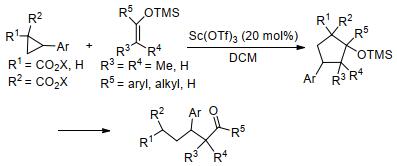
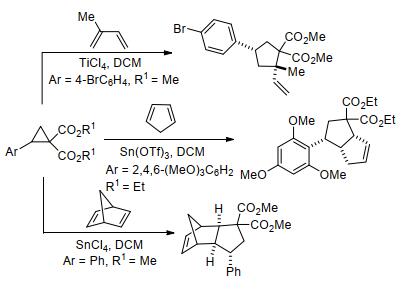
图式 1.
芳基环丙烷与烯醇甲硅烷基醚的[3+2]扩环反应
Scheme 1.
[3+2] ring-expansion reaction of aryl cyclopropanes with enol silyl ethers

环丙烷是结构最简单的环烷烃, 由于其环C—C的键角远远小于正常C(sp3)—C(sp3)的键角, 导致其键角的弯曲程度增大, 因此具有很强的环张力[1~3](应变能为121.4 kJ•mol-1 [3]).环丙烷可被具有不同电子特性的基团取代, 并生成一系列环丙烷衍生物.经实验测定和理论计算[2-4]表明, 环丙烷上的取代基对反应活性有很大影响.相对于无取代基的环丙烷而言, 引入具有吸电子特性或给电子特性的基团可增加环丙烷的活性[4~7].尤其是具有这两种电子特性的基团在邻位取代会增加环丙烷中环碳键的极性, 使C—C键的断裂变得更加容易.这一类被邻位吸电子基团和给电子基团活化的环丙烷通常被称为“供体-受体环丙烷”(D-A型环丙烷).在催化剂的作用下, D-A型环丙烷能与醛酮、亚胺、烯烃等不饱和化合物发生分子间或分子内的[3+2]扩环反应, 生成相应的五元碳杂环[1, 6, 8~18].常见的[3+2]扩环过程分为两种:一是环丙烷先开环生成1, 3-偶极子, 随后与不饱和化合物发生[3+2]扩环; 二是环丙烷在亲核试剂的进攻下发生开环和环加成, 随后进行[3+2]关环[9, 13~17].此类反应因具有快速有效构建复杂环状骨架的优点, 目前已被广泛应用于天然产物的全合成路线中[1, 19~24].近几年, 随着高效催化剂的不断开发和应用, 环丙烷化合物的[3+2]扩环反应得到了迅速发展, 环丙烷底物的范围也不断扩大.除了传统的D-A型环丙烷外, 单取代的供体环丙烷或受体环丙烷, 以及多环稠合环丙烷等也能发生扩环反应, 所构筑得到的五元碳环的种类日益丰富, 结构也更加复杂[1~18].从反应中所使用的催化剂种类出发, 对近十年来有代表性的环丙烷衍生物与不饱和化合物的[3+2]扩环反应的研究进展进行综述, 以加深人们对环丙烷作为三碳合成子在构建五元环化合物中所起的重要作用的认识, 为发展新的催化和反应体系提供帮助.
最近十年, 路易斯酸被广泛用于环丙烷的[3+2]扩环反应中, 它通过与环丙烷上的吸电子基团作用来活化环丙烷, 并稳定生成的阴离子.常见的路易斯酸包括氯化铝(AlCl3)、氯化铁(FeCl3)、三氟化硼(BF3)以及镧系元素的三氟甲磺酸盐等.
在与环丙烷发生[3+2]扩环反应的含C=C不饱和键的底物中, 烯醇醚因具有较高的反应活性而占有重要的地位.唐勇课题组曾对环丙烷与烯醇醚的[3+2]扩环反应展开过一系列研究, 并分别于2013年和2018年对自己的研究内容做过系统报道[25, 26].此外, Grover等[16]对唐勇课题组的相关研究成果也进行过综述.为避免重复, 本文对唐勇课题组的研究内容不再赘述.
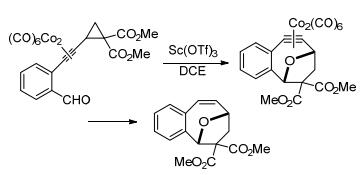
除唐勇课题组外, 其他很多课题组也对该领域的发展作出了重要贡献.如2008年王忠文等[27]报道了在Sc(OTf)3催化下, 芳基环丙烷与烯醇甲硅烷基醚的[3+2]扩环反应实例(Scheme 1).实验表明, 该反应先后经历了[3+2]扩环、开环的过程, 最终以中等至极好的产率制备得到链状羰基化合物; 除带有双给电子基团的环丙烷外, 单个给电子基团活化的环丙烷也能顺利生成链状产物.
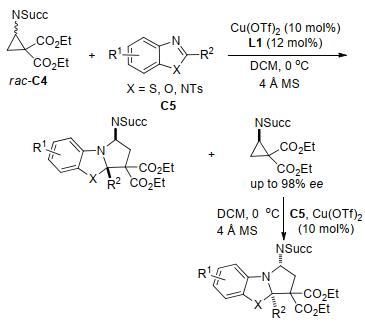
2011年, Waser等[28]报道了邻苯二甲酰亚胺(phth- alimide, NPhth)取代的环丙烷-1, 1-二酯与烯醇醚的非对映选择性[3+2]扩环反应(Eq. 1).反应以SnCl4为催化剂, 在较温和的条件下, 简便地获得了一系列高产率和高非对映选择性的氨基环戊烷衍生物.该反应的底物范围极广:各种醛类和酮类衍生的烯醇醚, 甚至是环状烯醇醚均能够很好地与氨基环丙烷发生反应. 2014年, Waser课题组[29]扩大对该类反应的研究范围, 用琥珀酰亚胺(succinimide, NSucc)取代的环丙烷-1, 1-二酯与烯醇醚进行动力学不对称[3+2]环化反应研究.反应在Cu(II)/双噁唑啉型配体L1的催化下, 以优异的产率和高对映选择性以及非对映选择性得到了氨基环戊烷类化合物(Scheme 2).反应所用的催化剂体系同样适用于醛类反应底物, 所生成的产物也具有优异的对映选择性和非对映选择性.这是首个使用氨基环丙烷进行动力学不对称[3+2]环化的例子, 也是第一个用对映选择性催化体系来合成环戊烷和四氢呋喃的反应实例.
|
|
(1) |
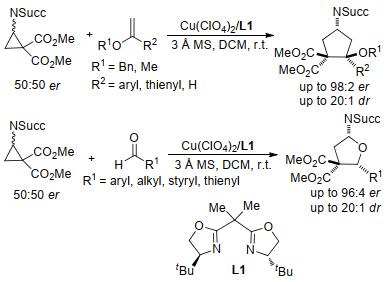
通过对上述反应的研究, Waser等[30]发现在环丙烷上引入氨基可以提高环丙烷的反应活性, 同时提高其反应产率.受到以上实验成果的启发, 该小组对氨基环丙烷的合成及其在[3+2]扩环反应中的应用进行了更深入的研究(Scheme 3).他们首先以邻苯二甲酰亚胺和乙酸乙烯酯为起始原料, 经过钯催化乙烯基化和铑催化环丙烷化两个步骤制得高产率的氨基环丙烷.随后, 氨基环丙烷与烯醇醚在二氯甲烷溶剂中进行[3+2]扩环反应, 得到一系列氨基环戊烷衍生物.除邻苯二甲酰亚胺基(NPhth)外, 马来酰亚胺(maleimide, NMalei)或琥珀酰亚胺(NSucc)取代的环丙烷也能够参与此类反应.
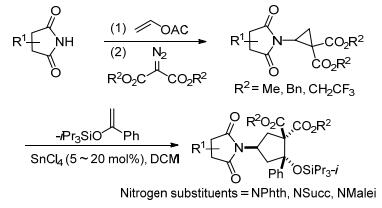
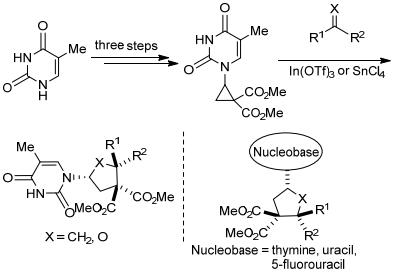
此外, Waser等[31]还报道了在铟和锡催化下, 核碱基取代的环丙烷-1, 1-二酯与醛、酮和烯醇醚的[3+2]扩环反应(Scheme 4).他们首先通过钯催化乙烯基化、叔丁氧基羰基保护和铑催化环丙烷化三个步骤制备得到核碱基环丙烷-1, 1-二酯.随后在铟或锡的催化下, 环丙烷分别与醛、酮和烯醇醚发生[3+2]扩环反应, 生成相应的四氢呋喃衍生物或环戊烷衍生物.虽然其反应底物局限于嘧啶类核碱基取代的环丙烷, 但反应产物可以通过脱羧、还原等几个步骤合成核苷类似物, 在合成生物活性化合物方面具有重要的应用价值.
上述反应均可以合成氨基取代的五元环类化合物.此外, Johnson课题组[32]通过Sc(OTf)3催化的环丙烷-1, 1-二酯与炔胺发生[3+2]扩环反应, 也能合成带有氨基的磺酰胺基环戊烯(Eq. 2).该反应适用于富电子芳族取代的环丙烷, 且能以良好至极好的产率生成相应产物.
|
|
(2) |
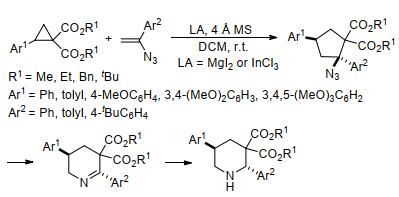
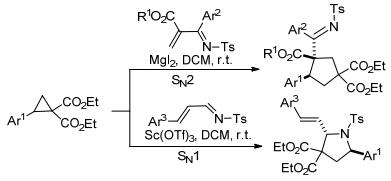
2016年, Banerjee等[33]也报道了通过芳基环丙烷-1, 1-二酯与烯胺的[3+2]不对称扩环快速合成氨基环戊烷的方法(Eq. 3).在该反应的基础上, Banerjee等[34]以MgI2或InCl3为催化剂, 将乙烯基叠氮化物作为烯胺, 继续与芳基环丙烷-1, 1-二酯发生[3+2]扩环反应, 最终以良好的非对映选择性合成了叠氮基环戊烷衍生物(Scheme 5).该反应的产物还可以进一步扩环得到四氢吡啶, 并被继续还原成哌啶衍生物.同年, Banerjee课题组[35]还开发了芳基环丙烷-1, 1-二酯与氮杂二烯的扩环反应(Scheme 6).其中, 氮杂二烯上取代基的种类和位置对反应产物具有重要影响, 它决定了该反应以SN2还是SN1方式进行, 从而分别获得环戊烷和吡咯烷衍生物.
|
|
(3) |
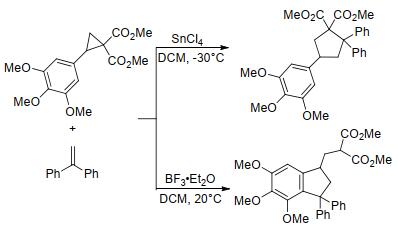
在上述环加成反应中, 环丙烷化合物均以三碳组分参与构建五元环. 2013年, Budynina等[36]发现在强路易斯酸催化下, 当环丙烷的供体位置为富电子芳基或杂芳基时, 环丙烷化合物能以不同的反应活性位点与烯烃发生扩环反应, 分别生成环戊烷衍生物和茚满衍生物(Scheme 7).作者认为, 该反应的化学选择性受多种因素影响, 其中环戊烷衍生物主要是动力学控制产物, 而茚满衍生物则主要是热力学控制产物.
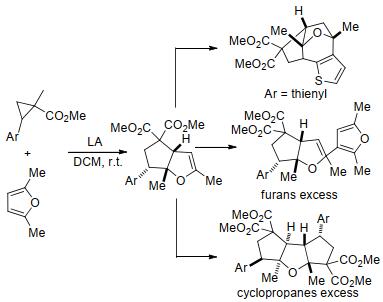
除脂肪烃外, 环丙烷化合物还可以与环状烯烃发生[3+2]扩环反应, 构建结构更为复杂的多环化合物. 2009年, Budynina等[37]报道了芳基环丙烷-1, 1-二酯与2, 5-二甲基呋喃在Yb(OTf)3或SnCl4催化下发生的[3+2]扩环反应(Scheme 8).该反应的产物还可以进一步转化为噻吩并[2, 3-d]吡喃、2-呋喃基环戊二烯并[b]呋喃、环戊二烯并[b, d]呋喃等多种产物, 且催化剂、起始原料用量和环丙烷中芳族取代基的亲核性都会对该反应带来影响.如果芳族取代基的亲核性强, 初始产物会发生分子内烷基化反应, 进一步生成相应的四环产物; 若呋喃过量, 则第二个呋喃分子会与初始产物的双键发生加成; 如果环丙烷过量, 则会与初始产物进行第二次[3+2]扩环.因此, 可以通过控制上述关键因素来获得不同类型的产物.
除呋喃化合物外, Budynina等[38]又报道了环丙烷与1, 3-二烯类化合物的[3+2]扩环反应(Scheme 9).在适宜的路易斯酸催化下, 反应以较高的非对映选择性合成了环戊烷骨架.其中, 二烯类化合物的底物范围广, 除链状1, 3-二烯以及共轭环状二烯外, 不含共轭双键体系的降冰片二烯也能与环丙烷发生[3+2]扩环反应.
唐勇课题组也对该类反应开展了研究, 并于2013年[39]报道了在Cu(II)/双噁唑啉型配体L3催化下、环丙烷-1, 1-二酯与吲哚类化合物发生的[3+2]扩环反应.该反应能生成一系列具有优异对映选择性和非对映选择性的环戊二烯并二氢吲哚类产物(Eq. 4).之后, 该课题组[40]继续以Cu(II)/双噁唑啉型配体L4为催化剂, 实现了2, 2-二取代环丙烷-1, 1-二酯与吲哚的[3+2]扩环反应(Eq. 5).该反应的底物范围宽泛, 能以优良的产率和中等至优异的非对应选择性一步构建吲哚并五元碳环结构, 并在碳环上一次性构建三个季碳中心, 对于简洁高效合成一些天然产物核心骨架具有十分重要的意义.
|
|
(4) |
|
|
(5) |
除环状烯烃外, Kaicharla等[41]还开发了环丙烷-1, 1-二酯与2-萘酚的[3+2]扩环反应(Eq. 6).该反应在Bi(OTf)3催化下能以良好的产率生成环戊烷衍生物, 同时对多种取代2-萘酚底物都具有极好的兼容性.而当使用Sc(OTf)3作为路易斯酸时, 则发生Friedel-Crafts加成, 生成官能化的2-萘酚.两种反应都具有很好的区域选择性和较高的产率.作者推测造成这种差异的主要原因是金属铋Bi(III)能够同时活化环丙烷C1和2-萘酚C2, 使二者得以发生[3+2]扩环反应; 而Sc(III)仅仅活化环丙烷C1, 因此发生Friedel-Crafts加成反应.
|
|
(6) |
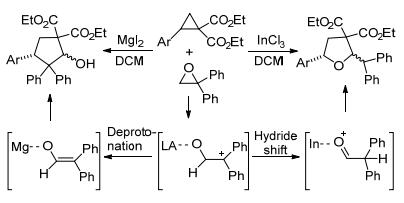
2017年, Banerjee等[42]报道了芳基环丙烷-1, 1-二酯与环氧化物的[3+2]扩环反应(Scheme 10).反应始于环氧化物的开环裂解, 然后裂解的中间体发生质子转移生成醛或者发生去质子化生成烯醇, 随后醛和烯醇分别与芳基环丙烷-1, 1-二酯进行[3+2]环加成, 生成四氢呋喃和环戊烷衍生物.与Kaicharla等[41]报道的反应类似, 该反应中路易斯酸的种类同样会影响产物的形成. MgI2和InCl3分别有利于形成环戊烷和四氢呋喃衍生物, 而BF3•OEt2的参与则生成两种产物的混合物.
环丙烷与烯烃、炔烃的[3+2]扩环反应除能以分子间的形式进行外, 还能以分子内的形式发生反应. 2012年, 梁永民等[43]报道了Sc(OTf)3催化下、环丙烷与炔烃的分子内[3+2]扩环反应(Eq. 7).该反应以中等至极好的产率一步构建出环戊二烯并[c]色烯骨架, 具有高效性、良好的官能团耐受性等优点, 为进一步制备巴比妥酸衍生物提供了一个简单有效的途径.
|
|
(7) |
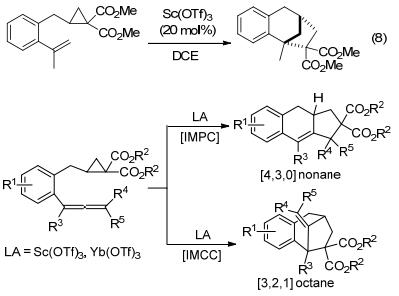
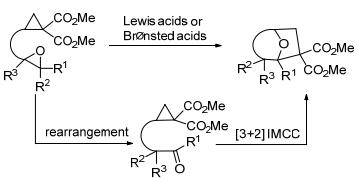
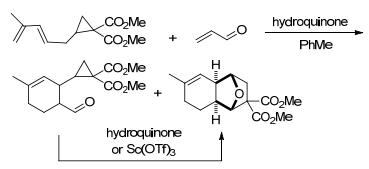
王忠文等[44]开发了Sc(OTf)3催化下环丙烷-1, 1-二酯与烯烃的新型分子内[3+2]交叉扩环反应(Eq. 8).作者通过在烯烃部分引入适当的基团, 改善了反应底物的活性和区域选择性, 最终制备得到了具有良好立体选择性和区域选择性的环加成产物, 为构建结构复杂的桥接[n.2.1] (n=2, 3, 4)碳环骨架提供了一种通用的有效策略.这是第一个路易斯酸催化的环丙烷与烯烃的分子内[3+2]扩环反应, 也是第一个路易斯酸催化环丙烷与非活化烯烃的[3+2]扩环反应.同一年, 王忠文等[45]又开发了路易斯酸催化的环丙烷-1, 1-二酯与丙二烯的分子内[3+2]扩环反应(Scheme 11).与上一个反应[44]不同的是, 该反应在不同的催化剂和温度条件下, 可以选择性进行分子内平行环加成(Intramolecular parallel cycloaddition, IMPC)或分子内交叉环加成(Intramolecular cross-cycloaddition, IMCC)反应, 从而合成[4.3.0]壬烷和[3.2.1]辛烷骨架.
|
|
(8) |
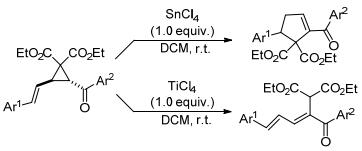
2018年, Thangamani等[46]也报道了环丙烷-1, 1-二酯与烯烃的分子内扩环反应(Scheme 12).该反应同样受催化剂种类的影响:可以在SnCl4催化下通过串联开环/闭环过程得到[3+2]扩环产物环戊烯, 也可以在TiCl4催化下通过开环和质子消除过程得到开环产物E, E-1, 3二烯.
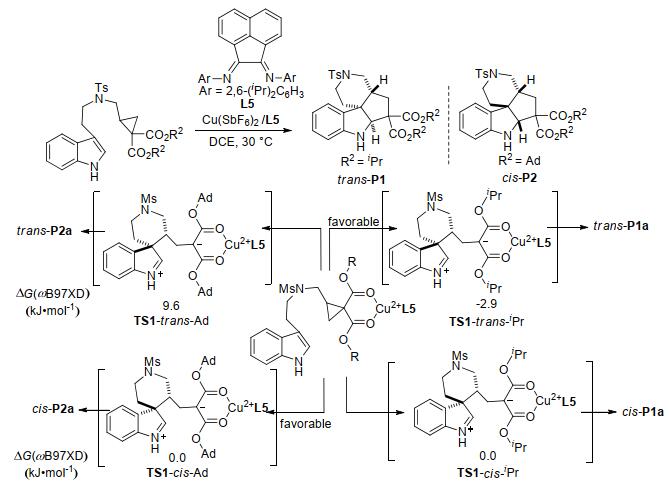
除烯烃、炔烃能与环丙烷化合物发生分子内环加成外, 唐勇课题组[47]还报道了吲哚与环丙烷-1, 1-二酯的分子内[3+2]扩环反应(Scheme 13).反应以Cu(II)和二亚胺配体L5为催化剂, 以优异的非对映选择性生成了四环螺吲哚啉骨架.研究表明:环丙烷的远端酯基对反应的非对映选择性有显著影响.当酯基为异丙酯(iPr)时, 主要生成反式产物trans-P1; 而当酯基为金刚烷基酯(Ad)时, 则主要生成顺式产物cis-P2.为了确定远端酯基对非对映选择性的影响, 作者运用密度泛函理论(DFT)对反应进行了计算.结果表明:反应底物在催化剂的活化下, 将分别经过顺式过渡态TS-trans和反式过渡态TS-cis进行开环, 该步骤为不可逆步骤, 决定了产物的立体构型.在开环过渡态中, 当基团为异丙酯时, TS1-trans-iPr中的酯基和芳烃之间存在明显的非共价作用, 其相对能量比TS1-cis-iPr低, 因此反应倾向于经过反式过渡态路径生成反式产物trans-P1a.而当基团为金刚烷基酯时, 过渡态TS1-trans-Ad中的空间位阻急剧增大, 导致反式过渡态的相对能量升高, 因此反应倾向于通过顺式过渡态路径生成顺式产物cis-P2a.
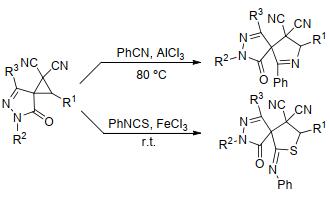
腈是含C≡N键不饱和化合物的典型代表, 常用于环丙烷的[3+2]扩环反应中构建含氮的五元碳杂环. Pagenkopf等[15]曾于2017年对腈类与D-A型环丙烷的[3+2]扩环反应进行过综述.同年, Mukherjee等[48]也曾对环丙烷与腈的环加成反应进行过报道:反应以AlCl3或FeCl3为催化剂, 通过螺吡唑啉酮基环丙烷与苯甲腈和异硫氰酸酯的[3+2]扩环反应, 合成了具有高度非对映选择性的吡咯啉骨架和2-亚胺基噻吩衍生物(Scheme 14).
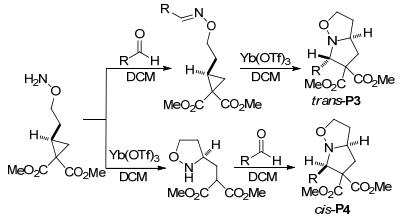
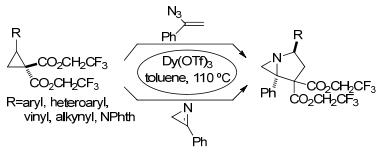
除腈类外, 其它含C、N不饱和键的化合物也能与环丙烷化合物发生[3+2]扩环反应.如2008年, Kerr等[49]报道了环丙烷-1, 1-二酯和肟醚的分子内[3+2]扩环反应(Scheme 15).该反应以高度的非对映选择性和立体选择性得到了吡咯并异噁唑烷, 并通过改变催化剂和底物的加入顺序, 选择性地生成了反式产物trans-P3或顺式产物cis-P4.之后Kerr等[50]进一步扩大环丙烷与亚胺反应的底物范围, 开发了环丙烷-1, 1-二(三氟乙基酯)与乙烯基叠氮化物和氮丙啶的[3+2]扩环反应(Scheme 16).反应以Dy(OTf)3为催化剂, 以中等至良好的产率得到了氮杂双环骨架.
2010年, Johnson等[51]以MgI2/吡啶型双噁唑啉配体L6作为催化剂, 实现了外消旋环丙烷-1, 1-二酯(rac-C3)与反式(E)-醛亚胺的动态动力学不对称环化, 并制备得到了2, 5-顺式-二取代吡咯烷(Eq. 9).反应中选择2-甲氧基苄基作为醛亚胺的N-苄基保护基团可以提高反应的对映选择性, 选择富电子和中性芳基醛亚胺也能得到较好的结果(产率高达86%, er值=98:2).
|
|
(9) |
2016年, Alajarin等[52]开发了Sc(OTf)3催化的环丙烷-1, 1-二酯与乙烯酮亚胺的[3+2]扩环反应(Eq. 10).该反应能在比较温和的条件下、以中等至良好的产率生成一系列高度取代的2-亚烷基吡咯烷.研究发现, 乙烯基、芳基和苯乙烯基取代的环丙烷-1, 1-二酯, 以及与苯并吲哚和苯并吡喃环稠合的环丙烷均适用于该反应.
|
|
(10) |
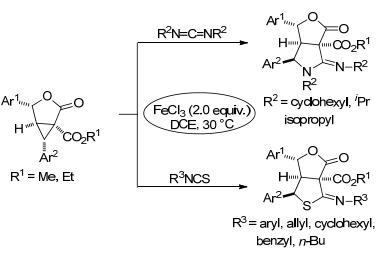
2017年, 杨高升等[53]报道了FeCl3催化的γ-丁内酯稠合环丙烷与碳二亚胺和异硫氰酸酯的[3+2]扩环反应(Scheme 17).该反应条件温和且具有宽泛的底物范围, 能够以极好的产率得到单一立体选择性的γ-丁内酯稠合脒以及双环/多环γ-丁内酯稠合的硫代亚胺酸酯.
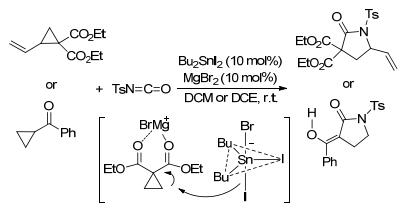
除亚胺和腈类外, 异氰酸酯、甚至吡啶、亚胺基吲哚也常以C=N不饱和键参与环丙烷的[3+2]扩环反应. 2016年, Tsunoi等[54]合成了有机锡碘配合物MgBr+[Bu2-SnBrI2]-活性催化剂, 并将其应用于环丙烷与异氰酸酯的[3+2]扩环反应中, 成功合成了γ-丁内酰胺(Scheme 18).这种催化体系的最大特点是, 既包含酸性的镁阳离子, 又包含亲核性的Sn—I键.在反应过程中, Sn—I键能有效进攻环丙烷促成开环, 同时MgBr+又可以活化环丙烷的二羧酸酯部分, 加速开环过程.鉴于该催化剂较强的催化活性, 在标准条件下, 除2-取代环丙烷-1, 1-二酯外, 单取代环丙烷、甚至酰基环丙烷都能发生[3+2]扩环反应.
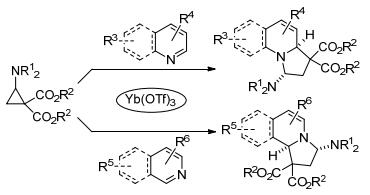
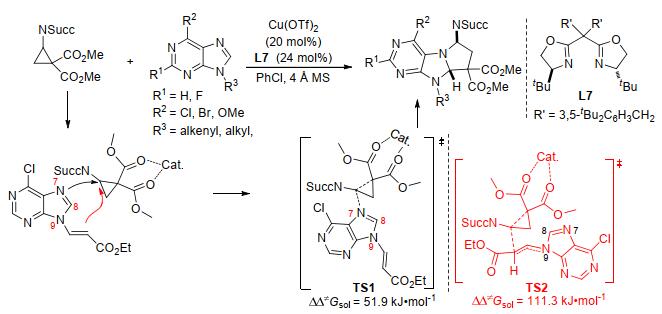
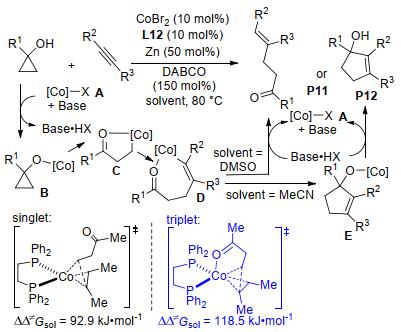
2017年, Waser及其同事[55]报道了Yb(OTf)3催化亚胺基环丙烷-1, 1-二酯与吡啶等氮杂环的脱芳构化[3+2]扩环反应(Scheme 19).该反应具有宽泛的底物范围:吡啶、喹啉和异喹啉等都能参与反应.其中亚氨基取代的环丙烷是反应成功的关键, 生成的四氢中氮茚衍生物具有优异的产率和很好的非对映选择性.两年后, 郭海明课题组分别使用Cu(OTf)2与双噁唑啉配体L1/L7作催化剂, 实现了氨基环丙烷与苯并唑类(Scheme 20)[56]和嘌呤类(Scheme 21)[57]的首个对映选择性脱芳构化[3+2]扩环反应.两个反应均以较高的产率和优异的对映选择性分别合成了一系列氢吡咯并苯并噁唑衍生物和氢吡咯并嘌呤衍生物.在前一个反应中, 作者在相同的催化条件下, 通过动力学拆分的方式获得了相应环加成产物的两个非对映异构体.而在氨基环丙烷与嘌呤类的[3+2]扩环反应中, 作者运用密度泛函理论对反应机理和化学选择性进行了计算(Scheme 21).其结果表明, 环丙烷在开环的同时与嘌呤的N7进行成键的过渡态TS1的自由能垒为51.9 kJ•mol-1, 而与N9上烯基成键的过渡态TS2的能垒高达111.3 kJ•mol-1, 所以反应优先通过与N7成键的反应路径生成氢吡咯并嘌呤衍生物.该结论很好地解释了反应底物没有在嘌呤的N9位上发生反应, 而是在嘌呤的N7、C8位置进行环加成的实验事实.
与此同时, Budynina等[58]以Yb(OTf)3和Sc(OTf)3作为催化剂, 实现了环丙烷-1, 1-二酯与亚胺基吲哚的[3+2]扩环反应(Eq. 11), 并以高达96%的产率和极好的非对映选择性合成了螺[羟吲哚-3, 2'-吡咯烷].
|
|
(11) |
路易斯酸催化环丙烷与含羰基不饱和化物的[3+2]扩环反应具有高效、原子经济性等优点, 因此一直是构建多取代氧杂环的常用方法之一.其中, 人们对2-取代环丙烷参与的[3+2]扩环反应进行了大量的研究并取得了一些卓有成效的结果.
2008年, Johnson课题组[59]开发了Sn(OTf)2催化下、环丙烷-1, 1-二酯与醛的非对映选择性[3+2]扩环反应, 并合成了顺式-2, 3, 5-四取代四氢呋喃衍生物(Eq. 12).该反应能兼容一系列烷基醛和芳基醛, 同时, 简单烷基作为环丙烷的供电子基团也可以参与底物的活化, 最后以很好的产率和非对映选择性制备得到顺式四氢呋喃衍生物.受该实验的启发, Werz等[60]在类似的反应条件下, 通过使用环丙基作为环丙烷的供体基团, 实现了环丙基环丙烷-1, 1-二酯与醛类的[3+2]扩环反应(Eq. 13).该反应以中等至优异的产率生成了四氢呋喃衍生物.动力学实验研究和计算结果表明, 这种特殊的环丙基给电子体的给电子性质比中性的芳基给电子体强得多.
|
|
(12) |
|
|
(13) |
2009年, Johnson课题组[61]以MgI2和吡啶型双噁唑啉配体L8作为手性催化剂, 成功实现了外消旋环丙烷-1, 1-二酯(rac-C6)与醛类的不对称[3+2]环化反应.该反应适用于各种脂族醛和富电子的芳基醛底物, 同样能以较高的产率和非对映选择性得到顺式2, 3, 5-四取代四氢呋喃衍生物(Eq. 14).值得注意的是, 缺电子芳基醛由于其亲核性差, 相应的反应产率往往较低.除以上反应外, Johnson课题组[62]还以Sn(OTf)2等路易斯酸为催化剂, 成功实现了2, 2-二取代环丙烷-1, 1-二酯与醛的非对映选择性[3+2]扩环反应, 并合成了一系列2, 4, 5-五取代四氢呋喃衍生物(Eq. 15).作者发现, 许多具有不同电子效应的芳香醛、杂芳族醛、支链脂族醛等都能发生反应, 且以极好的产率得到顺式产物; γ-丁内酯稠合环丙烷也能与芳香醛发生反应, 产物同样具有很好的收率和非对映选择性.在此之前, Christie及其同事[63]曾以溴化锌为催化剂, 实现了铁-二烯基环丙烷-1, 1-二酯与醛类的[3+2]扩环反应, 以中等至良好的产率合成了一系列2, 3, 5-四取代四氢呋喃(Eq. 16).
|
|
(14) |
|
|
(15) |
|
|
(16) |
2011年, Waser课题组[64]首次报道了Fe(III)催化2-氨基环丙烷-1, 1-二酯与醛的[3+2]扩环反应, 以极好的产率和优良的非对映选择性合成了2-氨基四氢呋喃衍生物(Eq. 17).该反应代表了铁催化氨基环丙烷与醛发生[3+2]扩环反应的第一个实例.由于此前对氨基环丙烷的研究主要集中在其制备方法的改进与发展上, 该反应对氨基环丙烷在开环/环加成反应中的应用进行了很好的补充.同年, 该课题组[65]继续探索2-氨基环丙烷与羰基化合物的[3+2]扩环反应, 并报道了SnCl4催化下、2-氨基环丙烷与酮类化合物的第一个对映特异性[3+2]扩环反应(Eq. 18).该反应以较高的产率得到了高对映选择性的C5-二取代氨基四氢呋喃, 且底物范围宽泛:具有各种不同电子效应的芳族、杂芳族、脂族酮(包括不对称脂族酮)等都能进行反应.
|
|
(17) |
|
|
(18) |
2013年, Haubenreisser等[66]开发了一种新型钙催化剂, 并将其作为路易斯酸应用于2, 2-二取代环丙烷-1, 1-二酯与醛的[3+2]扩环反应中, 以高达95%的产率和极好的反式非对映选择性得到四氢呋喃衍生物(Eq. 19).在比较温和的反应条件下, 该反应能将供体位置为季碳且带有炔烃的新型D-A环丙烷快速地转化为高度取代的反式四氢呋喃衍生物.作者经底物筛选还发现, 在环丙烷供体位点上的炔烃部分和第二个取代基的电子效应对反应性影响较大, 若炔烃上连有给电子基团, 则会导致副反应的发生.
|
|
(19) |
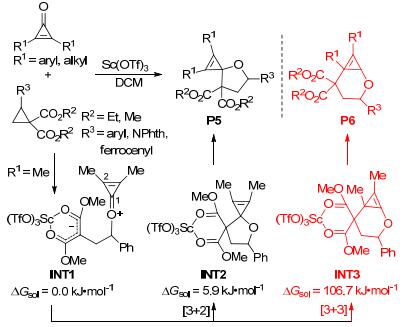
2014年, Rivero等[67]通过使用具有特殊反应性的环丙烯酮, 也实现了2-单取代环丙烷-1, 1-二酯与酮类的[3+2]扩环(Scheme 22), 并以良好至极好的产率得到了4-氧杂螺[2.4]庚-1-烯衍生物P5.反应产率主要受环丙烷供体位置处基团的电子性质的影响, 其中吸电子基团会导致较低的产率, 而给电子基团则会提高反应产率.带有二茂铁取代基的环丙烷也能在特定条件下发生[3+2]扩环反应并形成二茂铁基环状化合物, 这是制备带有有机金属片段的螺环化合物的少数反应实例之一.基于密度泛函理论计算, 作者提出了该反应的可能机理.首先, 环丙烷上的酯基与过渡金属配位形成亲电中心; 随后, 环丙烯酮羰基中的孤对电子对该亲电中心进行攻击, 形成两性离子中间体INT1; 最后, INT1闭环生成相应的[3+2]环状产物.为了更好地理解该反应的微观机理, Rivero等还对2-单取代环丙烷-1, 1-二酯与酮类可能发生的[3+3]环加成做了对比研究.计算结果表明: INT1在C1处环化形成[3+2]扩环中间体INT2的能垒是28.9 kJ•mol-1, 同时INT2相对于INT1能量略有升高(+5.9 kJ•mol-1).而在C2处发生环化形成[3+3]环加成中间体INT3的能垒高达126.4 kJ•mol-1, 且INT3相对于INT1能量升高了106.7 kJ•mol-1.因此, 无论从动力学角度还是从热力学角度考虑, 中间体INT1在C1处发生亲核进攻形成[3+2]扩环产物都是非常有利的.该结果很好地解释了2-单取代环丙烷-1, 1-二酯与酮类主要发生[3+2]扩环反应生成4-氧杂螺[2.4]庚-1-烯衍生物P5的实验事实.
从前文的叙述中可以看出:大部分D-A型环丙烷的受体基团多为酯基, 而酯基在[3+2]扩环产物的后续转化中较难转化, 为此, Sabbatani等[68]开发了缩醛取代的环丙烷与醛的[3+2]扩环反应(Eq. 20).该反应以三丁基甲硅烷基三氟甲磺酸酯(TBSOTf)为催化剂, 在温和的条件下以良好的非对映选择性形成了具有三个手性中心的四氢呋喃产物, 其中反式异构体占主导, 且产物中的缩醛部分在后续反应中很容易被去除.另一方面, 上述例子主要集中在2-取代环丙烷与醛的环加成反应中.与2-取代环丙烷相比, 2, 3-二取代环丙烷由于其环上所带的活性基团更多, 因此更能增加产物结构的多样性与复杂性, 为构建结构复杂的四氢呋喃环提供便捷的渠道.
|
|
(20) |
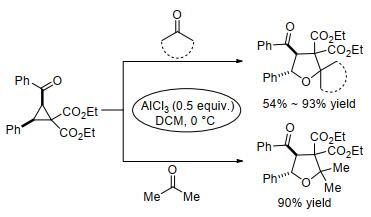
2011年, 杨高升及其同事[69]报道了反式-2, 3-二取代环丙烷(trans-C7)与醛在AlCl3催化下通过[3+2]扩环反应生成2, 3, 3, 4, 5-五取代四氢呋喃的反应(Eq. 21).该反应具有优异的区域选择性和非对映选择性, 且反式- 2, 3-二取代的环丙烷-1, 1-二酯在形成产物时受底物醛类和温度的影响:当底物为中性电子和缺电子芳香醛时, 主产物是顺式-五取代四氢呋喃(cis-P7); 当底物为富电子醛时, 反应需要更高的温度, 且产物变为反式-五取代四氢呋喃(trans-P8).
|
|
(21) |
为了比较反式和顺式-2, 3-二取代的环丙烷-1, 1-二酯的反应性, 杨高升等[70]继续研究了AlCl3催化下、顺式-2, 3-二取代环丙烷(cis-C8)与醛类的[3+2]扩环反应(Eq. 22).除强吸电子芳香醛不适合该反应外, 大部分底物均能以良好至极好的产率生成类似的高度取代的四氢呋喃衍生物, 且得到单一的非对映异构体.这说明D-A型环丙烷取代基的类型对反应效率和立体选择性具有非常大的影响.
|
|
(22) |
在以上两个反应的基础上, 杨高升课题组[71]于2015年开发了AlCl3催化顺式-2, 3-二取代环丙烷-1, 1-二酯与环状/无环脂族酮的[3+2]扩环反应(Scheme 23), 发现即使活性相对较低的酮也能在非常温和的条件下参与该反应, 并以中等至极好的产率快速转化为高度取代的反式-(螺)四氢呋喃衍生物.
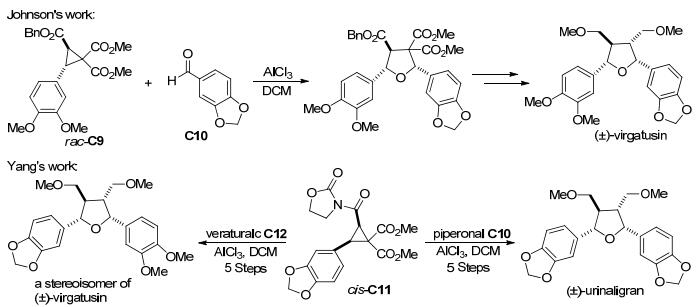
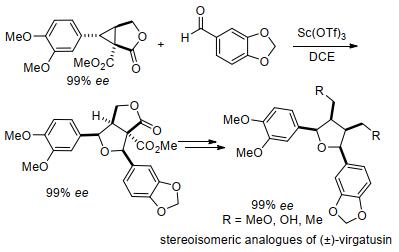
随着人们对2, 3-二取代环丙烷与羰基化合物[3+2]扩环反应研究的深入, 其在天然产物合成方面的应用也逐渐引起了广泛关注. 2009年, Johnson课题组[72]通过手性反式-2, 3-二取代环丙烷-1, 1-二酯(rac-C9)与胡椒醛C10 (piperonal)的[3+2]扩环, 以一定的选择性和高收率合成了呋喃木脂素virgatusin的骨架结构(Scheme 24).与此不同的是, 2013年杨高升课题组[73]通过顺式-2, 3-二取代环丙烷-1, 1-二酯(cis-C11)与胡椒醛C10和亚藜芦基C12 (veratral)的[3+2]环化, 以非对映选择性的方式合成了呋喃木脂素(±)-urinaligran和(±)-virgatusin (Scheme 24).经过一系列实验, 杨高升等证明了在路易斯酸催化的[3+2]环化反应中, 顺式-2, 3-二取代的环丙烷-1, 1-二酯比它们的反式异构体具有更高的反应性.
尽管单环环丙烷可以进一步反应生成稠合环丙烷, 但已有实验证明:在路易斯酸催化的[3+2]扩环反应中, 多环稠合环丙烷往往比未稠合环丙烷具有更高的反应性[53]. 2011年, Zhang等[74]首次报道了Sc(OTf)3催化手性γ-丁内酯稠合环丙烷与苯甲醛之间的对映特异性[3+2]扩环反应, 以良好的产率制备得到了单一的四氢呋喃非对映异构体, 且光学纯度几乎无损失(Eq. 23).同年, Johnson等[62]报道了γ-丁内酯稠合环丙烷与芳香醛的[3+2]扩环反应(Eq. 24), 发现该反应不仅实验条件温和, 而且同样具有非常好的非对映选择性.
|
|
(23) |
|
|
(24) |
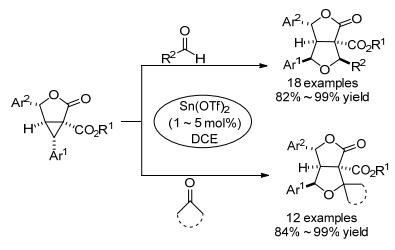
此后, 杨高升等[75]继续对该类反应进行了深入研究, 于2018年报道了Sn(OTf)2催化3, 4, 5, 6-四取代γ-丁内酯稠合环丙烷与醛类和酮类的[3+2]扩环反应(Scheme 25).该反应以优异的产率和非对映选择性制备得到了具有多个相邻立体中心的γ-丁内酯稠合四氢呋喃, 且均为单一非对映异构体.该反应的底物范围宽泛:除醛类外, 各种环酮、脂族酮甚至苯乙酮均表现出良好的反应活性.由于稠合环丙烷的高活性, 该反应的Sn(OTf)2催化剂最佳负载量(1~5 mol%)明显低于之前报道的单环环丙烷与醛的[3+2]扩环反应的催化剂负载量(50 mol%)[69, 70].
同年, 为了探究γ-丁内酯稠合环丙烷与醛类[3+2]扩环反应产物的实用性, 杨高升课题组[76]以Sc(OTf)3为催化剂实现了手性γ-丁内酯稠合环丙烷与芳香醛的[3+2]扩环反应(Scheme 26), 所得到的四氢呋喃骨架可进一步转化为具有光学活性的手性呋喃木脂素类似物.该反应操作简单, 条件温和且产率高, 为构建结构多样化的手性呋喃木脂素开辟了新途径.
除了杨高升课题组, 邵华武课题组[77]也对InCl3催化的稠合环丙烷与醛类的[3+2]扩环反应开展过研究.作者发现, 呋喃基稠合环丙烷与吡喃基稠合环丙烷都具有优良的反应活性, 能分别以中等至良好的产率和极好的非对映选择性获得一系列多取代双四氢呋喃或全氢呋喃并[2, 3-b]吡喃骨架(Eq. 25).该反应所具有的高官能团兼容性和操作简便性等优点, 为其它类似物的合成提供了一个新颖而有效的策略.之后, 在该实验的基础上, 邵华武课题组[78]以SnCl4为催化剂, 实现了类似的稠合环丙烷与酮类的[3+2]扩环反应, 并获得了一系列多取代双四氢呋喃和全氢呋喃并[2, 3-b]吡喃骨架(Eq. 26).该类反应同样具有良好的官能团兼容性和很高的非对映选择性, 是关于稠合环丙烷与酮类[3+2]扩环反应的又一成功实例.
|
|
(25) |
|
|
(26) |
由于酮羰基的反应活性与醛类化合物相比普遍较低, 人们对环丙烷与酮羰基发生[3+2]扩环反应的研究相对较少.为了解决酮羰基反应活性低的问题, 王忠文课题组设计了一系列底物, 让环丙烷以分子内环加成的方式与酮和醛发生[3+2]扩环反应.在该课题组所设计的环丙烷分子内[3+2]扩环策略中, 环丙烷与羰基化物的加成方式有两种:一种是分子内平行环加成(IMPC), 另一种是分子内交叉环加成(IMCC). 2012年, 王忠文等[24]曾基于对环丙烷的分子内交叉环化反应研究成果进行过综述.为避免重复, 下面将主要对该课题组在2012年之后的研究成果进行介绍.
在先前报道的环丙烷与羰基的分子内交叉环加成反应中, 苯乙醛由于在反应过程中不够稳定, 难以参与到[3+2]环化反应中合成相应的氧杂骨架[24]. 2014年, 王忠文等[79]开发了环丙烷1, 1-二酯与环氧化物的多米诺Pinacol重排/分子内[3+2]交叉环加成反应(Scheme 27).在该反应中, 通过环氧化物Pinacol重排产生的苯乙醛衍生物能与环丙烷发生[3+2]交叉环加成, 构建出桥联氧杂-[3.2.1]/-[4.2.1]骨架.除路易斯酸外, 布朗斯特酸TfOH也能有效促进该反应, 这对于开发D-A环丙烷与羰基环加成反应的新型催化体系具有重要意义.
同一年, 王忠文等[80]进一步开发了二烯基环丙烷- 1, 1-二酯与亲二烯体的分子内扩环反应(Scheme 28).该反应是一个Diels-Alder型[4+2]环加成和分子内[3+2]交叉扩环的串联过程, 可以同时得到[4+2]环加成产物环己烯基醛/酮衍生物和[3+2]扩环产物氧杂碳环, 且反应所得到的环己烯基醛/酮类可在苯酚或Sc(OTf)3催化下进一步生成氧杂碳环.该反应虽然产率偏低, 但具有不错的非对映选择性, 且能以“一锅法”构建得到6, 6-和6, 7-稠合碳环骨架, 在构筑稠环化合物方面具有重要的应用前景.
2015年, 王忠文等[81]通过在环丙烷上引入钴-炔部分, 进一步实现了钴-炔基环丙烷-1, 1-二酯与醛的分子内[3+2]交叉扩环反应(Scheme 29).该反应在Sc(OTf)3催化下、室温条件下就能顺利合成相应的桥联氧杂[n.2.1]碳环骨架, 其中钴配合物部分可进一步除去, 生成烯烃产物.该研究组还发现: α, β-不饱和酮/醛的顺式(Z-)异构体也能与环丙烷发生[3+2]扩环反应, 但立体选择性地合成(Z)-α, β-不饱和酮/醛还存在困难.为了克服这一难题, 王忠文等人[82]使用紫外可见光对α, β-不饱和酮/醛的反式(E-)异构体进行催化异构化, 使其异构化为E-和Z-异构体的混合物, 随后与环丙烷1, 1-二酯进行分子内[3+2]交叉环化(Eq. 27).整个反应在光和路易斯酸的协同催化下, 以中等至极好的产率制得了中型碳环骨架和基于碳环的桥联氧杂[n.2.1] (n=4, 5, 6)双环骨架, 这对于后续合成其它带有桥氧双环的天然产物骨架结构具有积极作用.
|
|
(27) |
除上述几类常见的不饱和化合物外, 环丙烷还能与含C=S键的不饱和化合物如异硫氰酸酯、硫代酮等发生[3+2]扩环反应.此类反应能够快速构建含S的五元杂环, 是环丙烷与不饱和化合物发生[3+2]扩环反应的重要类型之一.
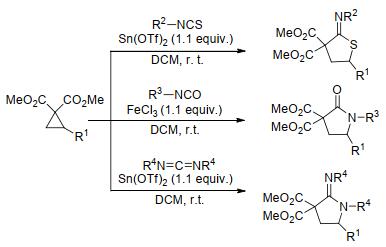
2012年, 王卫等[83]以FeCl3为催化剂, 通过芳基环丙烷-1, 1-二酯与芳基异硫氰酸酯的[3+2]扩环反应, 合成了硫代内酰胺(Eq. 28).该反应具有较宽的底物范围和高度区域选择性, 然而, 其产物很快被Stoltz课题组[84]校正为硫代亚胺. Stoltz及其同事在研究环丙烷- 1, 1-二酯与异硫氰酸酯的[3+2]扩环反应时发现, 在与王卫等[83]报道的相似的反应条件下, 反应的产物为硫代亚胺而不是硫代内酰胺(Scheme 30).他们通过一系列表征数据确证了自己和王卫课题组之前报道的实验结果, 并在这一发现的基础上, 以异氰酸酯和碳二亚胺为亲偶极体, 实现了路易斯酸催化的环丙烷-1, 1-二酯与异氰酸酯、碳二亚胺的[3+2]扩环反应.
|
|
(28) |
2013年, 杨高升等[85]发现顺式-2, 3-二取代环丙烷比反式异构体更具反应性, 之后他们以异硫氰酸酯作为新的亲偶极体, 实现了与顺式-2, 3-二取代环丙烷的[3+2]扩环反应(Eq. 29).该反应在AlCl3催化下, 以中等至优异的产率, 在极短时间内生成了单一的多取代2-亚氨基二氢噻吩非对映异构体.作者通过底物筛选发现:环丙烷上芳基取代基的电子性质对反应结果具有显著影响, 其中吸电子基取代的顺式环丙烷的反应性明显高于给电子基或中性基团取代的顺式环丙烷的反应性.作者还通过与反式环丙烷进行对比研究, 发现顺式-2, 3-二取代环丙烷的反应性比反式异构体的更好.
|
|
(29) |
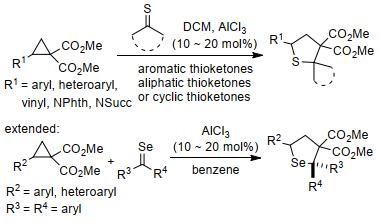
由于硫代酮的不稳定性, 过去几年中很少有团队报道环丙烷与其发生[3+2]扩环反应的实例. 2017年, Werz等[86]报道了AlCl3催化下2-取代环丙烷-1, 1-二酯与硫代酮的[3+2]扩环反应(Scheme 31).该反应可在温和条件下以立体特异性方式获得一系列多取代的四氢噻吩, 且具有较好的官能团耐受性:环状或非环状硫代酮都能获得较高产率的产物; 反应还能以分子内扩环的方式构建[n.2.1]双环骨架, 并扩展至环丙烷与硒酮的[3+2]扩环, 用以制备中等至良好产率的四氢硒吩衍生物.紧接着, 该课题组[87]将酮类底物扩展至3-硫代环丁酮, 并以优良的产率得到一系列四氢噻吩衍生物(Eq. 30).该反应同样具有底物范围宽泛、反应条件温和等特点.
|
|
(30) |
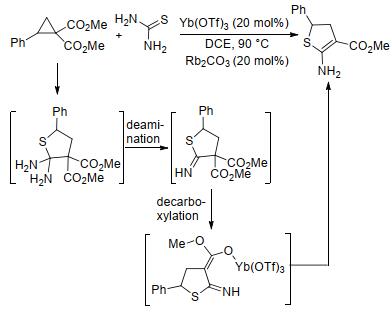
2019年, 郭海明课题组[88]报道了2-取代环丙烷-1, 1-二酯与硫脲的[3+2]扩环反应.该反应在Yb(OTf)3催化下, 以中等至良好的产率获得了2-氨基-4, 5-二氢噻吩(Scheme 32).基于实验结果, 研究人员推测该反应经历了先发生[3+2]扩环、随后发生脱氨基和脱羧的反应历程, 其中硫脲同时扮演了反应底物和脱羧试剂的双重角色.
除异硫氰酸酯、硫代酮之外, 还有一些含N、O等杂原子的不饱和化合物也能与路易斯酸活化的环丙烷发生[3+2]扩环反应.如2014年, Chakrabarty等[89]报道了环丙烷与亚硝基芳烃的[3+2]扩环反应(Eq. 31).该反应在二氯乙烷溶剂中在MgBr2催化下以优异至极好的产率制备得到了异噁唑烷.其中, 缺电子的亚硝基芳烃对该反应更有效, 而生成的异噁唑烷可通过后续的化学过程转化为各种氨基内酯. 2018年, 罗永春等[90]报道了Yb(OTf)3催化的环丙烷-1, 1-二酯与二噁唑类衍生物的[3+2]扩环反应, 以中等至良好的产率得到了一系列环状硝酮(Eq. 32), 为合成环状硝酮提供了一种新方法.
|
|
(31) |
|
|
(32) |
除路易斯酸外, 低价过渡金属如钯(Pd)、铱(Ir)、铑(Rh)、钌(Ru)、镍(Ni)等也极易活化环丙烷产生1, 3-偶极子.但与路易斯酸的活化位点不同, 过渡金属常与环丙烷上的给电子基团相互作用.其中, 乙烯基环丙烷是该类反应最常见的底物之一:过渡金属催化剂可与环丙烷上的乙烯基作用, 生成π-烯丙基阳离子, 从而使环丙烷的[3+2]扩环反应更加容易进行.
关于过渡金属催化的环丙烷与烯烃、炔烃的[3+2]扩环反应, 余志祥课题组曾对铑催化环丙烷的分子内扩环反应开展过系列研究. 2008年, 该课题组[91]报道了第一个铑催化反式-乙烯基环丙烷(trans-VCP)与烯烃的分子内[3+2]扩环反应(Scheme 33).该反应在[Rh(CO)2-Cl]2催化下, 以中等至良好的产率制备得到了5, 5-双环化合物, 且未被吸电子基团活化的乙烯基环丙烷也能顺利参与反应.不过, 该反应的底物仅限于反式-乙烯基环丙烷-烯烃(trans-VCP-enes), 若使用顺式-乙烯基环丙烷-烯烃(cis-VCP-enes), 则发生分子内[5+2]扩环反应.
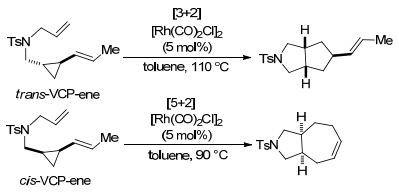
为了克服上述限制, 余志祥课题组[92]对该反应的底物和反应条件进行了优化, 并于2010年报道了乙烯基环丙烷与烯烃或炔烃的分子内新型[3+2]扩环反应(Eq. 33).该反应以Rh(I)/双膦配体L9为催化剂, 在80~110 ℃温度条件下生成了相应的桥碳带有立体中心的碳双环或杂双环骨架.反应的底物范围宽泛, 各种烷基取代的烯烃、炔烃和丙二烯都能很好地与乙烯基环丙烷发生[3+2]扩环反应, 系链上还允许存在O、N等杂原子, 为构建碳杂双环化合物提供了一种有效的途径.同年, 该课题组[93]还通过Rh(I)/双膦配体L10催化的α-乙烯基-环丙烷与烯烃的分子内[3+2]扩环反应, 合成了[4.3.0]壬烷骨架和[5.3.0]癸烷骨架(Eq. 34).该反应不受底物烯烃和乙烯基环丙烷单元之间的系链长短的影响, 即使系链上存在苯基和烷基取代基, 也能获得优异的非对映选择性.但它的缺点在于烯烃内部碳或乙烯基环丙烷单元的C1位置上带有取代基会导致反应性丧失, 且系链上只能兼容N杂原子.
|
|
(33) |
|
|
(34) |
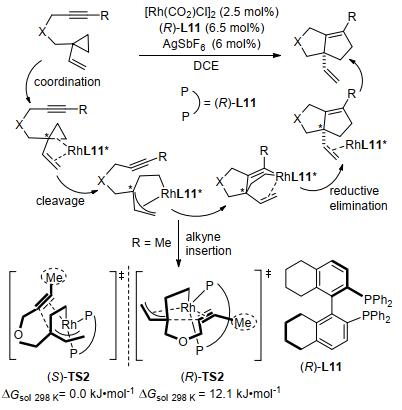
此后, 余志祥课题组[94]开发了铑催化的乙烯基环丙烷与炔烃的分子内不对称[3+2]扩环反应(Scheme 34).该反应以Rh(I)/手性双膦配体(R)-L11为催化剂, 在1, 2-二氯乙烷溶剂中以中等至优良的产率获得了含手性季碳立体构型中心的双环[3.3.0]化合物.该反应具有优异的ee值, 也是未活化的乙烯基环丙烷首次以三碳组分参与过渡金属催化的不对称扩环反应的实例.作者基于实验结果推测反应可能经历底物与催化剂络合、环丙烷裂解、炔烃插入和还原消除等四个过程, 并进一步应用密度泛函理论对反应进行了计算.研究结果表明, 第一个立体构型中心产生于Rh(I)与底物络合的步骤, 对应生成R和S两种构型的中间体, 而随后的炔烃插入过程则是整个反应的速率和立体化学决定步骤.在炔烃插入的过渡态中, (R)-TS2由于受底物炔基部分的R基团与H8-BINAP骨架之间的斥力影响, 导致其相对于(S)-TS2能量高出12.1 kJ•mol-1, 因此反应生成S构型的产物, 并且R基团越大, 产物的对映选择性越高.
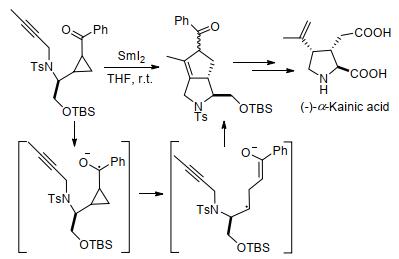
除余志祥课题组外, 其他课题组也报道了一些环丙烷与烯、炔发生分子内[3+2]扩环反应的实例.如2012年, 周兵和李援朝等[95]报道了第一个SmI2催化的酮基环丙烷与炔烃的分子内[3+2]扩环反应, 并通过该反应, 以较少的步骤对映选择性地合成了(-)-α-红藻氨酸(Scheme 35).作者推测反应遵循自由基历程, SmI2作为单电子还原剂, 与底物作用生成中间体, 然后环丙烷快速裂解, 经过环加成生成产物.
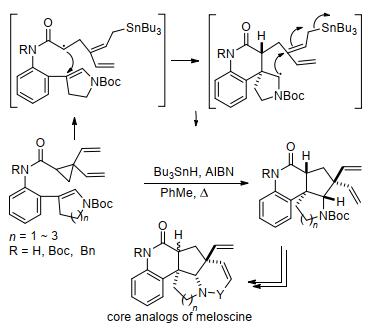
2014年, LaPorte等[96]报道了二乙烯基环丙烷与环烯烃的分子内[3+2]扩环反应(Scheme 36).他们认为该反应也遵循自由基机理, 即在三丁基氢化锡(Bu3SnH)和偶氮二丁腈(AIBN)存在下, 锡基团与底物中的一个乙烯基配位, 导致环丙烷断裂, 之后经过分子内[3+2]扩环反应得到相应的内酰胺产物.该反应的产物可以在几个步骤内转化为天然产物meloscine的核心骨架, 对于开发简洁的天然产物合成途径具有积极意义.之后, 王志刚等[97]以PPh3AuCl和AgSbF6为催化剂, 实现了环丙烷-1, 1-二酯与炔烃的分子内[3+2]扩环反应(Scheme 37).该反应可在温和的实验条件下, 经开环-环化-消除过程制备得到一系列高度官能化的亚烷基环戊烯.
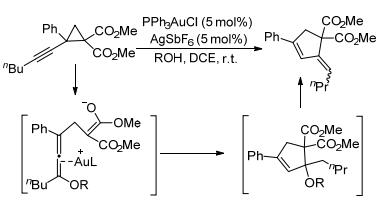
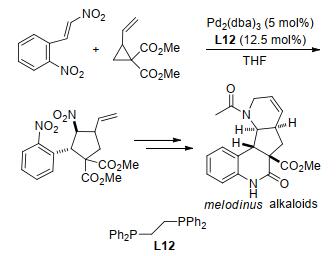
上述基本都是关于过渡金属催化环丙烷分子内[3+2]扩环反应的实例.与此同时, 人们也对过渡金属催化的环丙烷分子间[3+2]扩环反应开展了大量研究.其中, 金属钯(Pd)由于具有催化活性高、廉价易得、能与手性配体组合控制反应的立体选择性等特点而成为此类反应中最常用的过渡金属催化剂之一.如2011年, Stoltz及其同事[98]以钯和双膦配体L12为催化剂, 通过β, 2-二硝基苯乙烯和乙烯基环丙烷-1, 1-二酯的[3+2]扩环反应, 在四氢呋喃溶剂中合成了乙烯基环戊烷骨架, 并在后续的几个步骤中快速合成了Melodinus生物碱的四环骨架(Scheme 38).
2015年, 刘利等[99]以钯和手性双叔胺型配体L13为催化剂, 以1, 3-茚满二酮衍生的乙烯基环丙烷和硝基烯烃为底物, 通过分子间[3+2]扩环反应, 以高产率顺利地合成了带有2-螺环戊烷单元的1, 3-茚满二酮结构.该反应具有良好的非对映选择性和对映选择性, 且配体中氮原子上的取代基位阻对反应的对映选择性有显著影响(Eq. 35).刘全忠等[100]则以钯和手性双膦配体L14作催化剂, 实现了乙烯基环丙烷与芳基硝基烯烃的对映选择性[3+2]扩环反应(Eq. 36).该反应的产率和ee值可分别高达96%和92%, 且能形成具有三个连续手性立体构型中心的硝基环戊烷衍生物.作者经底物优化发现, 烯烃芳环上取代基的电子性质对反应的产率和对映选择性有极大影响, 其中具有强吸电子特性的基团会使底物失去反应性.
|
|
(35) |
|
|
(36) |
除上述硝基烯烃外, 带有其它吸电子基团的烯烃也能作为亲偶极体与环丙烷发生[3+2]扩环反应, 用以构建五元碳环骨架. 2011年, Trost等[101]将手性膦配体L15 与钯组成催化剂, 应用于外消旋乙烯基环丙烷-1, 1-二酯(rac-C13)与亚烷基氮杂内酯之间的[3+2]扩环反应中, 并成功制备出具有高对映选择性和非对映选择性的氨基酸衍生物(Eq. 37).该反应条件温和, 底物具有良好的取代基耐受性, 且能在单个合成步骤中形成具有三个立体异构中心的产物.这是第一个使用外消旋乙烯基环丙烷以不对称方式发生[3+2]扩环反应的实例.
|
|
(37) |
紧接着, 该课题组[102]用类似的催化体系, 成功实现了米氏酸取代的乙烯基环丙烷与米氏酸亚烷基之间的[3+2]扩环反应, 以极好的对映选择性制备得到了高度取代的环戊烷产物(Eq. 38).研究人员认为, 催化剂配体L16的构型对该反应的产物选择性起到了远程诱导作用, 使反应物通过π-σ-π互变减小了空间位阻, 从而生成具有高度立体选择性的产物.同年, 施敏等[103]以钯和手性咪唑啉膦配体L17为催化剂, 成功实现了乙烯基环丙烷与β, γ-不饱和α-酮基酯的不对称[3+2]扩环反应.该反应能兼容一系列富电子芳环取代的β, γ-不饱和α-酮基酯, 且能以很好的产率制备出含有多个立体中心、非对映选择性和对映选择性优异的官能化环戊烷(Eq. 39).
|
|
(38) |
|
|
(39) |
后来, 郭海明[104]和Ma等[105]也分别以钯和手性膦配体L15、L18为催化体系, 实现了乙烯基环丙烷与α-核碱基取代的丙烯酸酯(Eq. 40)和对苯醌甲基化物(Eq. 41)的[3+2]扩环反应.在乙烯基环丙烷与α-核碱基取代的丙烯酸酯的[3+2]扩环反应中, 各种嘌呤和嘧啶取代的丙烯酸酯均能充当合适的反应底物, 且生成的核苷类似物产率高, 具有良好的对映选择性.外消旋乙烯基环丙烷(rac-C14)与对苯醌甲基化物的[3+2]扩环反应同样具有反应底物宽泛、产率高、非对映选择性和对映选择性好等特点.
|
|
(40) |
|
|
(41) |
2017年, Hyland等[106]以Pd(0)/红菲啰啉配体L19为催化剂, 在四丁基碘化铵(Bu4NI)卤素添加剂存在条件下, 通过3-硝基-1-甲苯磺酰基吲哚与乙烯基环丙烷的去芳构化[3+2]扩环反应, 以高收率和良好的非对映选择性制备得到了反式环戊二烯并[b]吲哚啉(Eq. 42).研究发现, 卤素添加剂的存在对该反应的非对映选择性有巨大影响, 除去卤素添加剂甚至会使产物的非对映选择性发生逆转.作者推测卤素添加剂能够增加过渡态的π-σ-π转化速率, 从而促成构型转换.
|
|
(42) |
最近, 丁昌华等[107]以Pd(0)/手性双膦配体L20为催化剂, 实现了乙烯基环丙烷与炔烃的不对称[3+2]扩环反应, 以良好的产率和对映选择性合成了环戊烯P9, 同时得到少量四氢呋喃副产物P10 (Eq. 43).经研究发现, 温度对这两种产物的选择性形成有显著影响:高温有利于环戊烯产物的生成, 且该反应在一定条件下是可逆的, 四氢呋喃副产物能经逆[3+2]扩环过程进一步转化为环戊烯.该反应是第一个钯催化炔烃与乙烯基环丙烷的不对称[3+2]扩环反应, 同时也是过渡金属催化乙烯基环丙烷扩环反应中发生逆[3+2]扩环的反应实例之一.
|
|
(43) |
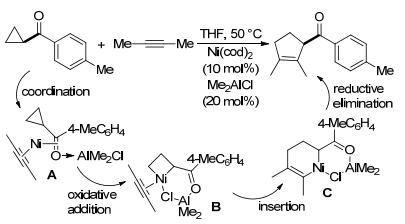
镍也是促进环丙烷发生[3+2]扩环反应的常用过渡金属催化剂之一. 2011年, Ogoshi等[108]在镍催化的酮基环丙烷与炔烃的[3+2]扩环反应中, 通过加入有机铝试剂Me2AlCl, 以中等至良好的产率顺利制得了环戊烯衍生物(Scheme 39), 甚至反应活性较低的单取代酮基环丙烷也能获得不错的收率.作者推测反应的可能历程为, 底物与催化剂形成络合物A, 随后Ni氧化加成到环丙烷的C—C键中形成中间体B, B经过炔烃插入形成中间体C, 最后C还原消除得到产物.其中有机铝试剂的关键作用在于增强了酮基环丙烷对镍的配位能力, 且通过Me、Cl基团与镍的螯合配位稳定反应中间体.
2014年, Tombe等[109]以镍作为催化剂, 实现了乙烯基环丙烷与丙二烯的分子间[3+2]扩环反应, 以中等至极好的产率、良好的区域和非对映选择性合成了多取代的环戊烷衍生物(Scheme 40).得益于催化剂的高活性, 反应中丙二烯可作为富电子的亲核烯烃与环丙烷进行环加成, 且带有羟基的丙二烯也能参与该反应.
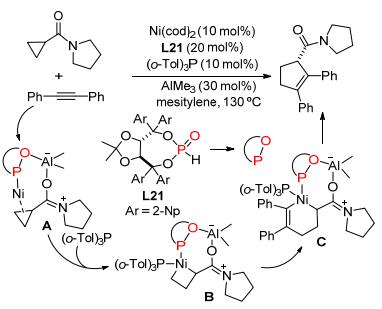
之后, 叶萌春等[110]使用镍-铝双金属催化剂, 实现了惰性的酰胺基环丙烷与炔烃的[3+2]扩环反应, 并通过额外加入膦配体L21改善了反应的对映选择性, 最终以中等至极好的产率和高达94%的ee值制得了环戊烯基甲酰胺(Scheme 41).作者推测该反应的机理与上一个反应类似, 其中双功能配体能够帮助镍-铝催化剂与底物配位, 而邻三甲苯基膦配体(o-Tol)3P可加速镍氧化加成到环丙烷的C—C键上并生成四元环中间体B.
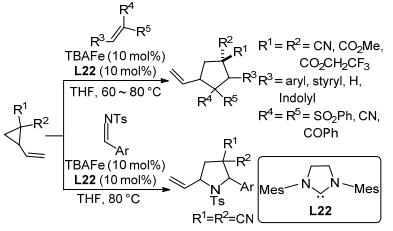
除铑、钯、镍以外, 其它的过渡金属如铁(Fe)、钴(Co)、钛(Ti)等催化的环丙烷[3+2]扩环反应也在持续研发中. 2012年, Plietker等[111]报道了在铁络合物Bu4N[Fe-(CO)3(NO)](TBAFe)催化下, 乙烯基环丙烷与吸电子烯烃在四氢呋喃溶剂中发生的[3+2]扩环反应(Scheme 42).该反应以良好到极好的产率和中等的区域选择性制得了多取代环戊烷.乙烯基环丙烷与N-甲苯磺酰基芳基亚胺在相同的条件下也能发生[3+2]扩环反应, 并以同样优良的产率生成吡咯烷.
2018年, Lin等[112]以钛配合物为催化剂实现了酮基环丙烷与烯烃的[3+2]扩环反应, 并以极好的产率、优异的非对映选择性和对映选择性制得了高度取代的环戊烷产物(Eq. 44).该反应的特别之处在于实现了自由基反应历程中的立体选择性.
|
|
(44) |
SMD
基于上述研究成果, Yoshikai等[114]继续用Co(II)/二膦配体L10作为催化剂, 实现了单取代环丙烷与丙二烯之间的非对映选择性[3+2]扩环反应(Eq. 45).该反应底物范围宽泛, 各种环状或链状的丙二烯均能参与反应, 而且以不错的产率制得一系列含有四取代碳中心的单环和稠合环戊醇衍生物.
|
|
(45) |
与C=C类不饱和化合物相比, 人们对过渡金属催化的环丙烷与C=O类不饱和化合物的[3+2]扩环反应研究相对较少. 2008年, Johnson等[115]开发了钯与红菲啰啉配体L19催化下乙烯基环丙烷与醛的[3+2]扩环反应, 以中等至优异的产率合成了2, 5-顺式二取代的四氢呋喃衍生物(Eq. 46).该反应对缺电子特性的醛类化合物最有效, 能在短时间内达到最大产率.
|
|
(46) |
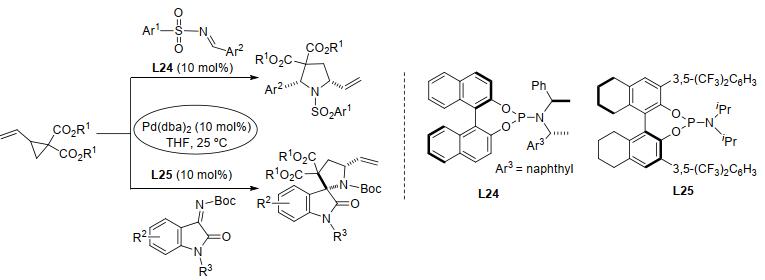
2013年, 施敏等[116]将新开发的手性咪唑啉膦配体(aS, R, R)-L23应用于钯催化的乙烯基环丙烷与靛红的对映选择性[3+2]扩环反应中, 以良好的产率、中等至优异的非对映选择性和对映选择性制得了螺二氢呋喃羟吲哚骨架(Eq. 47).该反应条件温和, 且具有良好的底物耐受性.不久前, 刘全忠课题组[117]也以钯/手性亚磷酰胺配体L24、L25为催化体系, 实现了乙烯基环丙烷与亚胺和酮亚胺的对映选择性[3+2]扩环反应, 以极好的产率和对映选择性制得了吡咯烷或螺[吡咯烷-3, 2'-羟吲哚]衍生物(Scheme 44).
|
|
(47) |
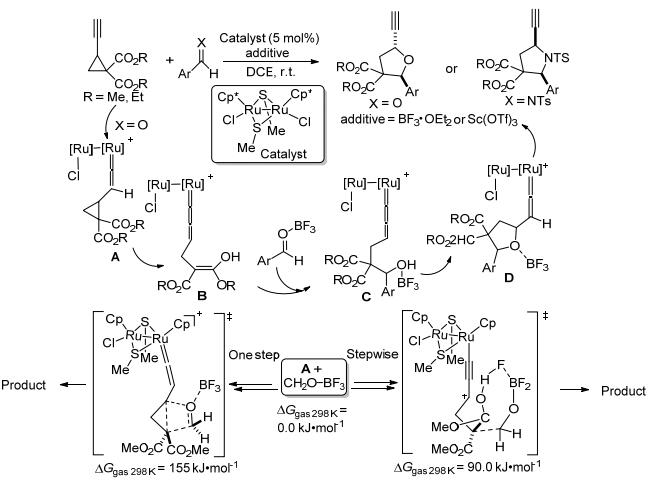
除钯外, 关于钌、镍等过渡金属催化的环丙烷与C=O类不饱和化合物的[3+2]扩环反应也有零星报道.如2013年, Nishibayashi等[118]报道了二钌配合物催化的乙炔基环丙烷与醛和亚胺的[3+2]扩环反应(Scheme 45).该反应对醛类和各种N-甲苯磺酰亚胺均有很好的兼容性, 能分别以中等至极好的产率得到相应的四氢呋喃衍生物和乙炔基吡咯烷.作者在实验基础上提出了一个经历亚烯基钌络合物的分步反应机理:二钌化合物首先与环丙烷的炔基配位, 形成钌亚乙烯基络合物A, 随后A开环异构化为丙二烯基络合物B, 同时B亲核进攻BF3活化的醛, 形成新的丙二烯基络合物C. C经过分子内环化得到亚乙烯基络合物D, 最后D与另一分子环丙烷底物之间发生配体交换得到相应的[3+2]扩环产物, 同时实现A的再生循环.为了验证上述反应机理, 作者运用密度泛函理论的B3LYP方法对乙炔基环丙烷与醛的[3+2]扩环反应进行了理论研究, 其计算结果表明:该反应有分步环加成和协同环加成两条反应路径, 其中分步环加成路径上的决速步能垒为90.0 kJ•mol-1, 这在室温下是完全可以实现的; 而协同环加成路径上的协同过渡态的能垒高达155 kJ•mol-1, 远高于分步环加成路径中过渡态的能垒, 说明该反应不太可能按照协同方式发生[3+2]扩环反应.该结果很好地确证了作者所提出的涉及亚烯基钌络合物的分步反应机理.
几乎与此同时, Matsubara等[119]也发现, 在Ni(0)/双膦配体L26催化下, 乙烯基环丙烷能对亚胺进行亲核进攻, 发生立体选择性环加成, 以良好的区域选择性和非对映选择性合成一系列吡咯烷(Eq. 48).
|
|
(48) |
从上述反应实例可以看出:一些过渡金属和手性配体的搭配使用, 已经能够很好地实现环丙烷[3+2]扩环反应的区域选择性和对映选择性, 但铂、钌、钛等金属通常价格昂贵且不易获得, 极大地限制了过渡金属催化剂在环丙烷[3+2]扩环反应中的广泛使用.因此, 寻找廉价高效的过渡金属催化剂, 并进一步降低催化剂负载量等仍然是该领域发展中亟待解决的一个难题.
随着对环丙烷[3+2]扩环反应研究的不断深入, 传统的催化剂越来越不能满足高效、简单易得等需求, 一些研究者开始尝试在传统催化体系中引入可见光催化、微波加热等条件来改善催化剂的活性, 并开发了一些新型的有机碱等催化剂.其中, 部分新型催化剂已经在促进环丙烷[3+2]扩环反应中表现出很好的催化活性.
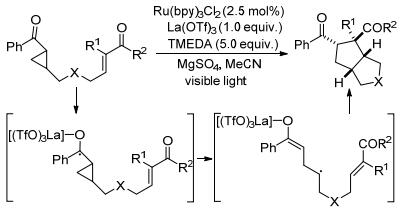
2011年, Yoon课题组[120]将路易斯酸与光催化剂组合应用于酮基环丙烷与烯烃的[3+2]扩环反应中, 成功合成了相应的多环化合物(Scheme 46).该反应的关键步骤是由催化剂引发酮基环丙烷的单电子还原过程, 并生成相应的自由基阴离子.同时, 使用MgSO4或其它干燥剂可提高该反应的产率和重现性.五年后, 该课题组[121]再次将这种双催化剂策略应用于酮基环丙烷与烯烃的[3+2]扩环反应中, 以不对称方式合成了环戊烷骨架(Eq. 49).该反应的优点在于实现了光催化环加成过程中的立体控制, 并能以高达99%的ee值得到相应的五元环产物.
|
|
(49) |
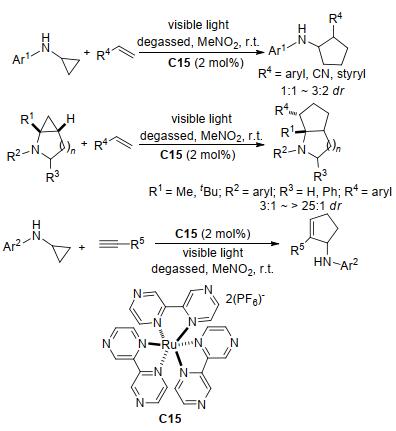
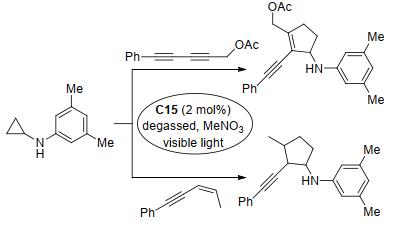
2012年, Maity等[122]也开发了一种由钌络合物C15与可见光组合的新型光催化体系, 并将其应用于单取代氨基环丙烷与烯烃的分子间[3+2]扩环反应中, 以中等至良好的产率得到了相应的环戊烷产物(Scheme 47).该反应能很好地兼容双环氨基环丙烷底物生成稠合双杂环骨架, 且得益于双环底物中较高的环应变力, 其非对映选择性明显优于环戊烷产物的非对映选择性.两年后, Maity研究组[123]将反应的底物范围扩大至炔烃, 并在相同的催化条件下得到了环戊烯衍生物.该反应的底物耐受性与之前的反应相比进一步增强, 但产率并没有明显提高(Scheme 47).稍后, 该研究组[124]将底物范围进一步扩大至1, 3-共轭二炔和1, 3-共轭烯炔, 这两种底物均能顺利发生[3+2]扩环反应生成相应的产物, 部分产物甚至具有完全的区域选择性(Scheme 48).研究表明, 该反应对带有取代基的环丙烷也能很好兼容.
除上述光催化的环丙烷[3+2]扩环反应外, Sasaki等[125]开发了一种双三甲基二硅基氨基锂(LiHMDS)介导的环丙烷[3+2]扩环反应(Scheme 49).与其它反应不同的是, 该反应主要通过活化环丙烷的供体基团来促进反应:首先, 底物在碱基的诱导下产生α-对甲苯磺酰碳负离子, 该碳负离子进一步诱导环丙烷开环并与烯烃发生[3+2]扩环反应.其中碳负离子可通过后续的四元环中间体去除, 最终生成环戊烯产物.该反应是碳负离子诱导环丙烷发生[3+2]扩环反应的第一个实例.
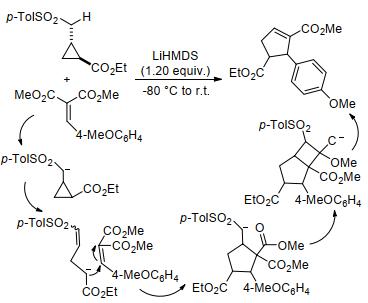
2017年, Blom等[126]使用具有光学活性的布朗斯特碱, 实现了外消旋二氰基酮基环丙烷(rac-C16)与硝基烯烃的[3+2]扩环反应, 制备得到带有三个连续立体中心的多取代环戊烷(Eq. 50).该反应具有优异的官能团耐受性, 反应产率高, 且产物具有优异的对映选择性和非对映选择性.这是第一个有机催化D-A型环丙烷发生立体选择性[3+2]扩环反应的实例, 为有机催化活化环丙烷开辟了新的途径.
|
|
(50) |
Kamlar等[127]开发了钯和手性仲胺(S-TMS-DPP)协同催化的乙烯基环丙烷二氢唑酮与烯醛的[3+2]扩环反应(Eq. 51).该反应能以良好至极好的非对映选选择性和对映选择性合成螺环二氢唑酮骨架, 为构建含季碳中心的手性环戊烷骨架提供了一条简便途径.
|
|
(51) |
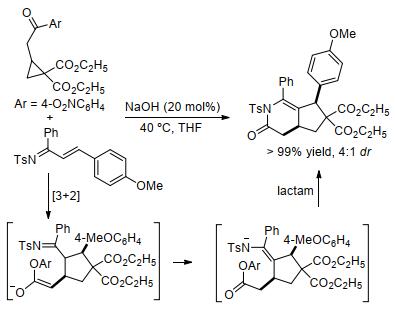
最近, 金智超等[128]通过NaOH催化的酯基环丙烷与α, β-不饱和亚胺的串联环化反应, 以极好的产率制得了环戊二烯并[c]吡啶衍生物(Scheme 50).基于此前Blom等[126]观察到的胺催化环丙烷与烯烃发生[3+2]扩环的实验事实, 作者推测该反应先后经历了[3+2]扩环和分子内酰胺化过程并最终生成环戊二烯并[c]吡啶衍生物.
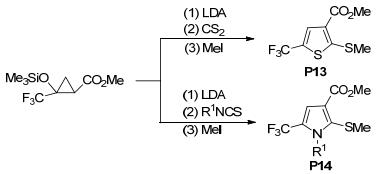
2013年, 王忠文等[129]报道了三氟甲磺酸(TfOH)催化环丙烷-1, 1二酯与腈类的[3+2]扩环反应(Eq. 52), 并以中等至极好的产率制得了1-吡咯啉衍生物.与常规的路易斯酸作催化剂的反应相比, 该反应的速率更快(仅需5 min), 底物范围更宽泛, 而且反应不需要溶剂.稍后, Reissig等[130]以二异丙基氨基锂(LDA)为催化剂, 实现了2-三氟甲基-2-甲硅烷氧基环丙烷与二硫化碳或异硫氰酸的[3+2]扩环反应, 以“一锅法”合成了二氢噻吩P13或吡咯衍生物P14 (Scheme 51).
|
|
(52) |
2018年, Budynina等[131]则以氰酸根离子作为含氮亲核试剂, 与羟吲哚取代的环丙烷发生[3+2]扩环反应, 顺利制得了一系列螺[吡咯烷酮-3, 3'-羟吲哚]产物(Eq. 53).但该反应条件较为苛刻:除需要Et3N•HCl催化和微波加热外, 还需要加入两倍当量的KNCO.
|
|
(53) |
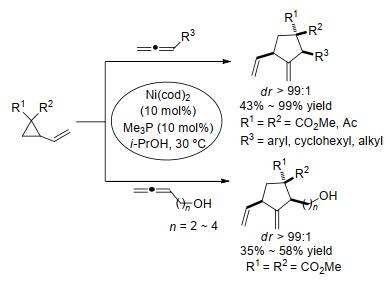
最近十年, 人们在通过环丙烷与不饱和化合物的[3+2]扩环反应构建五元碳环和五元杂环骨架方面取得了长足进步, 尤其是在环丙烷与烯烃、炔烃的分子间/分子内[3+2]扩环反应方面呈现出了一片繁荣景象.就反应底物而言, 环丙烷已从最初的D-A型环丙烷发展到多取代环丙烷, 从活化环丙烷发展到未活化或低活性环丙烷; 不饱和底物的类型则从最初的活化烯烃、炔烃发展到非活化烯烃、炔烃, 并进一步扩展到醛、酮、硫代酮、腈等.就催化剂而言, 除经典的路易斯酸催化和过渡金属催化剂外, 一些新兴的催化手段如光催化、微波催化等也已经被成功应用于部分环丙烷的[3+2]扩环反应中; 与此同时, 一些廉价高效的金属催化剂也不断被开发利用.就反应选择性而言, 随着各种新型底物和催化体系的开发和应用, 很多反应的产率、反应选择性尤其是立体选择性和非对映选择性得到了极大的改善.未来一段时间内, 继续优化实验条件、拓宽反应底物范围、提高反应效率和反应选择性、开发新型催化体系、降低催化剂负载量等依然是该领域发展的重中之重.同时, 有效发挥此类反应在合成天然产物领域的应用, 以及合理发挥理论研究对于解决上述问题的启迪和辅助作用也将是重要的努力方向.
Wong, H. N. C.; Hon, M.-Y.; Tse, C.-W.; Yip, Y.-C.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165. doi: 10.1021/cr00091a005
Khoury, P. R.; Goddard, J. D.; Tam, W. Tetrahedron 2004, 60, 8103. doi: 10.1016/j.tet.2004.06.100
Souillart, L.; Cramer, N. Chem. Rev. 2015, 115, 9410. doi: 10.1021/acs.chemrev.5b00138
Bach, R. D.; Dmitrenko, O. J. Org. Chem. 2002, 67, 2588. doi: 10.1021/jo016241m
王磊, 徐建华, 有机化学, 2003, 23, 750. http://sioc-journal.cn/Jwk_yjhx/CN/Y2003/V23/I8/750Wang, L.; Xu, J.-H. Chin. J. Org. Chem. 2003, 23, 750(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2003/V23/I8/750
Yu, M.; Pagenkopf, B. L. Tetrahedron 2005, 61, 321. doi: 10.1016/j.tet.2004.10.077
Rubin, M.; Rubina, M.; Gevorgyan, V. Chem. Rev. 2007, 107, 3117. doi: 10.1021/cr050988l
袁美斐, 陈雅丽, 丁维钰, 曹卫国, 有机化学, 2003, 23, 901. http://www.cnki.com.cn/Article/CJFDTOTAL-YJHU200309000.htmYuan, M.-F.; Chen, Y.-L.; Ding, W.-Y.; Cao, W.-G. Chin. J. Org. Chem. 2003, 23, 901(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-YJHU200309000.htm
De Simone, F.; Waser, J. Synthesis 2009, 3353. doi: 10.1055/s-0029-1216998
Lebold, T. P.; Kerr, M. A. Pure Appl. Chem. 2010, 82, 1797. doi: 10.1351/PAC-CON-09-09-28
Mel'nikov, M. Y.; Budynina, E. M.; Ivanova, O. A.; Trushkov, I. V. Mendeleev Commun. 2011, 21, 293. doi: 10.1016/j.mencom.2011.11.001
Cavitt, M. A.; Phun, L. H.; France, S. Chem. Soc. Rev. 2014, 43, 804. doi: 10.1039/C3CS60238A
De Nanteuil, F.; de Simone, F.; Frei, R.; Benfatti, F.; Serrano, E.; Waser, J. Chem. Commun. 2014, 50, 10912. doi: 10.1039/C4CC03194F
Pandey, A. K.; Ghosh, A.; Banerjee, P. Isr. J. Chem. 2016, 56, 512. doi: 10.1002/ijch.201500100
Pagenkopf, B. L.; Vemula, N. Eur. J. Org. Chem. 2017, 2017, 2561. doi: 10.1002/ejoc.201700201
Grover, H. K.; Emmett, M. R.; Kerr, M. A. Org. Biomol. Chem. 2015, 13, 655. doi: 10.1039/C4OB02117G
Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504. doi: 10.1002/anie.201309886
Rassadin, V. A.; Six, Y. Tetrahedron 2016, 72, 4701. doi: 10.1016/j.tet.2016.06.014
Brackmann, F.; de Meijere, A. Chem. Rev. 2007, 107, 4493. doi: 10.1021/cr078376j
Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151. doi: 10.1021/cr010016n
Carson, C. A.; Kerr, M. A. Chem. Soc. Rev. 2009, 38, 3051. doi: 10.1039/b901245c
De Simone, F.; Waser, J. Synlett 2011, 589. doi: 10.1055/s-0030-1259553
Tang, P.; Qin, Y. Synthesis 2012, 44, 2969. doi: 10.1055/s-0032-1317011
Wang, Z. Synlett 2012, 23, 2311. doi: 10.1055/s-0032-1317082
Liao, S.; Sun, X.-L.; Tang, Y. Acc. Chem. Res. 2014, 47, 2260. doi: 10.1021/ar800104y
Wang, L.; Zhou, J.; Tang, Y. Chin. J. Chem. 2018, 36, 1123. doi: 10.1002/cjoc.201800373
Fang, J.; Ren, J.; Wang, Z. Tetrahedron Lett. 2008, 49, 6659. doi: 10.1016/j.tetlet.2008.09.028
De Nanteuil, F.; Waser, J. Angew. Chem., Int. Ed. 2011, 50, 12075. doi: 10.1002/anie.201106255
De Nanteuil, F.; Serrano, E.; Perrotta, D.; Waser, J. J. Am. Chem. Soc. 2014, 136, 6239. doi: 10.1021/ja5024578
Waser, J.; Serrano, E.; de Nanteuil, F. Synlett 2014, 25, 2285. doi: 10.1055/s-0034-1378512
Racine, S.; de Nanteuil, F.; Serrano, E.; Waser, J. Angew. Chem., Int. Ed. 2014, 53, 8484. doi: 10.1002/anie.201404832
Mackay, W. D.; Fistikci, M.; Carris, R. M.; Johnson, J. S. Org. Lett. 2014, 16, 1626. doi: 10.1021/ol500256n
Verma, K.; Banerjee, P. Adv. Synth. Catal. 2016, 358, 2053. doi: 10.1002/adsc.201600221
Dey, R.; Banerjee, P. Org. Lett. 2017, 19, 304. doi: 10.1021/acs.orglett.6b03276
Verma, K.; Banerjee, P. Adv. Synth. Catal. 2017, 359, 3848. doi: 10.1002/adsc.201700744
Volkova, Y. A.; Budynina, E. M.; Kaplun, A. E.; Ivanova, O. A.; Chagarovskiy, A. O.; Skvortsov, D. A.; Rybakov, V. B.; Trushkov, I. V.; Melnikov, M. Y. Chem.-Eur. J. 2013, 19, 6586. doi: 10.1002/chem.201300731
Chagarovskiy, A. O.; Budynina, E. M.; Ivanova, O. A.; Grishin, Y. K.; Trushkov, I. V.; Verteletskii, P. V. Tetrahedron 2009, 65, 5385. doi: 10.1016/j.tet.2009.04.061
Budynina, E. M.; Ivanova, O. A.; Chagarovskiy, A. O.; Grishin, Y. K.; Trushkov, I. V.; Melnikov, M. Y. J. Org. Chem. 2015, 80, 12212. doi: 10.1021/acs.joc.5b02146
Xiong, H.; Xu, H.; Liao, S.; Xie, Z.; Tang, Y. J. Am. Chem. Soc. 2013, 135, 7851. doi: 10.1021/ja4042127
严文广, 王盼, 王丽佳, 孙秀丽, 唐勇, 化学学报, 2017, 75, 783.Yan, W.; Wang, P.; Wang, L.; Sun, X.; Tang, Y. Acta Chim. Sinica 2017, 75, 783(in Chinese).
Kaicharla, T.; Roy, T.; Thangaraj, M.; Gonnade, R. G.; Biju, A. T. Angew. Chem., Int. Ed. 2016, 55, 10061. doi: 10.1002/anie.201604373
Pandey, A. K.; Varshnaya, R. K.; Banerjee, P. Eur. J. Org. Chem. 2017, 2017, 1647. doi: 10.1002/ejoc.201601549
Xia, X.-F.; Song, X.-R.; Liu, X.-Y.; Liang, Y.-M. Chem. Asian J. 2012, 7, 1538. doi: 10.1002/asia.201200104
Zhu, W.; Fang, J.; Liu, Y.; Ren, J.; Wang, Z. Angew. Chem., Int. Ed. 2013, 52, 2032. doi: 10.1002/anie.201206484
Wang, Z.; Ren, J.; Wang, Z. Org. Lett. 2013, 15, 5682. doi: 10.1021/ol402662j
Thangamani, M.; Srinivasan, K. J. Org. Chem. 2018, 83, 571. doi: 10.1021/acs.joc.7b02335
Zhu, J.; Liang, Y.; Wang, L.; Zheng, Z.-B.; Houk, K. N.; Tang, Y. J. Am. Chem. Soc. 2014, 136, 6900. doi: 10.1021/ja503117q
Mukherjee, P.; Das, A. R. J. Org. Chem. 2017, 82, 2794. doi: 10.1021/acs.joc.7b00089
Jackson, S. T.; Karadeolian, A.; Driega, A. B.; Kerr, M. A. J. Am. Chem. Soc. 2008, 130, 4196.
Curiel Tejeda, J. E.; Irwin, L. C.; Kerr, M. A. Org. Lett. 2016, 18, 4738. doi: 10.1021/acs.orglett.6b02409
Parsons, A. T.; Smith, A. G.; Neel, A. J.; Johnson, J. S. J. Am. Chem. Soc. 2010, 132, 9688. doi: 10.1021/ja1032277
Alajarin, M.; Egea, A.; Orenes, R.-A.; Vidal, A. Org. Biomol. Chem. 2016, 14, 10275. doi: 10.1039/C6OB02005D
Feng, M.; Yang, P.; Yang, G.; Chen, W.; Chai, Z. J. Org. Chem. 2017, 83, 174.
Tsunoi, S.; Maruoka, Y.; Suzuki, I.; Shibata, I. Org. Lett. 2016, 47, 4010. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM26247349
Preindl, J.; Chakrabarty, S.; Waser, J. Chem. Sci. 2017, 8, 7112. doi: 10.1039/C7SC03197A
Zhang, M.-C.; Wang, D.-C.; Xie, M.-S.; Qu, H.-M.; Guo, H.-M.; You, S.-L. Chem. 2019, 5, 156. doi: 10.1016/j.chempr.2018.10.003
Hao, E.-J.; Fu, D.-D.; Wang, D.-C, Zhang, T.; Qu, G.-R.; Li, G.-X.; Lan, Y.; Guo, H.-M. Org. Chem. Front. 2019, 6, 863. doi: 10.1039/C9QO00039A
Akaev, A. A.; Bezzubov, S. I.; Desyatkin, V. G.; Vorobyeva, N. S.; Majouga, A. G.; Melnikov, M. Y.; Budynina, E. M. J. Org. Chem. 2019, 84, 3340. doi: 10.1021/acs.joc.8b03208
Pohlhaus, P. D.; Sanders, S. D.; Parsons, A. T.; Li, W.; Johnson, J. S. J. Am. Chem. Soc. 2008, 130, 8642. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM18543924
Kreft, A.; Jones, P. G.; Werz, D. B. Org. Lett. 2018, 20, 2059. doi: 10.1021/acs.orglett.8b00603
Parsons, A. T.; Johnson, J. S. J. Am. Chem. Soc. 2009, 131, 3122. doi: 10.1021/ja809873u
Smith, A. G.; Slade, M. C.; Johnson, J. S. Org. Lett. 2011, 13, 1996. doi: 10.1021/ol200395e
Christie, S. D. R.; Cummins, J.; Elsegood, M. R. J.; Dawson, G. Synlett 2009, 257. doi: 10.1002/chin.200922103
Benfatti, F.; de Nanteuil, F.; Waser, J. Org. Lett. 2012, 14, 386. doi: 10.1021/ol203144v
Benfatti, F.; de Nanteuil, F.; Waser, J. Chem.-Eur. J. 2012, 18, 4844. doi: 10.1002/chem.201103971
Haubenreisser, S.; Hensenne, P.; Schröder, S.; Niggemann. M. Org. Lett. 2013, 15, 2262. doi: 10.1021/ol400809n
Rivero, A. R.; Fernandez, I.; Arellano, C. R. D.; Sierra, M. A. J. Org. Chem. 2014, 80, 1207.
Sabbatani, J.; Maulide, N. Angew. Chem., Int. Ed. 2016, 55, 6780. doi: 10.1002/anie.201601340
Yang, G.; Shen, Y.; Li, K.; Sun, Y.; Hua, Y. J. Org. Chem. 2010, 76, 229.
Yang, G.; Sun, Y.; Shen, Y.; Chai, Z.; Zhou, S.; Chu, J.; Chai, J. J. Org. Chem. 2013, 78, 5393.
Yang, G.; Wang, T.; Chai, J.; Chai, Z. Eur. J. Org. Chem. 2015, 1040. https://www.ingentaconnect.com/content/bpl/chem/2015/00000021/00000047/art00007
Sanders, S. D.; Ruiz-Olalla, A.; Johnson, J. S. Chem. Commun. 2009, 34, 5135. https://www.ingentaconnect.com/content/rsoc/13597345/2009/00002009/00000034/art00022
Shen, Y.; Chai, J.; Yang, G.; Chen, W.; Chai, Z. J. Org. Chem. 2018, 83, 12549. doi: 10.1021/acs.joc.8b01798
Xu, X.; Lu, H.; Ruppel, J. V.; Cui, X.; Lopea de Mesa, S.; Wojtas, L.; Zhang, X. J. Am. Chem. Soc. 2011, 133, 15292. doi: 10.1021/ja2062506
Yang, P.; Shen Y.; Feng, M.; Yang, G.; Chai, Z. Eur. J. Org. Chem. 2018, 4103. https://pubmed.ncbi.nlm.nih.gov/29992702/
Shen, Y.; Yang, P.-F.; Yang, G.; Chen, W.-L.; Chai, Z. Org. Biomol. Chem. 2018, 16, 2688. doi: 10.1039/C8OB00455B
Ma, X.; Tang, Q.; Ke, J.; Yang, X.; Zhang, J.; Shao, H. Org. Lett. 2013, 15, 5170. doi: 10.1021/ol402192f
Ma, X.; Zhang, J.; Tang, Q.; Ke, J.; Zou, W.; Shao, H. Chem. Commun. 2014, 50, 3505. doi: 10.1039/C3CC48963A
Zhu, W.; Ren, J.; Wang, Z. Eur. J. Org. Chem. 2014, 2014, 3561. doi: 10.1002/ejoc.201402160
Ren, J.; B, J.; Ma, W.; Wang, Z. Synlett 2014, 25, 2260. doi: 10.1055/s-0034-1378897
Zhang, J.; Xing, S.; Ren, J.; Jiang, S.; Wang, Z. Org. Lett. 2014, 17, 218.
Wang, Z.; Chen, S.; Ren, J.; Wang, Z. Org. Lett. 2015, 17, 4184. doi: 10.1021/acs.orglett.5b01928
Wang, H.; Yang, W.; Liu, H.; Wang, W.; Li, H. Org. Biomol. Chem. 2012, 10, 5032. doi: 10.1039/c2ob25682g
Goldberg, A. F. G.; O'Connor, N. R.; Craig II, R. A.; Stoltz, B. M. Org. Lett. 2012, 14, 5314. doi: 10.1021/ol302494n
Sun, Y.; Yang, G.; Chai, Z.; Mu, X.; Chai, J. Org. Biomol. Chem. 2013, 11, 7859. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM24129618
Augustin, A. U.; Sensse, M.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2017, 56, 14293. https://www.ingentaconnect.com/content/bpl/anie/2017/00000056/00000045/art00077
Augustin, A. U.; Busse, M.; Jones, P. G.; Werz, D. B. Org. Lett. 2018, 20, 820. doi: 10.1021/acs.orglett.7b03961
Xie, M.-S.; Zhao, G.-F.; Qin, T.; Suo, Y.-B.; Qu, G.-R.; Guo, H.-M. Chem. Commun. 2019, 55, 1580. doi: 10.1039/C8CC09595G
Chakrabarty, S.; Chatterjee, I.; Wibbeling, B.; Daniliuc, C. G.; Studer, A. Angew. Chem., Int. Ed. 2014, 53, 5964. doi: 10.1002/anie.201400885
Wang, Z.; Zhang, H.; Xu, P; Luo, Y. Chem. Commun. 2018, 54, 10128. doi: 10.1039/C8CC04656E
Jiao, L.; Ye, S.; Yu, Z.-X. J. Am. Chem. Soc. 2008, 130, 7178. doi: 10.1021/ja8008715
Jiao, L.; Lin, M.; Yu, Z.-X. Chem. Commun. 2010, 46, 1059. doi: 10.1039/B922417C
Li, Q.; Jiang G.; Jiao, L.; Yu, Z.-X. Org. Lett. 2010, 12, 1332. doi: 10.1021/ol100237h
Lin, M.; Kang, G.-Y.; Guo, Y.-A.; Yu, Z.-X. J. Am. Chem. Soc. 2012, 134, 398. doi: 10.1021/ja2082119
Luo, Z.; Zhou, B.; Li, Y. Org. Lett. 2012, 14, 2540. doi: 10.1021/ol3008414
Zhang, H.; Jeon, K. O.; Hay, E.; Geib, S.; Curran, D.; LaPorte, M. Org. Lett. 2014, 16, 94. doi: 10.1021/ol403078e
Chen, H.; Zhang, J.; Wang, D. Z. Org. Lett. 2015, 17, 2098. doi: 10.1021/acs.orglett.5b00671
Goldberg, A. F. G.; Stoltz, B. M. Org. Lett. 2011, 13, 4474. doi: 10.1021/ol2017615
Wei, F.; Ren, C.-L.; Wang, D.; Liu, L. Chem.-Eur. J. 2015, 21, 2335. doi: 10.1002/chem.201405407
Li, W.-K.; Liu, Z.-S.; He, L.; Kang, T.-R.; Liu, Q.-Z. Asian. J. Org. Chem. 2015, 4, 28. doi: 10.1002/ajoc.201402219
Trost, B. M.; Morris, P. J. Angew. Chem., Int. Ed. 2011, 50, 6167. doi: 10.1002/anie.201101684
Trost, B. M.; Morris, P. J.; Sprague, S. J. J. Am. Chem. Soc. 2012, 134, 17823. doi: 10.1021/ja309003x
Mei, L.-Y.; Wei, Y.; Xu, Q.; Shi, M. Organometallics 2012, 31, 7591. doi: 10.1021/om300896z
Xie, M.-S.; Wang, Y.; Li, J.-P.; Du, C.; Zhang, Y.-Y.; Hao, E.-J.; Zhang, M.-Z.; Qu, G.-R.; Guo, H.-M. Chem. Commun. 2015, 51, 12451. doi: 10.1039/C5CC04832J
Ma, C.; Huang, Y.; Zhao, Y. ACS Catal. 2016, 6, 6408. doi: 10.1021/acscatal.6b01845
Gee, Y. S.; Rivinoja, D. J.; Wales, S. M.; Gardiner, M. G.; Ryan, J. H.; Hyland, C. J. T. J. Org. Chem. 2017, 82, 13517. doi: 10.1021/acs.joc.7b02624
Ding, W.-P.; Zhang, G.-P.; Jiang, Y.-J.; Du, J.; Liu, X.-Y.; Chen, D.; Ding, C.-H.; Deng, Q.-H.; Hou, X.-L. Org. Lett. 2019, 21, 6805. doi: 10.1021/acs.orglett.9b02431
Tamaki, T.; Ohashi, M.; Ogoshi, S. Angew. Chem., Int. Ed. 2011, 50, 12067. doi: 10.1002/anie.201106174
Tombe, R.; Iwamoto, T.; Kurahashi, T.; Matsubara, S. Synlett 2014, 25, 2281. doi: 10.1055/s-0034-1378371
Liu, Q.-S.; Wang, D.; Yang, Z.; Luan, Y.; Yang, J.; Li, J.; Pu, Y.; Ye, M. J. Am. Chem. Soc. 2017, 139, 18150. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM21722266
Dieskau, A. P.; Holzwarth, M. S.; Plietker, B. J. Am. Chem. Soc. 2012, 134, 5048. doi: 10.1021/ja300294a
Hao, W.; Harenberg, J. H.; Wu, X.; MacMillan, S. N.; Lin, S. J. Am. Chem. Soc. 2018, 140, 3514. doi: 10.1021/jacs.7b13710
Yang, J.; Shen, Y.; Lim, Y. J.; Yoshikai, N. Chem. Sci. 2018, 9, 6928. doi: 10.1039/C8SC02074D
Yang, J.; Sun, Q.; Yoshikai, N. ACS Catal. 2019, 9, 1973. doi: 10.1021/acscatal.8b05114
Parsons, A. T.; Campbell, M. J.; Johnson, J. S. Org. Lett. 2008, 10, 2541. doi: 10.1021/ol800819h
Mei, L.-Y.; Wei, Y.; Xu, Q.; Shi, M. Organometallics 2013, 32, 3544. doi: 10.1021/om400473p
Huang, X.-B.; Li, X.-J.; Li, T.-T.; Chen, B.; Chu, W.-D.; He, L.; Liu, Q.-Z. Org. Lett. 2019, 21, 1713. doi: 10.1021/acs.orglett.9b00274
Miyake, Y.; Endo, S.; Moriyama, T.; Sakata, K.; Nishibayashi, Y. Angew. Chem., Int. Ed. 2013, 52, 1758. doi: 10.1002/anie.201207801
Tombe, R.; Kurahashi, T.; Matsubara, S. Org. Lett. 2013, 15, 1791. doi: 10.1021/ol4005068
Lu, Z.; Shen, M.; Yoon, T. P. J. Am. Chem. Soc. 2011, 133, 1162. doi: 10.1021/ja107849y
Amador, A. G.; Sherbrook, E. M.; Yoon, T. P. J. Am. Chem. Soc. 2016, 138, 4722. doi: 10.1021/jacs.6b01728
Maity, S.; Zhu, M.; Shinabery, R. S.; Zheng, N. Angew. Chem., Int. Ed. 2012, 51, 222. doi: 10.1002/anie.201106162
Nguyen, T. H.; Maity, S.; Zheng, N. Beilstein J. Org. Chem. 2014, 10, 975. doi: 10.3762/bjoc.10.96
Nguyen, T. H.; Morris, S. A.; Zheng, N. Adv. Synth. Catal. 2014, 356, 2831. doi: 10.1002/adsc.201400742
Sasaki, M.; Kondo, Y.; Nishio, T.; Takeda, K. Org. Lett. 2016, 18, 3858. doi: 10.1021/acs.orglett.6b01865
Blom, J.; Vidal-Albalat, A.; Jørgensen, J.; Barløse, C. L.; Jessen, K. S.; Iversen, M. V.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2017, 56, 11831. doi: 10.1002/anie.201706150
Kamlar, M.; Franc, M.; Císařová, I.; Gyepes, R.; Veselý, J. Chem. Commun. 2019, 55, 3829. doi: 10.1039/C8CC06500D
Pan, D.; Mou, C.; Zan, N.; Lv, Y.; Song, B.-A.; Chi, Y. R.; Jin, Z. Org. Lett. 2019, 21, 6624. doi: 10.1021/acs.orglett.9b02088
Cui, B.; Ren, J.; Wang, Z. J. Org. Chem. 2014, 79, 790. doi: 10.1021/jo402383a
Gladow, D.; Reissig, H.-U. J. Org. Chem. 2014, 79, 4492. doi: 10.1021/jo500534t
Zaytsev, S. V.; Ivanov, K. L.; Skvortsov, D. A.; Bezzubov, S. I.; Melnikov, M. Y.; Budynina, E. M. J. Org. Chem. 2018, 83, 8695. doi: 10.1021/acs.joc.8b00922

图式 1 芳基环丙烷与烯醇甲硅烷基醚的[3+2]扩环反应
Scheme 1 [3+2] ring-expansion reaction of aryl cyclopropanes with enol silyl ethers

图式 2 氨基环丙烷与烯醇醚类和醛类的不对称[3+2]扩环反应
Scheme 2 Asymmetric [3+2] ring-expansion reaction of aminocyclopropanes with enol ethers and aldehydes

图式 3 氨基环丙烷的合成及其与烯醇甲硅烷基醚类的[3+2]扩环反应
Scheme 3 Synthesis of aminocyclopropanes and their application in [3+2] ring-expansion reaction with enol silyl ethers

图式 4 核碱基环丙烷的合成及其与烯醇醚类、醛类和酮类的[3+2]扩环反应
Scheme 4 Synthesis of nucleobase cyclopropanes and their application in [3+2] ring-expansion reaction with enol ethers, aldehydes and ketones

图式 5 芳基环丙烷与乙烯基叠氮化物的[3+2]扩环反应及其产物的后续转化
Scheme 5 [3+2] Ring-expansion reaction of aryl cyclopropanes with vinyl azides and chemical conversion of its products

图式 6 芳基环丙烷与氮杂二烯的[3+2]扩环反应
Scheme 6 [3+2] ring-expansion reaction of aryl cyclopropanes with azadienes

图式 7 芳基环丙烷和烯烃的[3+2]扩环反应
Scheme 7 [3+2] ring-expansion reaction of aryl cyclopropanes with alkenes

图式 8 芳基环丙烷和2, 5-二甲基呋喃的[3+2]扩环反应及其产物的后续转化
Scheme 8 [3+2] ring-expansion reaction of aryl cyclopropanes with 2, 5-diphenylfurans and chemical conversion of its products

图式 9 芳基环丙烷与1, 3-二烯类的[3+2]扩环反应
Scheme 9 [3+2] ring-expansion reaction of aryl cyclopropanes with 1, 3-dienes

图式 10 芳基环丙烷与环氧化物的[3+2]扩环反应
Scheme 10 [3+2] ring-expansion reaction of aryl cyclopropanes with epoxides

图式 11 环丙烷与丙二烯的分子内[3+2]扩环反应
Scheme 11 Intramolecular [3+2] ring-expansion reaction of aryl cyclopropanes with allenes

图式 12 环丙烷与烯烃的分子内[3+2]扩环反应
Scheme 12 Intramolecular [3+2] ring-expansion reaction of aryl cyclopropanes with alkenes

图式 13 环丙烷与吲哚的分子内[3+2]扩环反应
Scheme 13 Intramolecular [3+2] ring-expansion reaction of aryl cyclopropanes with indoles

图式 14 环丙烷与腈和异硫氰酸酯的[3+2]扩环反应
Scheme 14 [3+2] ring-expansion reaction of cyclopropanes with nitriles and isothioncyanates

图式 15 环丙烷和肟醚的分子内[3+2]扩环反应
Scheme 15 Intramolecular [3+2] ring-expansion reaction of cyclopropane with oxime ethers

图式 16 环丙烷-1, 1-二(三氟乙基酯)与乙烯基叠氮化物和氮丙啶的[3+2]扩环反应
Scheme 16 [3+2] ring-expansion reaction of cyclopropane- 1, 1-bis(trifluoroethyl ester) with vinyl azides and azirine

图式 17 γ-丁内酯稠合的环丙烷与碳二亚胺和异硫氰酸酯的[3+2]扩环反应
Scheme 17 [3+2] ring-expansion reaction of γ-butyrolactone fused cyclopropanes with carbodiimides and isothiocyanates

图式 18 环丙烷与异氰酸酯的[3+2]扩环反应
Scheme 18 [3+2] ring-expansion reaction of cyclopropanes with isocyanates

图式 19 亚胺基环丙烷与吡啶、喹啉和异喹啉的[3+2]扩环反应
Scheme 19 [3+2] ring-expansion reaction of iminocyclopropanes with pyridines, quinolines and isoquinolines

图式 20 氨基环丙烷与苯并唑类的[3+2]扩环反应
Scheme 20 [3+2] ring-expansion reaction of aminocyclopropanes with benzazoles

图式 21 氨基环丙烷与嘌呤的[3+2]扩环反应
Scheme 21 [3+2] ring-expansion reaction of aminocyclopropanes with purines

图式 22 2-单取代环丙烷与酮类的[3+2]扩环反应
Scheme 22 [3+2] ring-expansion reaction of 2-monosub- stituted cyclopropanes with ketones

图式 23 顺式-2, 3-二取代环丙烷与环状/无环脂族酮的[3+2]扩环反应
Scheme 23 [3+2] ring-expansion reaction of cis-2, 3-disub- stituted cyclopropanes with cyclic/acyclic aliphatic ketones

图式 24 反式/顺式-2, 3-二取代环丙烷与胡椒醛的[3+2]扩环反应
Scheme 24 [3+2] ring-expansion reaction of trans/cis-2, 3-disubstituted cyclopropanes with piperonal

图式 25 γ-丁内酯稠合环丙烷与醛类和酮类的[3+2]扩环反应
Scheme 25 [3+2] ring-expansion reaction of γ-butyrolactone fused cyclopropanes with aldehydes and ketones

图式 26 手性γ-丁内酯稠合环丙烷与芳香醛的[3+2]扩环反应
Scheme 26 [3+2] ring-expansion reaction of chiral γ-butyrol- actone fused cyclopropanes with aromatic aldehydes

图式 27 环丙烷和环氧化物的串联重排/分子内[3+2]扩环反应
Scheme 27 Tandem rearrangement/intramolecular [3+2] ring-expansion reaction of cyclopropanes with epoxides

图式 28 二烯基环丙烷和亲二烯体的串联分子间[4+2]环加成和分子内[3+2]扩环反应
Scheme 28 Tandem intermolecular [4+2] cycloaddition and intramolecular [3+2] ring-expansion reaction of dienylcyclopropanes with dienophiles

图式 29 钴-炔基环丙烷与醛的分子内[3+2]扩环反应
Scheme 29 Intramolecular [3+2] ring-expansion reaction of cobalt-alkynylcyclopropanes with aldehydes

图式 30 2-取代环丙烷与异(硫)氰酸酯和碳二亚胺的[3+2]扩环反应
Scheme 30 [3+2] ring-expansion reaction of 2-substituted cyclopropanes with isothiocyanates and carbodiimides

图式 31 2-取代环丙烷与硫代酮和硒酮的[3+2]扩环反应
Scheme 31 [3+2] ring-expansion reaction of 2-substituted cyclopropanes with thioketones and selenoketones

图式 32 2-取代环丙烷与硫脲的[3+2]扩环反应
Scheme 32 [3+2] ring-expansion reaction of 2-substituted cyclopropanes with thioureas

图式 33 乙烯基环丙烷与烯烃的分子内[3+2]/[5+2]扩环反应
Scheme 33 Intramolecular [3+2]/[5+2] ring-expansion reaction of vinylcyclopropanes with olefins

图式 34 乙烯基环丙烷与炔烃的分子内[3+2]扩环反应及其可能的反应机理
Scheme 34 Intramolecular [3+2] ring-expansion reaction of vinylcyclopropanes with alkynes and its proposed mechanism

图式 35 酮基环丙烷与炔烃的分子内[3+2]扩环反应
Scheme 35 Intramolecular [3+2] ring-expansion reaction of ketocyclopropanes with alkynes

图式 36 二乙烯基环丙烷与环烯烃的分子内[3+2]扩环反应
Scheme 36 Intramolecular [3+2] ring-expansion reaction of divinylcyclopropanes with cyclic olefins

图式 37 环丙烷与炔烃的分子内[3+2]扩环反应
Scheme 37 Intramolecular [3+2] ring-expansion reaction of cyclopropanes with alkynes

图式 38 乙烯基环丙烷和β, 2-二硝基苯乙烯的[3+2]扩环反应
Scheme 38 [3+2] ring-expansion reaction of vinylcyclopropanes with β, 2-dinitrostyrene

图式 39 酮基环丙烷与炔烃的[3+2]扩环反应及其可能的反应机理
Scheme 39 [3+2] ring-expansion reaction of ketocyclopropanes with alkynes and its proposed mechanism

图式 40 乙烯基环丙烷与丙二烯的[3+2]扩环反应
Scheme 40 [3+2] ring-expansion reaction of vinylcyclopropanes with allenes

图式 41 酰胺基环丙烷与炔烃的[3+2]扩环反应及其可能的反应机理
Scheme 41 [3+2] ring-expansion reaction of acylamide cyclopropanes with alkynes and its proposed mechanism

图式 42 乙烯基环丙烷与烯烃和N-甲苯磺酰基芳基亚胺的[3+2]扩环反应
Scheme 42 [3+2] ring-expansion reaction of vinylcyclopropanes with olefins and N-toluenesulfonyl arylimines

图式 43 单取代环丙烷与炔烃的开环偶联反应和[3+2]扩环反应
Scheme 43 Ring-opening coupling reaction and [3+2] ring-expansion reaction of monosubstituted cyclopropanes with alkynes

图式 44 乙烯基环丙烷与亚胺和酮亚胺的[3+2]扩环反应
Scheme 44 [3+2] ring-expansion reaction of vinylcyclopropanes with imines and ketimines

图式 45 乙炔基环丙烷与醛和亚胺的[3+2]扩环反应及其可能的反应机理
Scheme 45 [3+2] ring-expansion reaction of ethynylcyclopropanes with aldehydes and imines, and their proposed mechanism

图式 46 酮基环丙烷与烯烃的[3+2]扩环反应
Scheme 46 [3+2] ring-expansion reaction of ketocyclopropane with olefins

图式 47 氨基环丙烷与烯烃、炔烃的分子间[3+2]扩环反应
Scheme 47 Intermolecular [3+2] ring-expansion reaction of aminocyclopropanes with olefins and alkynes

图式 48 单取代氨基环丙烷与1, 3-共轭二炔和1, 3-共轭烯炔的分子间[3+2]扩环反应
Scheme 48 Intermolecular [3+2] ring-expansion reaction of monosubstituted aminocyclopropanes with 1, 3-conjugated diynes and enynes

图式 49 α-对甲苯磺酰基环丙烷与烯烃的[3+2]扩环反应及其可能的反应机理
Scheme 49 [3+2] ring-expansion reaction of α-p-toluenesul- fonylcyclopropanes with olefins and its proposed mechanism

图式 50 环丙烷与α, β-不饱和亚胺的串联[3+2]扩环反应
Scheme 50 Trandem [3+2] ring-expansion reaction of cyclopropanes with α, β-unsaturated imines

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





















































 下载:
下载:

 下载:
下载: