基导向的铱催化C—H活化反应, 发展了以苯乙酮30和二苯基乙炔29为底物, [Ir(cod)2]BF4 (5 mol%)为催化剂, 邻位C—H键活化构建邻烯基苯乙酮片段.当铱催化体系改变为[Ir(cod)2]OTf (5 mol%), 得到环化的主产物苯并富烯和茚醇[22], 拓展底物后以中等至优秀的收率得到了一系列环化的苯并富烯产物(Eq. 1).但对于能够产生环外构型的底物, 作者没有进行充分研究, 仅考察了一个底物, 尽管最终得到了单一构型的产物, 但是没有确定构型.该课题组又尝试了其它类型的铱络合物, 不能得到环化的苯并富烯产物, 而且这种方法需要以氯苯为溶剂并长时间回流才能得到产物.
|
|
(1) |
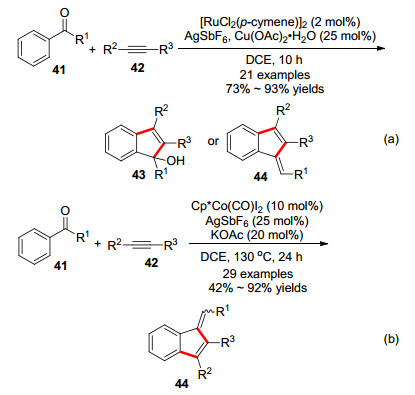
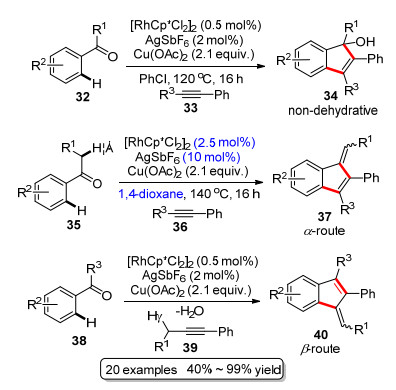
2011年Glorius课题组[23]报道了一例铑催化C—H键活化构建苯并富烯衍生物的反应(Scheme 6).在优化条件下, 以[RhCp*Cl2]2和AgSbF6为共同催化剂, 同时有等物质的量的Cu(OAc)2存在时, 以高达99%的分离收率得到环化产物.通过对反应条件的微调, 改变溶剂和反应温度分别得到苯并富烯或者茚醇(non-dehydra- tive vs α-route, β-route).研究发现醋酸铜对这个反应很重要.作者根据实验现象推测, 认为铜在催化循环中通过转金属化释放铑催化剂物种, 促进催化循环的进行.作者还发现, 底物的取代基不同会导致最终产物的不同, 也就是取代基会影响终产物.在非脱水反应模式(non-dehydrative)中, 当R1为异丙基, 叔丁基等大位阻的取代基, R2为拉电子基团时得到茚醇. R1为甲基或者氢原子且R2为推电子基团时则得到苯并富烯的产物(α-route). Glorius等发展的这一方法的官能团容忍性很好, 无论是强的吸电子或者给电子取代基或者杂环化合物的底物, 反应均可以进行, 还可以控制反应停留在茚醇阶段.但对于环外双键的构型, 该方法无法有效的控制.
2012年, 利用类似的反应底物, Jeganmohan课题组[24a]报道了一类较廉价的钌催化的C—H键活化构建苯并富烯的方法.反应与铑催化的C—H活化类似, 利用类似的底物41在[RuCl2(p-cymene)]、Cu(OAc)2·H2O (25 mol%)作用下, 生成茚醇类产物43 (Scheme 7).其它条件不变, 将AgSbF6的用量增加至20 mol%, 得到苯并富烯类产物44.但该反应在环外双键的构型方面并没有取得进展, 这是苯乙酮类底物构建苯并富烯的缺点之一, 另一个缺点是反应会不可避免地产生副产物茚醇.作者推测其反应机理与铑催化的C—H键活化构建苯并富烯的机理类似. 2019年谭泽和朱钢国课题组使用钴催化实现了同样的反应[24b].
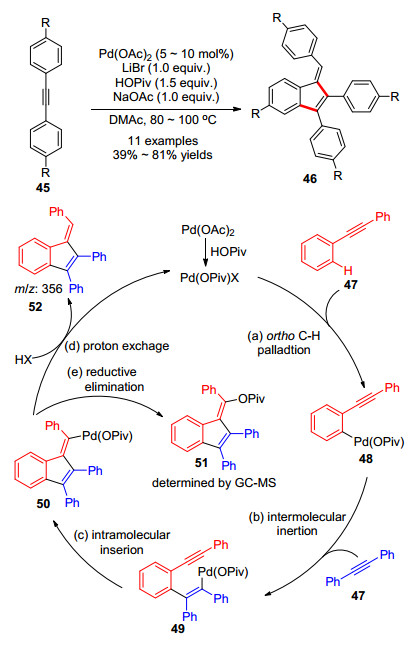
2015年, 华瑞茂课题组[25]用新的底物二苯基乙炔45发展了一类钯催化的C—H键活化构建苯富烯46的方法(Scheme 8).新底物的使用有效地避免了茚醇类副产物的生成.最终, 二苯基乙炔在醋酸钯的催化下, 在N, N-二甲基乙酰胺中加入溴化锂、特戊酸铯等添加剂, 以中等偏上的收率得到了一系列苯基取代苯并富烯化合物.
2019年, 莊士卿课题组[26]报道了一例类似的反应, 也是由两分子二芳基乙炔构建出苯并富烯骨架(Eq. 2).
|
|
(2) |
除了C—H活化引发的串联环化反应, 过渡金属催化的多种反应类型都可以引发相应的反应.
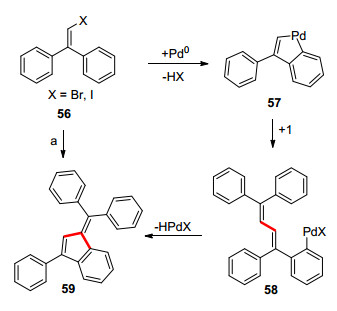
1996年, Dyker等[27]报道了一例以卤代1, 1-二苯基- 2-溴代乙烯(56)为底物, 多米诺的Heck反应构建苯并富烯的方法(Scheme 9), 该反应先经历分子间Heck反应, 再发生分子内的Heck反应构建苯并富烯.作者没有对底物的官能团进行考察, 只从构建大环的角度对底物的进行了拓展合成了一系列苯并富烯并环的结构.
2004年, Singer课题组[28]开发了用2-卤代苯乙腈60与乙氧基代丙烯酸乙酯(61)反应构建氰基取代苯并富烯衍生物62的方法(Eq. 3).作者合成苯并富烯仅作为合成其他化合物的一个关键中间体, 并没有对底物的官能团兼容性进行深入考察, 仅考察了几个简单的底物.这种方法合成了氰基取代的苯并富烯衍生物, 有利于苯并富烯骨架的官能团化, 而且氯代的底物也同样适用于该反应.
|
|
(3) |
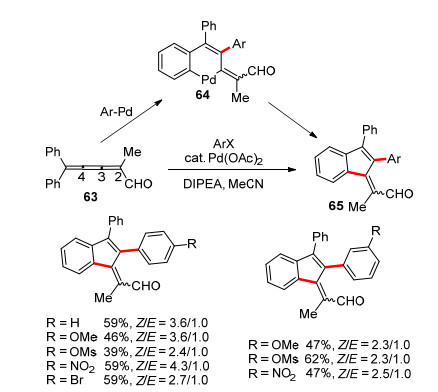
2006年, Kan课题组[29]报道了一例钯催化的多步Heck反应, 由累积三烯构建苯并富烯的方法(Scheme 10).尽管在63中有共轭醛基的存在, 但芳基钯物种仍会高选择性地插入到C3-C4之间, 形成六元环钯物种64, 最后还原消除得到苯并富烯的衍生物65.
2006年, Tobe课题组[30]合成了1, 6-二取代炔二烯结构的底物66, 在钯的催化下构建出桥联苯并富烯衍生物67 (Eq. 4).这种刚性的大共轭桥联苯并富烯骨架的分子, 是一类具有优异二阶非线性光学发色团(Second Order Nonlinear Optical Chromophores)的化合物, 可以作为一些先进光学器件的核心原料.
|
|
(4) |
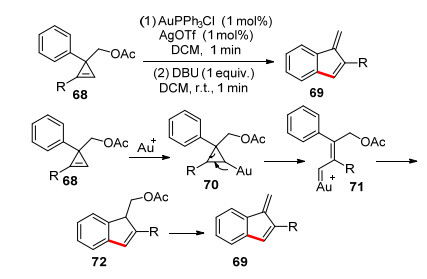
2009年, 王剑波课题组[31]报道了一例金催化的不饱和三元环扩环反应构建苯并富烯的方法(Scheme 11).不饱和底物68的碳碳双键与金离子进行配位, 然后三元环开环形成金卡宾71, 接着发生卡宾插入反应得到72, 再在碱的作用下消除一分子乙酸得到产物69.该方法研究的是简单的苯并富烯有衍生的合成, 并不涉及环外双键的考察.
2009年, 吴劼课题组[32]报道了醋酸钯催化的串联反应构建氯代苯并富烯74 (Eq. 5).作者认为该反应由二价钯启动, 与炔基进行配位, 氯离子进攻C-C叁键, 接着进行双键插入反应, 还原消除后形成产物74和零价钯物种.零价钯在铜离子的氧化再生二价钯, 继续参与下一轮催化循环.这种方法采用的是E构型的底物, 经Heck反应双键插入后顺式消除, 不会产生环外双键为混合物的情况.这种方法为合成环外双键为单一构型的苯并富烯衍生物提供了一个可借鉴的思路.
|
|
(5) |
2010年, 该课题组随后又报道了两例偕二溴取代烯基苯合成苯并富烯的方法.分别使用75[33a]和77[33b]为底物.在醋酸钯的催化下, 以三苯基膦为配体, 加入碱, 室温下发生分子内的Heck反应, 接着与芳基硼酸发生suzuki反应构建苯并富烯衍生物(Scheme 12a).这种方法反应条件温和, 收率等达到中等偏上, 而且还避免了环外双键产生混合物的情况.
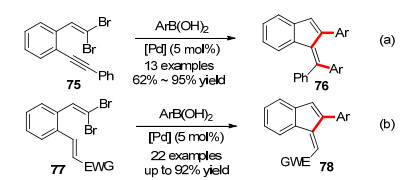
同年, Lautens课题组[34]利用类似的溴代二烯基的底物77以Pd2dba3为钯源, 碳酸铯为碱, 三呋喃基膦为配体, 在60 ℃的二氧六环和水的混合溶剂中得到苯并富烯的衍生物78 (Scheme 12b).
2010年, Kim课题组[35]以Baylis-Hillman加成产物为原料经过一步反应得到产物79, 然后通过Barbier反应得到80, 接着发生分子内Heck反应消除构建顺反异构体的苯并富烯混合物81 (Scheme 13).
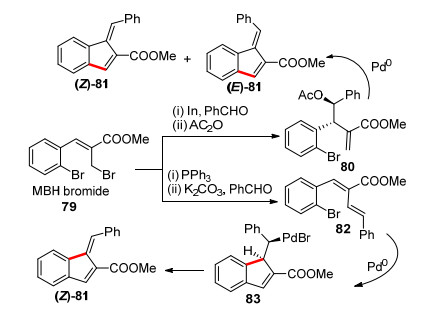
2013年, 该课题组[36]改进了上述工作, 采用反式共轭二烯结构底物82, 通过控制碱的不同(三乙胺和碳酸铯), 实现了对几个不同官能团底物环外双键的构型的基本控制, 分别得到主产物不同的81.
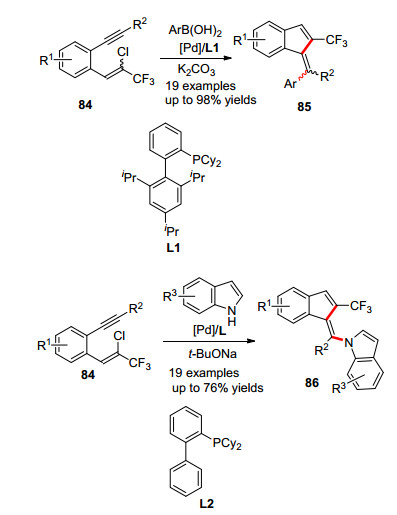
2012年, 张兴国课题组[37a]报道了一例钯催化的苯并富烯衍生物的合成方法(Scheme 14).该工作直接使用氯代底物84为底物, 与钯发生氧化加成、炔烃插入接着与硼酸发生Suzuki-Miyaura偶联反应, 得到三氟甲基化的苯并富烯.该反应以串联的环化-偶联反应提供了一个构建苯并富烯以及合成三氟甲基化苯并富烯的新方法. 2018年他们[37b]进一步地拓展了这一反应, 直接使用有机胺为偶联片段合成吲哚或吡咯取代的苯并富烯.
2012年, Sanz课题组[38a]发展了一例金催化环化串联反应构建苯并富烯衍生物的方法.该方法也需要经历金正离子物种, 环化之后通过消除反应得到环外双键(Eq. 6), 最终以较高的收率得到了(E)-89为主产物. 2015年他们[38b]使用对甲苯磺酸催化实现了该反应的转化, 无需金属催化剂.
|
|
(6) |
2014年, 陈知远课题组[39]发展了一例铂催化环化串联反应构建苯并富烯衍生物的方法(Eq. 7).作者认为该反应首先经历了酰基[3, 3]-重排反应生成联烯中间体, 然后在金属铂的催化作用下关环, 最后发生[1, 5]-酰基迁移得到了环外双键为Z构型的产物.
|
|
(7) |
2013年, 姚祝军课题组[40]也报道了一例金催化的苯并富烯衍生的合成方法(Eq. 8).他们设计了一类含羧基的二炔基苯为底物, 以5 mol% Ph3PAuNTf2为催化剂, 在二氯甲烷溶液中, 得到了氧杂六元环并的苯并富烯衍生物.
|
|
(8) |
2015年, Hamze等[41]报道了一例一锅法串联合成苯并富烯的方法.该方法以邻炔基腙类化合物93为底物, 通过配体的调控, 可以依次实现腙与卤代烃的偶联和成环反应(Eq. 9).
|
|
(9) |
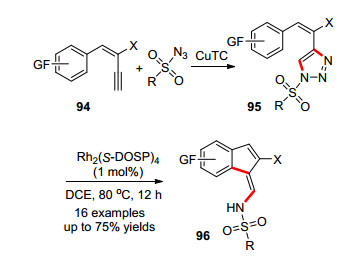
2016年, Lee课题组[42]报道了铑催化的三氮唑脱氮环化反应构建苯并富烯的方法(Scheme 15).三氮唑95在二价铑存在的条件下脱除一分子的氮气, 生成的铑-卡宾中间体亲电进攻苯环, 金属铑离去后重排得到苯并富烯96.三氮唑类化合物可以由炔烃与叠氮通过铜催化的Click反应得到. 94和磺酰叠氮在催化剂CuTC和Rh2(S-DOSP)4的催化下, 可以一锅法合成苯并富烯.这一工作采用一系列设计新颖的底物, 最终得到了一系列氨基官能团化的苯并富烯衍生物.
2018年, 张扬会课题组[43]报道了碘苯97、炔98和二溴甲烷99在钯催化下发生三组分串联反应制备苯并富烯的反应(Eq. 10).作者认为该反应经历了环钯中间体, 碘苯和炔烃与金属钯形成环钯物种后与二溴甲烷偶联得到茚, 茚在碱性条件下与另一分子的二溴甲烷缩合得到苯并富烯.
|
|
(10) |
2019年, 吴小锋课题[44]组报道了一例钯催化的四组分反应构建苯并富烯的反应(Eq. 11).该方法使用简单易得的原料, 经过一步反应以中等收率得到多取代的苯并富烯.
|
|
(11) |
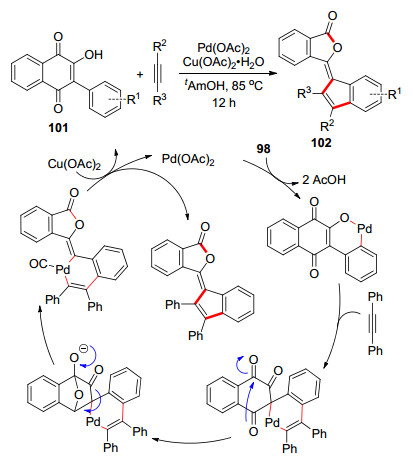
同年Gogoi等[45]报道了一例钯催化的C-H活化/炔烃插入的脱羰环化反应(Scheme 16).利用此方法, 可以很方便地合成含苯酞片段的苯并富烯.该反应以羟基萘醌101为底物, 与炔烃在乙酸钯催化下, 经过C-H活化, 炔烃插入, 重排脱羰, 还原消除得到产物102.钯在乙酸铜的氧化下再生二价钯实现了催化循环.
Hashmi课题组[46]报道了双金催化的非共轭二炔底物103构建刚性结构的的苯并富烯衍生物的方法(Eq. 12).该方法反应条件温和, 底物适用范围比较广泛.
|
|
(12) |
通过不同策略和合成砌块构建环戊二烯环是苯并富烯的主要合成方法. 2019年Blond等[47]报道了一例完全不同的策略, 通过直接构建苯环来合成苯并富烯(Eq. 13).使用预组织好的三炔化合物为底物在[Rh(CO)2]2的催化下, 炔烃芳构化构建苯环得到多环骨架的产物.反应还会生成环庚三烯酮骨架的副产物.该方法可以合成多取代的非对称的多环苯并富烯.
|
|
(13) |
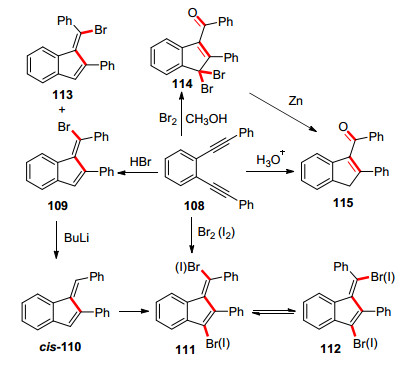
利用亲电或者亲核试剂进攻引发环化反应的苯并富烯合成研究很早就有报道. 1969年Reichardt课题组[48]研究了简单亲电试剂, 如溴化氢、水、碘或溴等对苯并烯二炔类底物108的影响(Scheme 17).当加入溴化氢气体或者溴化氢的乙酸溶液会产生两种异构体106、113, 加入溴或者碘单质则会生成二溴代或者二碘代的苯并富烯衍生物111, 而加入溴和甲醇得到的是溴代茚的产物114.
2003年, Schreiner课题组[49a]报道了一例构建多苯取代苯并富烯的方法(Eq. 14).该方法采用烯炔化合物116, 与单质溴反应得到多芳基取代的溴代苯并富烯. 2017年Sanz等[49b]利用相同的策略, 使用NIS为亲电试剂, 得到了碘代苯并富烯.
|
|
(14) |
2007年, 黄宪课题组[50]报道了三氯化铝促进的酰氯和亚烃基环丙烷开环/环化串联反应构建苯并富烯的反应(Eq. 15).该方法在温和的条件下可以方便地合成官能团化的苯并富烯, 而且高选择性地生成了Z-构型的环外双键.
|
|
(15) |
2014年, 陈知远课题组[51]报道了一例BiCl3或TsOH促进的串联反应合成三氟甲硫基取代的苯并富烯(Eq. 16).使用N-(三氟甲基硫代)苯胺提供亲电的三氟甲基硫正离子, 可以促进二炔的亲电关环, 并有利于氯离子或对甲苯磺酸离子的亲核反应.
|
|
(16) |
2014年, Martinelli等[52]先通过两步简单的反应合成1, 2-二芳基炔基苯, 然后用碘促进环化构建碘代苯并富烯, 通过偶联反应, 最终得到多取代苯并富烯的骨架的共轭分子(Eq. 17).
|
|
(17) |
2001年, Schreiner课题组[53]用末端为羰基取代的烯二炔为底物, 以TEMPO作为引发剂时, 在甲苯中回流得到了5-Exo-Dig型关环中间体, 再攫取氢后得到了富烯产物.但该方法用于苯并富烯衍生物的合成时, 只是发生了5-Exo-Dig型关环反应, 得到Bergman环化产物, 没有苯并富烯生成.
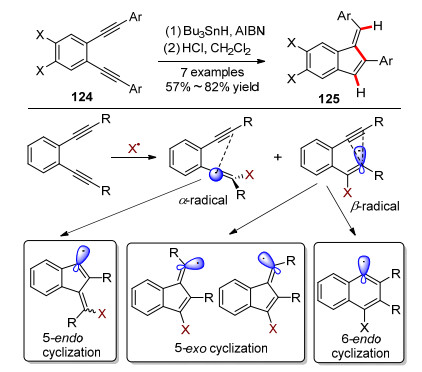
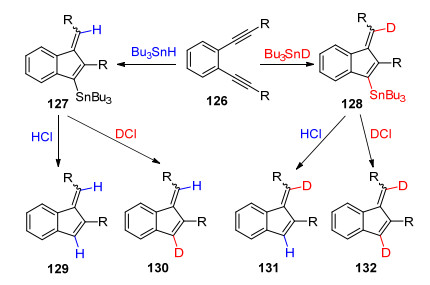
2004年, Alabugin课题组[54]尝试用自由基环化的方法合成苯并富烯.他们使用传统的烯二炔类化合物为底物, 在AIBN引发下以三丁基锡氢作为自由基源, 首次合成了锡取代的苯并富烯衍生物, 经过酸化得到中等产率的苯基取代苯并富烯(Scheme 18).这种方法不同于Bergman环化的双自由基机理, 是引发剂引发的自由基链式反应.而且该方法不能有效控制环外的双键的构型, 得到的是Z/E构型的混合物.
随后, Alabugin课题组[55]对苯并富烯产物中环外烯烃中氢原子的来源进行了研究(Scheme 19).通过氘代实验, 发现氢原子来源于叔丁基锡氢, 证实了该反应是自由基链式反应.该研究为合成稳定同位素标记的苯并富烯类化合物提供了一个有益的参考.
2019年, 姜波等[56]报道了一例自由基引发的构建苯并富烯的反应(Eq. 18).他们使用DABCO·(SO2)2复合物作为引发剂, 与重氮盐133发生单电子转移, 二氧化硫插入生成磺酰基自由基并释放出氮气, 磺酰基自由基进攻另一分子炔烃134, 进而发生串联的自由基环化反应生成具有苯并富烯骨架的多环砜类化合物135.
|
|
(18) |
2009年, Gandon课题组[57]报道了酸催化合成苯并富烯的方法(Eq. 19).该方法使用一类含有α-羟基联烯骨架的底物136, 在酸的作用下发生Nazarov-Type的环化构建苯并富烯.研究发现, 路易斯酸和布朗斯特酸, 如ZnCl2, Zn(OTf)2, Cu(OTf)2, AgOTf, TfOH, H3PMo12-O40等, 都可以催化该反应.
|
|
(19) |
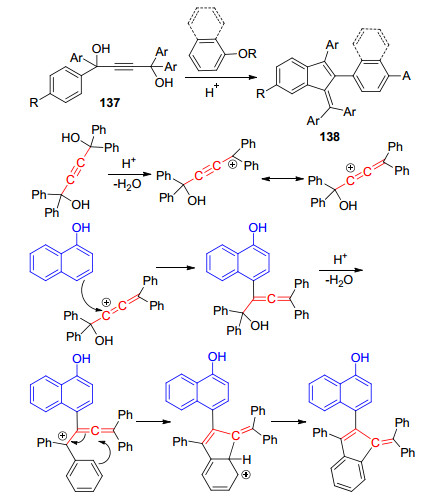
2014年, Coelho课题组[58]报道了一例p-TsOH催化的环化反应构建苯并富烯的方法(Scheme 20).该方法以四芳基-2-丁炔-1, 4-二醇137为底物, 在催化量的p- TsOH作用下, 与萘酚反应快速生成多取代的苯并富烯138.底物137可以由二苯基甲酮与乙炔基锂反应得到, 然后在酸的催化下与萘酚发生傅克烷基化反应后异构化为联烯的中间体, 然后进行环化反应得到最终的苯并富烯产物.这个方法特点是底物易于制备, 反应简单, 能得到高度取代的多芳基苯并富烯.
2014年, Matsubara课题组[59]发展了锂催化的Nazarov-Type环化构建苯并富烯的合成方法(Eq. 20), 相比过渡金属催化的反应, 锂催化对环境更加友好.
|
|
(20) |
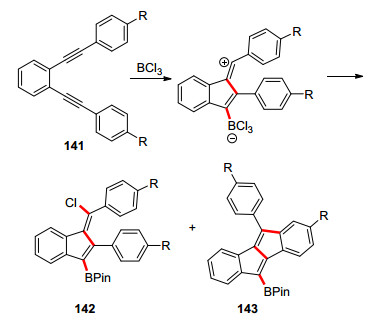
2019年Ingleson等[60]报道了二炔基苯硼化环化合成苯并富烯的硼酸衍生物(Scheme 21).该反应以路易斯酸BCl3引发炔烃环化, 同时又作为硼源.当两性离子生成后, 一个反应途径是攫取一个Cl离子生成氯代苯并富烯硼酸, 另一反应途径是进攻苯环, 生成二苯并戊搭烯.使用带正电荷的Cl2B(lutidine)+时, 反应更倾向于生成二苯并戊搭烯143, 而加入(NBu4)BCl4后, 则可以增加苯并富烯142的比例.
由于苯并富烯在不同领域的广泛用途, 吸引了众多有机化学家进行相关的合成研究.一直以来, 传统的碱催化苯并环戊二烯与醛酮缩合的方法存在底物的官能团兼容性比较差的问题.后来报道的以烯二炔和烯炔-联烯类化合物为底物合成苯并富烯的方法丰富了苯并富烯的合成途径.但早期, 这种底物合成难度比较大, 随着金属催化的偶联反应的发展, 这种底物的合成难度减小, 使这种方法又有了新的活力.利用烯二炔和烯炔-联烯类底物, 早期主要是引入亲电试剂来加速环化反应的进行, 后来逐渐发展了光催化或热引发的环化反应.但是利用这类底物进行环化反应, 不可避免的会产生C1-C6环化的苯环产物, 因为这类环化需要的能量较低.
21世纪之后, 随着各种偶联反应(Heck, Suzuki)和C—H键活化应用于苯并富烯骨架的合成, 用于苯并富烯合成的底物变得丰富多样.苯以酮类的底物可以通过过渡金属催化的C—H键活化与乙炔类的底物进行偶联构建苯并富烯, 不过这类方法总是会伴有茚醇类副产物生成.偕二溴烯炔类的底物通过分子内的偶联反应直接构建苯并富烯产物, 这类方法反应条件温和, 改善了以往苯并富烯骨架合成需要剧烈条件的状况, 然而底物实用性范围还有待进一步的研究.
近期合成苯并富烯骨架的方法更加丰富, 通过设计合适串联反应, 可以以简单的底物, 一锅合成结构复杂的苯并富烯产物.底物方面, 既有多联烯的底物应用于苯并富烯的合成, 又有累积烯烃类型的底物来合成苯并富烯骨架, 饱和三元环也可以通过金催化构建苯并富烯.
虽然苯并富烯的合成已发展出了许多的方法, 但仍有问题需要就解决.例如环外双键的构型控制问题就一直没有很好的解决办法.虽然对于一些特殊底物可以实现有效控制, 但是不具有普适性.发展一类可以控制苯并富烯环外双键构型, 而且对广泛底物适用的方法很有必要.另外合成苯并富烯的底物基本都含有碳碳叁键或者联烯, 对于仅含有碳碳双键的底物研究够少, 未来这方面将有望成为苯并富烯合成的趋势.
Thiele, J. Chem. Ber. 1900, 33, 666. doi: 10.1002/cber.190003301113
Yates, P. V. Adv. Alicyclic Chem. 1968, 2, 59. doi: 10.1016/B978-1-4831-9918-4.50008-9
Simple examples for application, see: (a) Kosaka, Y.; Kitazawa, K.; Inomata, S.; Ishizone, T. ACS Macro Lett. 2013, 2, 164.
(b) Cappelli, A.; Galeazzi, S.; Giuliani, G.; Anzini, M.; Grassi, M.; Lapasin, R.; Grassi, G.; Farra, R.; Dapas, B.; Aggravi, M.; Donati, A.; Zetta, L.; Boccia, A. C.; Bertini, F.; Samperi, F.; Vomero, S. Macromolecules 2009, 42, 2368.
(a) Douglas, A. W.; Larsen, R. D.; Verhoeven, T. R.; Reider, P. J. Tetrahedron Lett. 1995, 36, 3993.
(b) Shanmugam P.; Rajasingh, P. Chem. Lett. 2005, 34, 1494.
(a) Walters, M. J.; Blobaum, A. L.; Kingsley, P. J.; Felts, A. S.; Sulikowski, G. A.; Marnett, L. J. Bioorg. Med. Chem. Lett. 2009, 19, 3271.
(b) Felts, A. S.; Siegel, B. S.; Young, S. M.; Moth, C. W.; Lybrand, T. P.; Dannenberg, A. J.; Marnett, L. J.; Subbaramaiah, K. J. Med. Chem. 2008, 51, 4911.
(c) Korte, A.; Legros, J.; Bolm, C. Synlett 2004, 2397.
(d) Maguire, A. R.; Papot, S.; Ford, A.; Touhey, S.; O'Connor, R.; Clynes, M. Synlett 2001, 41.
(a) Snyder, S. A.; Zografos, A. L.; Lin, Y. Angew. Chem., Int. Ed. 2007, 46, 8186.
(b) Jeffrey, J. L.; Sarpong, R. Tetrahedron Lett. 2009, 50, 1969.
(c) Singer, R. A.; McKinley, J. D.; Barbe, G.; Farlow, R. A. Org. Lett. 2004, 6, 2357.
(a) Cappelli, A.; Galeazzi, S.; Giuliani, G.; Anzini, M.; Donati, A.; Zetta, L.; Mendichi, R.; Aggravi, M.; Giorgi, G.; Paccagnini, E.; Vomero, S. Macromolecules 2007, 40, 3005.
(b) Cappelli, A.; Galeazzi, S.; Giuliani, G.; Anzini, M.; Grassi, M.; Lapasin, R.; Grassi, G.; Farra, R.; Dapas, B.; Aggravi, M.; Donati, A.; Zetta, L.; Boccia, A. C.; Bertini, F.; Samperi, F.; Vomero, S. Macromolecules 2009, 42, 2368.
(c) Cappelli, A.; Paolino, M.; Grisci, G.; Giuliani, G.; Donati, A.; Mendichi, R.; Boccia, A. C.; Botta, C.; Mroz, W.; Samperi, F.; Scamporrino, A.; Giorgi, G.; Vomero, S. J. Mater. Chem. 2012, 22, 9611.
(d) Kosaka, Y.; Kitazawa, K.; Inomata, S.; Ishizone, T. ACS Macro Lett. 2013, 2, 164.
(e) Cappelli, A.; Villafiorita-Monteleone, F.; Grisci, G.; Paolino, M.; Razzano, V.; Fabio, G.; Giuliani, G.; Donati, A.; Mendichi, R.; Boccia, A. C.; Pasini, M.; Botta, C. J. Mater. Chem. C 2014, 2, 7897.
(f) Cappelli, A.; Razzano, V.; Paolino, M.; Grisci, G.; Giuliani, G.; Donati, A.; Mendichi, R.; Samperi, F.; Battiato, S.; Boccia, A. C.; Mura, A.; Bongiovanni, G.; Mroz, W.; Botta, C. Polym. Chem. 2015, 6, 7377.
(g) Kosaka, Y.; Kawauchi, S.; Goseki, R.; Ishizone, T. Macro-molecules 2015, 48, 4421.
(h) Wang, W.; Schlegel, R.; White, B. T.; Williams, K.; Voyloy, D.; Steren, C. A.; Goodwin, A.; Coughlin, E. B.; Gido, S.; Beiner, M.; Hong, K.; Kang, N.-G.; Mays, J. Macromolecules 2016, 49, 2646.
(i) Chen. S.-D.; Li, Q.-P.; Sun, S.-Y.; Ding, Y.; Hu, A.-H. Macromolecules 2017, 50, 534.
(a) Fischer, M.; Oswald, T.; Ebert, H.; Schmidtmann, M.; Beckhaus, R. Organometallics 2018, 37, 415.
(b) Rogers, J. S.; Lachicotte, R. J.; Bazan, G. C. Organometallics 1999, 18, 3976.
Habaue, S.; Sakamoto, H.; Okamoto, Y. Polym. J. 1997, 29, 384. doi: 10.1295/polymj.29.384
Jaquith, J. B.; Guan, J.; Wang, S.; Collins, S. Organometallics 1995, 14, 1079. doi: 10.1021/om00003a001
Yuki, K.; Susumu, K.; Raita, K.; Takashi, I. Macromolecules 2015, 48, 4421. doi: 10.1021/acs.macromol.5b00944
Grissom, J. W.; Gunawardena, G. U.; Klingberg, D.; Huang, D. Tetrahedron 1996, 52, 6453. doi: 10.1016/0040-4020(96)00016-6
(a) Mers, A. G.; Kuo, E. Y.; Finney, N. S. J. Am. Chem. Soc. 1989, 111, 8057.
(b) Nagata, R.; Yamanaka, H.; Okazaki, E.; Saito, I. Tetrahedron Lett. 1989, 30, 4995.
Matthias, P.; Alexander, W.; Peter, R. S. J. Phys. Chem. A 2001, 105, 9265. doi: 10.1021/jp0028002
Schmittel, M.; Strittmatter, M.; Kiau, S. Tetrahedron Lett. 1995, 36, 4975. doi: 10.1016/00404-0399(50)09378-
Schmittel, M.; Kiau, S.; Siebert, T.; Strittmatter, M. Tetrahedron Lett. 1996, 37, 7691.
(b) Schmittel, M.; Maywald, M.; Strittmatter, M. Synlett 1997, 165.
Igor, V. A.; Serguei, V. K. J. Am. Chem. Soc. 2002, 124, 9052 doi: 10.1021/ja026630d
Schmittel, M.; Mahajan, A. A.; Bucher, G.; Bats, J. W. J. Org. Chem. 2007, 72, 2166. doi: 10.1021/jo062448+
Vavilala, C.; Byrne, N.; Kraml, C. M.; Douglas, M.; Pascal, J. R. J. Am. Chem. Soc. 2008, 130, 13549. doi: 10.1021/ja803413f
Bucher, G.; Mahajan, A.; Schmittel, M. J. Org. Chem. 2008, 73, 8815. doi: 10.1021/jo801689w
Tsuchikama, K.; Kasagawa, M.; Hashimoto, Y.; Endo, K.; Shibata, T. J. Organomet. Chem. 2008, 693, 3939. doi: 10.1016/j.jorganchem.2008.09.065
Tsuchikama, K.; Kasagawa, M.; Endo, K.; Shibata, T. Synlett 2010, 1, 97.
Patureau, F, W.; Besset, T.; Kuhl, T.; Glorius, J. F. J. Am. Chem. Soc. 2011, 133, 2154. doi: 10.1021/ja110650m
(a) Chinnagolla, R. K.; Jeganmohan, M. Eur. J. Org. Chem. 2012, 417.
(b) Yu, Y.; Wu, Q.; Liu, D.; Hu, L.; Yu, L.; Tan, Z.; Zhu, G. J. Org. Chem. 2019, 84, 7449.
Guo, B.; Zheng, L.Y.; Zheng, Z. L.; Hua, R. M. J. Org. Chem. 2015, 80, 8430. doi: 10.1021/acs.joc.5b01304
Raju, S.; Hsiao, H.-C.; Thirupathi, S.; Chen, P.-L.; Chuang, S.-C. Adv. Synth. Catal. 2019, 361, 683. doi: 10.1002/adsc.201801352
Dyker, G.; Nerenz, F.; Siemsen, P.; Bubenitschek, P.; Jones, P. G. Chem. Ber. 1996, 1265. doi: 10.1002/chin.199652119
Singer, R. A.; McKinley, J. D.; Barbe, G.; Farlow, R. A. Org. Lett. 2004, 6, 2357. doi: 10.1021/ol049316s
Furuta, T.; Asakawa, T.; Iinuma, M.; Fujii, S.; Tanaka, K.; Kan, T. Chem. Commun. 2006, 3648. doi: 10.1002/chin.200703090
Abdur Rahman, S. M.; Sonoda, M.; Ono, M.; Miki, K.; Tobe, Y., Org. Lett. 2006, 8, 1197. doi: 10.1021/ol0600281
Li, C.-K.; Zeng, Y.; Wang, J.-B. Tetrahedron Lett. 2009, 50, 2956. doi: 10.1016/j.tetlet.2009.03.203
Ye, S.; Gao, K.; Zhou, H.; Yang, X.; Wu, J. Chem. Commun. 2009, 5406. https://www.ingentaconnect.com/content/rsoc/13597345/2009/00002009/00000036/art00020
(a) Ye, S.-Q.; Yang X.-D.; Wu, J. Chem. Commun. 2010, 46, 2950.
(b) Ye, S.-Q.; Ren, H.; Wu, J. J. Comb. Chem. 2010, 12, 670.
Bryan C. S.; Lautens, M. Org. Lett. 2010, 12, 2754. doi: 10.1021/ol100844v
Kim, K. H.; Kim, S. H.; Park, B. R.; Kim, J.-N. Tetrahedron Lett. 2010, 51, 3368. doi: 10.1016/j.tetlet.2010.04.110
Lim, C. H.; Kim, K. H.; Lim, J. W.; Kim, J. N. Tetrahedron Lett. 2013, 54, 5808. doi: 10.1016/j.tetlet.2013.08.053
(a) Wang, W.-Y.; Sun, L. L.; Deng, C. L.; Tang, R.-Y.; Zhang, X.-G. Synthesis 2013, 45, 118.
(b) Zhang, T.; Chen, F.; Zhang, X.-H.; Qian, P.-C.; Zhang, X.-G. J. Org. Chem. 2019, 84, 307.
(a) Álvarez, E.; Miguel, D.; García-García, P.; Fernández-Rodríguez, M. A.; Rodríguez, F.; Sanz, R. Synthesis 2012, 44, 1874.
(b) Alvarez, E.; Nieto Faza, O.; Silva Lopez, C.; Fernandez-Rodri-guez, M. A.; Sanz, R. Chem.-Eur. J. 2015, 21, 12889.
Chen, Z.; Jia, X.; Huang, J.; Yuan, J. J. Org. Chem. 2014, 79, 10674. doi: 10.1021/jo5020245
Hou, Q. W.; Zhang, Z. H.; Kong, F. J.; Wang, S. Z.; Wang, H. Q.; Yao, Z. J. Chem. Commun. 2013, 49, 695. doi: 10.1039/C2CC36245G
Aziz, J.; Brion, J.-D.; Alami, M.; Hamze, A. RSC Adv. 2015, 5, 74391. doi: 10.1039/C5RA14459K
Shin, S.; Son, J. Y.; Choi, C.; Kim, S.; Lee, P. H. J. Org. Chem. 2016, 81, 11706. doi: 10.1021/acs.joc.6b02140
Zhou, B.; Wu, Z.; Qi, W.; Sun, X.; Zhang, Y. Adv. Synth. Catal. 2018, 360, 4480. doi: 10.1002/adsc.201801141
Peng, J.-B.; Wu, F.-P.; Spannenberg, A.; Wu, X.-F. Chem. Eur. J. 2019, 25, 8696.
Borthakur, S.; Baruah, S.; Sarma, B.; Gogoi, S. Org. Lett. 2019, 21, 2768. doi: 10.1021/acs.orglett.9b00726
(a) Hashmi, A. S. K.; Braun, I.; Noesel, P.; Schaedlich, J.; Wieteck, M.; Rudolph, M.; Rominger, F. Angew. Chem., Int. Ed. 2012, 51, 4456.
(b) Plajer, A; Ahrens, L.; Wieteck, M.; Lustosa, D. M.; Babaahmadi, R.; Yates, B.; Ariafard, A.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Chem.-Eur. J. 2018, 24, 10766.
Salacz, L.; Girard, N.; Suffert, J.; Blond, G. Molecules 2019, 24, 595. doi: 10.3390/molecules24030595
Whitlock, H. W.; Sandvick, P. E.; Overman, L. E.; Reichardt, P. B. J Org. Chem. 1969, 34, 879. doi: 10.1021/jo01256a023
(a) Schreiner, P. R.; Prall, M.; Lutz, V. Angew. Chem., Int. Ed. 2003, 42, 5757.
(b) Garcia-Garcia, P.; Sanjuan, A. M.; Rashid, M. A.; Martinez-Cuezva, A.; Fernandez-Rodriguez, M. A.; Rodriguez, F.; Sanz, R. J. Org. Chem. 2017, 82, 1155.
Huang, X.; Yang, Y. Org. Lett. 2007, 9, 1667. doi: 10.1021/ol070331h
Xiao, Q.; Zhu, H.; Li, G.; Chen, Z. Adv. Synth. Catal. 2014, 356, 3809. doi: 10.1002/adsc.201400561
Martinelli, C.; Cardone, A.; Pinto, V.; Mastropasqua Talamo, M.; D'Arienzo, M. L.; Mesto, E.; Schingaro, E.; Scordari, F.; Naso, F.; Musio, R.; Farinola, G. M. Org. Lett. 2014, 16, 3424. doi: 10.1021/ol5015366
König, B.; Pitsch, W.; Klein, M.; Vasold, R.; Prall, M.; Schreiner, P. R. J. Org. Chem. 2001, 66, 1742. doi: 10.1021/jo001417q
Kovalenko, S. V.; Peabody, S.; Manoharan, M.; Clark, R. J.; Alabugin, I. V. Org. Lett. 2004, 6, 2457. doi: 10.1021/ol049122c
Peabody, S. W.; Breiner, B.; Kovalenko, S. V.; Patil, S.; Alabugin, I. V. Org. Biomol. Chem. 2005, 3, 218. doi: 10.1039/b417493n
Qin, X.-Y.; He, L.; Li, J.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Chem. Commun. 2019, 55, 3227. doi: 10.1039/C9CC00324J
Cordier, P.; Aubert, C.; Malacria, M.; Lacote, E.; Gandon, V. Angew. Chem., Int. Ed. 2009, 48, 8757. doi: 10.1002/anie.200903675
Sousa, C. M.; Berthet, J.; Delbaere, S.; Coelho P. J. J. Org. Chem. 2014, 79, 5781. doi: 10.1021/jo500907z
Sai, M.; Matsubara, S. Synlett 2014, 25, 2067. doi: 10.1055/s-0034-1378333
Warner, A. J.; Enright, K. M.; Cole, J. M.; Yuan, K.; McGough, J. S.; Ingleson, M. J. Org. Biomol. Chem. 2019, 17, 5520. doi: 10.1039/C9OB00991D

图 2 含苯并富烯骨架的生物活性分子和功能材料
Figure 2 Bioactive molecules and materials containing benzofulvene moiety

图式 2 烯二炔和烯炔-联烯类底物环芳香化反应的几种模式
Scheme 2 Cycloaromatization patterns of enediyne and enyne- allene substrates

图式 3 光或热引发联烯-烯炔环化构建苯并富烯
Scheme 3 Synthesis of benzofulvenes by photo/thermal cyclization of enyne-allenes

图式 4 热引发联烯-烯炔环化构建苯并富烯
Scheme 4 Synthesis of benzofulvenes by photo/thermal cyclization of enyne-allenes

图式 5 光引发联烯-烯炔环化构建苯并富烯
Scheme 5 Synthesis of benzofulvenes by photo cyclization of enyne-allenes

图式 7 钌催化的C—H键活化构建苯并富烯
Scheme 7 Synthesis of benzofulvenes by ruthenium catalyzed C—H bond activation

图式 8 钯催化的C—H键活化构建苯并富烯
Scheme 8 Palladium catalyzed synthesis of benzofulvenes via C—H activation

图式 10 不对称累积烯烃多米诺反应构建苯并富烯
Scheme 10 Synthesis of benzofulvenes via domino reaction with unsymmetrically substituted cumulene

图式 11 金催化三元环异构构建苯并富烯
Scheme 11 Au-catalyzed isomerization of cyclopropenes to build benzofulvene derivatives

图式 12 钯催化Suzuki/Heck串联反应构建苯并富烯
Scheme 12 Pd-catalyzed tandem Suzuki/Heck reaction of 1- (2, 2-dibromovinyl)-2-alkenylbenzenes

图式 13 由Baylis-Hillman加成产物构建苯并富烯
Scheme 13 Building benzofulvene derivatives from Baylis- Hillman adduct

图式 14 钯催化的串联环化-偶联反应构建苯并富烯
Scheme 14 Synthesis of 2-CF3-benzofulvenes via Pd-catalyzed tandem carbocyclization-coupling reactions

图式 17 引入亲电试剂促进环化反应构建苯并富烯
Scheme 17 Enediyne cyclization promoted by electrophiles to build benzofulvene derivatives

图式 20 酸催化构建多芳基取代苯并富烯衍生物
Scheme 20 Acid catalyzed synthesis of multi-substituted benzofulvenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





















 下载:
下载:








 下载:
下载: