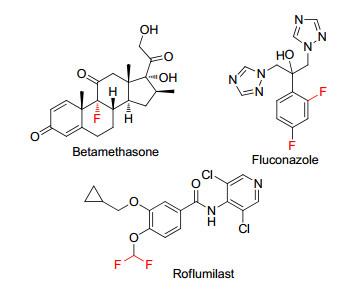
图 1.
Representative fluorine-containing drugs

Organofluorine compounds are widely applied in agrochemical, [1] pharmaceutical, [2] and material[3] fields because of the distinctive function of fluorine atom in the molecules.[4] Especially in medicinal chemistry, the introduction of fluorine or fluoro-containing moiety into molecules can optimize the pharmacological and pharmacokinetic property[5] of compounds. There are about 10%~12% known drugs[6] in clinic containing fluorine atom, such as betamethasone, fluconazole, roflumilast and so on (Figure 1). Therefore, over the past few years, the development of efficient and novel methods to synthesize organofluorine compounds has become a hot topic in organic chemistry.
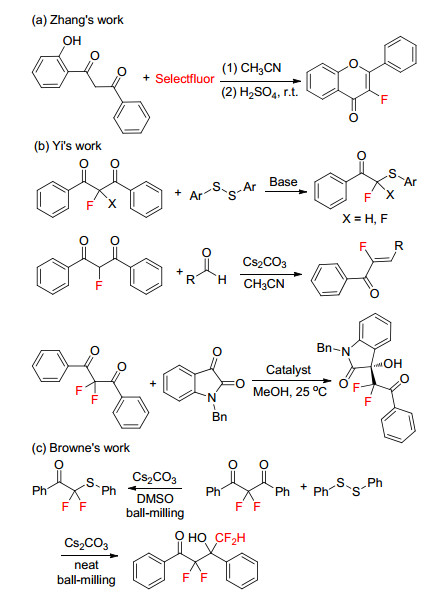
1, 3-Diketones are stable and widely available reagents in organic chemistry.[7] They usually served as useful building diverse heterocycles, [8] such as 2H-pyrans, [9] indoles, [10] blocks in organic synthesis, especially in the construction of pyridines, [11] and so on. In addition, 1, 3-diketones are easily fluorinated to get 2-fluoro-1, 3-diketones or 2, 2-difluoro-1, 3-diketones, which are used to prepare various organofluorine compounds.[12] For example, Zhang’s group[13] reported an efficient one-pot synthetic method of 3-fluoroflavones using in situ prepared 2-fluoro-1, 3- diketones (Scheme 1a). Yi’s group[14] revealed that 2- fluoro-1, 3-diketones and 2, 2-difluoro-1, 3-diketones can be used as nucleophilic mono(di)-fluoromethylation reagents, followed by reacting with diaryl disulfides, aldehyde and N-benzylisatins to achieve α-thioaryl-α, α-difluoroaceto- phenones, α-fluoro-α, β-unsaturated ketones, and 3-difluo- roalkyl-3-hydroxyoxindoles, respectively (Scheme 1b). More recently, Browne’s group[15] described the mechanochemistry reaction of 2, 2-difluoro-1, 3-diketones with phenyl disulfide using ball-milling, in which difluorothioether or tetrafluorohydroxyketone derivatives could be prepared in good yields (Scheme 1c).
Due to that the ball-milling is not widely available in organic chemistry laboratory, we tried to perform the reaction of 2, 2-difluoro-1, 3-diphenyl-1, 3-propanedione (1a) with Cs2CO3 using general stirring method. As expected, the desired tetrafluorohydroxyketone (2a) can be obtained in 31% yield when acetonitrile was used as solvent (Table 1, Entry 1). Then, other different solvents, including CH2Cl2, N, N-dimethylformamide (DMF), tetrahydrofuran (THF), 1, 4-dioxane and dimethyl sulfoxide (DMSO) were screened to improve the yields of corresponding product 2a (Table 1, Entries 2~6), and the results showed that DMSO was the optimal one among all these solvents with yield of 56% (Table 1, Entry 6). The replacement of Cs2CO3 with other bases, including K2CO3, KOH, EtONa, NaOAc and 1, 4-diazabicyclo[2.2.2]octane (DABCO) cannot improve the yields of 2a (Table 1, Entries 7~11). Interestedly, the neat reaction of 1a and cesium carbonate at room temperature for 10 h can produce 2a in excellent yield of 82% (Table 1, Entry 12).
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Entry | Solvent | Base | Yieldb/% |
| 1 | CH3CN | Cs2CO3 | 31 |
| 2 | CH2Cl2 | Cs2CO3 | 12 |
| 3 | DMF | Cs2CO3 | 39 |
| 4 | THF | Cs2CO3 | 51 |
| 5 | 1, 4-Dioxane | Cs2CO3 | 49 |
| 6 | DMSO | Cs2CO3 | 56 |
| 7 | DMSO | K2CO3 | 18 |
| 8 | DMSO | KOH | 0 |
| 9 | DMSO | EtONa | 26 |
| 10 | DMSO | NaOAc | 49 |
| 11 | DMSO | DABCO | 13 |
| 12 | Solvent-free | Cs2CO3 | 82 |
| a Reaction condition: 1a (0.5 mmol) and base (1.5 mmol) in 2 mL of solvent stirred at room temperature for 10 h; b isolated yields. | |||
With the optimized conditions in hand, the substrate scope of the reaction was investigated with a variety of 2, 2-difluoro-1, 3-diaryl-1, 3-diketones, and the results are summarized in Table 2. In general, for the para- or meta-sub- stituted substrates, the electronic effect did not affect the yields of products obviously, both electron-donating and electron-withdrawing substituted substrates 1b~1i afford the desired products 2b~2i in moderate to good yields (50%~82%). The heteroaryl substrate 1j was also suitable for this reaction and gave the corresponding product 2j in 60% yield, while the ortho-methoxy substituted substrate 1k only yielded trace of product 2k, indicating the importance of steric effect.
下载:
导出CSV
 |
||
| Entry | R | Yieldb/% |
| 1 | Ph (1a) | 82 |
| 2 | 4-FC6H4 (1b) | 76 |
| 3 | 4-ClC6H4 (1c) | 72 |
| 4 | 4-MeC6H4 (1d) | 50 |
| 5 | 4-CF3C6H4 (1e) | 71 |
| 6 | 4-CNC6H4 (1f) | 68 |
| 7 | 4-BrC6H4 (1g) | 65 |
| 8 | 3-MeOC6H4 (1h) | 53 |
| 9 | 3-ClC6H4 (1i) | 52 |
| 10 | Thiophene (1j) | 60 |
| 11 | 2-MeOC6H4 (1k) | Trace |
| a Reaction condition: 1 (0.5 mmol), Cs2CO3 (1.5 mmol), solvent-free, r.t. for 10 h. b isolated yields. | ||
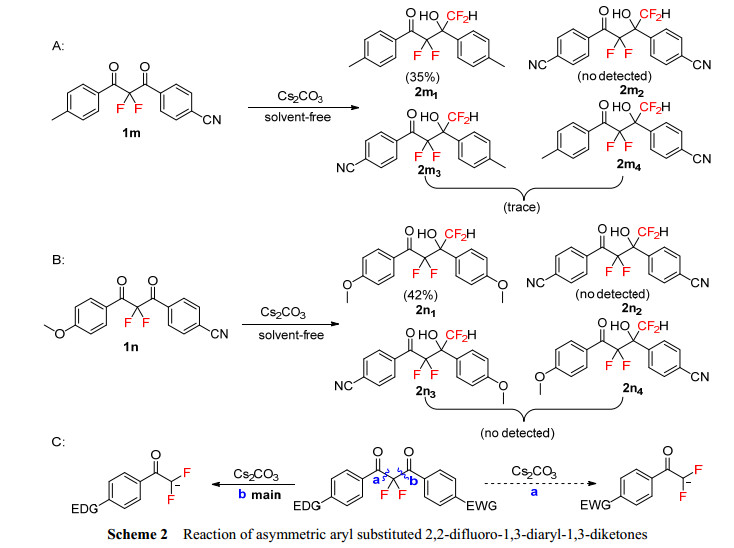
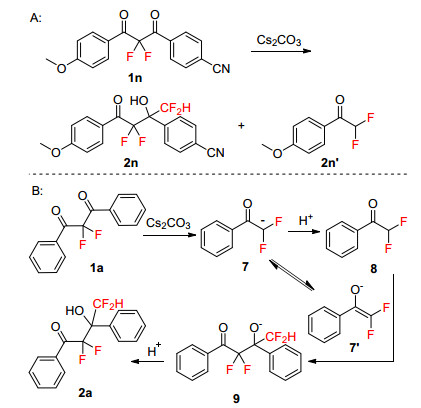
Then, asymmetric substrate 1m with different substituents on the phenyl ring was investigated under the standard conditions (Scheme 2A). The main product 2m1 which containing electron-donating substituents on the phenyl ring was obtained in 35% yield, and trace amount of 2m3 or 2m4 was detected in the reaction mixture with LC-MS analysis. The possible product 2m2 cannot be detected in the mixture. Similarly, when another asymmetric substrate 1n was performed under the same conditions (Scheme 2B), only product 2n1, which containing electron-donating substituents on the phenyl ring, was separated as main product, other three possible products 2n2, 2n3 and 2n4 cannot be found in the reaction mixture. These results show that the main way of carbon-carbon bond cleavage in this reaction was path b, which produced more nucleophilic difluoroenolate intermediates, as shown in Scheme 2C.
α-Fluoroacetophenones are frequently found in many bioactive molecules and can act as important intermediates in many organic reactions.[16] Many methods have been developed in the past few decades to get α-fluoroaceto- phenones. In general, they can be classified into two strategies.[17] The first one involves the introduction of fluorine atom into substrates with electrophilic fluorinating reagents, including N-fluorobenzenesulfonimide (NFSI), and Selectfluor®.[18] The second approach is the nucleophilic substitution of substrate with fluoride anion using KF/Ph3SnF, KF and so on.[19] We envisioned that 1, 3-diaryl-1, 3-diketones can also provide α-fluoroacetogation through decarbonyl fluorination.
Our investigation started with the reaction of 1, 3-di- phenylpropane-1, 3-dione (3a) with Selectfluor® under various conditions. Firstly, Hu’s protocol[20] for the decarboxylative fluorination of β-ketoacids in the CH3CN/H2O mixed solvent system was tried. As expected, 11% yield of desired product 4a can be detected in 19F NMR (Table 3, Entry 1). The reaction can hardly proceed when the water was removed (Table 3, Entry 2). Thus, the different co- solvents were studied for this transformation and the results showed that CH3CN/H2O (V:V=9:1) was optimal to furnish 4a in 25% yield (Table 3, Entry 3). Then, the temperature of reaction mixture was raised and the yields of 4a was improved accordingly (Table 3, Entries 5~7). Then, different bases and the equivalent of base were evaluated. When Cs2CO3 was replaced by other bases (KOH, K2CO3, NaOAc), a decrease in the yield of 4a was observed (Table 3, Entries 8~10), and 1.5 equiv. of Cs2CO3 is optimal for this reaction with yield of 69% (Table 3, Entry 13). In summary, these results showed that the optimal conditions for this reaction as follows: the mixture of 3a (1.0 equiv.) and Selectfluor® (1.05 equiv.) was dissolved in a mixture of 2.0 mL of CH3CN/H2O (V:V=9:1) and refluxed for 2.5 h, followed by the addition of 1.5 equiv. of Cs2CO3 and refluxed for another 5 h.
下载:
导出CSV
 |
||||
| Entry | Solvent (volume ratio) | t/℃ | Base (equiv.) | Yieldb/% |
| 1 | CH3CN/H2O (20:1) | r.t. | Cs2CO3 (3.0) | 11 |
| 2 | CH3CN | r.t. | Cs2CO3 (3.0) | Trace |
| 3 | CH3CN/H2O (9:1) | r.t. | Cs2CO3 (3.0) | 25 |
| 4 | CH3CN/H2O (5:1) | r.t. | Cs2CO3 (3.0) | 20 |
| 5 | CH3CN/H2O (9:1) | 40 | Cs2CO3 (3.0) | 29 |
| 6 | CH3CN/H2O (9:1) | 60 | Cs2CO3 (3.0) | 43 |
| 7 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (3.0) | 53 |
| 8 | CH3CN/H2O (9:1) | Reflux | KOH (3.0) | 45 |
| 9 | CH3CN/H2O (9:1) | Reflux | K2CO3 (3.0) | 43 |
| 10 | CH3CN/H2O (9:1) | Reflux | NaOAc (3.0) | 10 |
| 11 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (4.0) | 41 |
| 12 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (2.0) | 56 |
| 13 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (1.5) | 69 |
| 14 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (1.0) | 52 |
| a Reaction condition: The mixture of 3a (0.2 mmol) and Selectfluor® (0.21 mmol) was stirred with various solvents (2.0 mL) for 2.5 h at different temperature, followed by the addition of various bases. The resulting mixture was continuously stirred for 5 h; byields were determined by 19F NMR using PhCF3 as an internal standard. | ||||
Then, the reaction scope of 1, 3-diaryl-1, 3-diketones 3 was surveyed, and the results are summarized in Table 4. In general, the electronic effect on phenyl ring did not affect the yields obviously in this reaction. Different substituent on the phenyl ring, both electron-donating groups (methy, methoxy) and electron-withdrawing groups (fluoro, chloro, cyano, trifluoromethyl) can proceed this reaction smoothly and obtain corresponding products in good yields (53%~75%). The ortho-substituent on the aromatic ring of 1, 3-diaryl-1, 3-diketones (3k) provided relatively lower yields of the desired products in comparison with the meta-, para-substituted 1, 3-diaryl-1, 3-diketones (3h, 3l), probably due to the steric effects. The heterocycle substrate 3j was also suitable for this transformation and gave the respective product 4j in good yield.
下载:
导出CSV
 |
||
| Entry | R | Yieldb/% |
| 1 | Ph (3a) | 68 |
| 2 | 4-FC6H4 (3b) | 65 |
| 3 | 4-ClC6H4 (3c) | 75 |
| 4 | 4-MeC6H4 (3d) | 69 |
| 5 | 4-CF3C6H4 (3e) | 68 |
| 6 | 4-CNC6H4 (3f) | 70 |
| 7 | 4-BrC6H4 (3g) | 66 |
| 8 | 3-MeOC6H4 (3h) | 61 |
| 9 | 3-ClC6H4 (3i) | 61 |
| 10 | Thiophene (3j) | 65 |
| 11 | 2- MeOC6H4 (3k) | 53 |
| 12 | 4-MeOC6H4 (3l) | 62 |
| a Reaction condition: 3 (0.50 mmol), Selectfluor® (0.53 mmol) was dissolved in 4.0 mL CH3CN/H2O (9:1) and refluxed for 2.5 h, after which Cs2CO3 (0.75 mmol) was added. The resulting mixture was refluxed for 5 h. b isolated yields. | ||
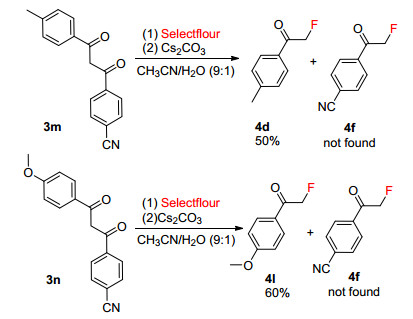
The asymmetric substrates of 3m and 3n with different susbstituents on phenyl ring were also investigated in this reaction (Scheme 3). We found that the main products were 4d and 4l, respectively. The other possible product 4f cannot be detected in the reaction system. These results showed that asymmetric substrates could selectively generate α-fluoroacetophenones which containing electron-donating substituents on the phenyl ring.
To further demonstrate the practicability and effectiveness of this methodology, two gram-scale synthesis has been performed as followed. When 4.0 mmol of 2, 2- difluoro-1, 3-diphenyl-1, 3-propanedione (1a) was reacted with 12.0 mmol of Cs2CO3, the corresponding product 2a can be obtained with yield of 73%, which was consistent with previous data (82%). We also carried out the gram- scale synthesis of 4a and gave yield of 61%, which was consistent with previous data (68%).
We then turned our attention on the mechanistic features of this reaction. When 1n was used as substrate under standard conditions, a side-product was found and confirmed as 2, 2-difluoro-1-(4-methoxyphenyl)ethan-1-one (2n') by 1H NMR spectrum. Therefore, we proposed a plausible mechanism[15] for the synthesis of 2a in Scheme 4. Initially, 2, 2-difluoro-1, 3-diphenyl-1, 3-propanedione (1a) is attacked by Cs2CO3 to form α, α-difluoroenolate intermediate 7, then 7 was protonated to form α, α-difluoro- acetophenone 8. The aldol reaction between 7' and 8 gave the intermediate 9 and protonated into product 2a.
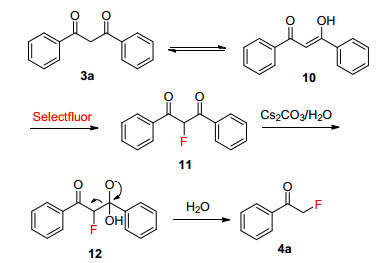
We proposed the possible mechanism for the synthesis of 4a as shown in Scheme 5. Firstly, 1, 3-diphenylpropane-1, 3- dione (3a) was transformed to enol intermediate 10, which was fluorinated by Selectfluor to form intermediate 11. Then, intermediate 11 was attacked by Cs2CO3/H2O to yield α-fluoroacetophenone 4a.
In summary, we have demonstrated a simple and efficient method to prepare α, α, γ, γ-tetrafluoro-β-hydroxy ketones and α-fluoroacetophenones from various 1, 3-diaryl-1, 3-diketones. With the modification of reaction conditions, two different products could be obtained in moderate to good yields, respectively.
All reactions were monitored by thin layer chromatography purchased from commercial suppliers. And the spots were visualized using UV radiation (254 nm). Flash chromatography was performed on 200~300 mesh silica gel. 1H NMR spectra were recorded on 400 or 500 MHz (100 or 126 MHz for 13C NMR, 376 or 471 MHz for 19F NMR) Zhongke-Niujin WNMR-I-400 or BRUCKER AVANCE III 500 NMR spectrometer with DMSO-d6 or CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard. Chemical shifts were reported downfield from TMS at δ 0.00 and referenced to CDCl3 (δ 7.26 in 1H NMR and δ 77.02 in 13C NMR) and DMSO-d6 (δ 2.50 in 1H NMR and δ 39.52 in 13C NMR). High resolution MS (HRMS) was performed on an Agilent 6224 TOF LC/MS spectrometer. All commercially available reagents and solvents were used as received unless otherwise specified. The substrates were readily prepared according to known methods. Substrates 1 and 3 were prepared according to literature methods.[21]
The mixture of 2, 2-difluoro-1, 3-diphenyl-propane-1, 3- dione (1a) (0.5 mmol, 1.0 equiv.) and cesium carbonate (1.5 mmol, 3.0 equiv.) was stirred in Schlenk tube by magnetic stirrer at room temperature for 10 h. Then, the reaction was extracted with ethyl acetate (15 mL×3), the combined organic layers were dried over anhydrous sodium sulfate and the solvent was evaporated in vacuo to give the crude product, which was purified carefully by column chromatography, ethyl acetate and petroleum ether as eluent to afford product 2a as yellow oil. Compounds 2b~2k were prepared in the similar manner.
2, 2, 4, 4-Tetrafluoro-3-hydroxy-1, 3-diphenylbutan-1-one (2a): 82% yield, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 7.96 (d, J=8.5 Hz, 2H), 7.66~7.61 (m, 3H), 7.46 (t, J=7.5 Hz, 2H), 7.42~7.39 (m, 3H), 6.50 (t, JH—F=54.5 Hz, 1H), 4.20 (s, 1H); 13C NMR (126 MHz, CDCl3) δ: 190.67 (t, J=29.7 Hz), 134.76, 132.56 (d, J=10.7 Hz), 130.33 (t, J=3.0 Hz), 129.33, 128.62, 128.46, 126.87, 117.14 (t, JC—F=267.4 Hz), 115.93 (t, JC—F=250.6 Hz), 78.24 (t, J=23.1 Hz); 19F NMR (471 MHz, CDCl3) δ: -105.11~-106.91 (m), -128.36~-130.64 (m). ESI-HRMS calcd for C16H12F4O2Na [M+Na]+ 335.0666, found 335.0671.
2, 2, 4, 4-Tetrafluoro-1, 3-bis(4-fluorophenyl)-3-hydroxy-butan-1-one (2b): 76% yield, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 8.04~8.01 (m, 2H), 7.65~7.62 (m, 2H), 7.16~7.12 (m, 2H), 7.11~7.07 (m, 2H), 6.47 (t, JH—F=54.5 Hz, 1H), 4.28 (s, 1H); 13C NMR (126 MHz, CDCl3) δ: 188.84 (t, J=29.7 Hz), 167.79, 165.72, 164.29, 162.31, 133.51 (dt, J=10.0, 3.7 Hz), 129.05 (d, J=8.1 Hz), 128.78 (q, J=2.6 Hz), 128.09 (t, J=2.9 Hz), 117.07 (t, JC—F=267.0 Hz), 116.17, 116.00, 115.77 (t, JC—F=250.9 Hz), 115.59, 115.42, 77.98 (t, J=22.9 Hz); 19F NMR (376 MHz, DMSO-d6) δ: -103.26, -105.49~-107.79 (m), -113.16, -127.30~-130.43 (m). ESI-HRMS calcd for C16H10F6O2Na[M+Na]+ 371.0477, found 371.0477.
1, 3-Bis(4-chlorophenyl)-2, 2, 4, 4-tetrafluoro-3-hydroxy-butan-1-one (2c): 72% yield, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 7.93 (d, J=8.5 Hz, 2H), 7.59 (d, J=8.5 Hz, 2H), 7.46~7.43 (m, 2H), 7.40~7.37 (m, 2H), 6.45 (t, JH—F=54.5 Hz, 1H), 4.22 (s, 1H); 13C NMR (126 MHz, CDCl3) δ: 189.16 (t, J=30.0 Hz), 141.86, 135.72, 131.78 (t, J=3.7 Hz), 130.74 (t, J=2.0 Hz), 130.59 (t, J=2.4 Hz), 129.12, 128.71, 128.39, 116.94 (t, JC—F=267.2 Hz), 115.63 (t, JC—F=251.1 Hz), 77.98 (t, J=23.3 Hz); 19F NMR (376 MHz, DMSO-d6) δ: -105.58~-108.16 (m), -127.21~-130.40 (m). ESI-HRMS calcd for C16H10Cl2F4O2Na [M+Na]+ 402.9886, found 402.9890.
2, 2, 4, 4-Tetrafluoro-3-hydroxy-1, 3-di-p-tolylbutan-1-one (2d): 50% yield, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 7.88 (d, J=8.5 Hz, 2H), 7.53 (d, J=8.0 Hz, 2H), 7.25 (d, J=8.0 Hz, 2H), 7.21 (d, J=8.5 Hz, 2H), 6.46 (t, JH—F= 54.5 Hz, 1H). 4.27 (s, 1H), 2.41 (s, 3H), 2.34 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 190.26 (t, J=29.6 Hz), 146.23, 139.20, 130.58 (t, J=3.4 Hz), 129.94 (t, J=2.4 Hz), 129.59, 129.37, 129.18, 126.76, 117.13 (t, JC—F=267.0 Hz), 115.96 (t, JC—F=250.4 Hz), 78.19 (t, J=22.9 Hz), 21.88, 21.13; 19F NMR (376 MHz, DMSO-d6) δ: -104.90~-106.80 (m), -128.32~-30.78 (m). ESI-HRMS calcd for C18H16F4O2Na[M+Na]+ 363.0979, found 363.0977.
2, 2, 4, 4-Tetrafluoro-3-hydroxy-1, 3-bis(4-(trifluorome-thyl)phenyl)butan-1-one (2e): 71% yield, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 8.08 (d, J=8.0 Hz, 2H), 7.80 (d, J=8.0 Hz, 2H), 7.74 (d, J=8.5 Hz, 2H), 7.69 (d, J=8.0 Hz, 2H), 6.51 (t, JH—F=54.5 Hz, 1H), 4.17 (s, 1H); 13C NMR (126 MHz, CDCl3) δ: 189.41 (t, J=30.0 Hz), 136.34 (q, J=33.1 Hz), 136.04, 135.15, 132.17 (q, J=32.9 Hz), 130.71 (t, J=3.5 Hz), 127.56, 125.78 (q, J=3.8 Hz), 125.52 (q, J=3.8 Hz), 124.86 (d, J=65.5 Hz), 122.69 (d, J=66.2 Hz), 117.01 (t, JC—F=267.2 Hz), 115.62 (t, J=251.4 Hz), 78.17 (t, J=23.2 Hz); 19F NMR (376 MHz, DMSO-d6) δ: -61.33(s), -61.96 (s), -105.75~-109.35 (m), -126.80~-130.37 (m). ESI-HRMS calcd for C36H19F20O4 [2M-H]- 895.0969, found 895.0973.
1, 3-Bis(4-cyano)-2, 2, 4, 4-tetrafluoro-3-hydroxybutan-1-one (2f): 68% yield, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 8.10 (d, J=8.0 Hz, 2H), 7.81~7.77 (m, 4H), 7.74~7.72 (m, 2H), 6.49 (t, JH—F=54.5 Hz, 1H), 4.22 (s, 1H); 13C NMR (126 MHz, CDCl3) δ: 189.22 (t, J=30.5 Hz), 137.41, 135.67 (t, J=2.4 Hz), 132.81, 132.56, 131.10 (t, J=3.5 Hz), 128.27, 118.46, 118.40, 117.72, 117.21 (t, JC—F=267.0 Hz), 115.75 (t, JC—F=251.2 Hz), 114.05, 78.48 (t, J=23.4 Hz); 19F NMR (376 MHz, DMSO-d6) δ: -105.38~-108.97 (m), -127.01~-130.31 (m). ESI- HRMS calcd for C18H9F4N2O2[M-H]- 361.0606, found 361.0602.
1, 3-Bis(4-bromophenyl)-2, 2, 4, 4-tetrafluoro-3-hydro-xybutan-1-one (2g): 65% yield, yellow oil. 1H NMR (500 MHz, DMSO-d6) δ: 7.90 (d, J=4.0 Hz, 2H), 7.88 (s, 1H), 7.79 (d, J=8.5 Hz, 2H), 7.65 (d, J=8.5 Hz, 2H), 7.52 (d, J=8.5 Hz, 2H), 6.94 (t, JH—F=53.0 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ: 187.91 (t, J=27.5 Hz), 132.39, 131.69 (t, J=3.4 Hz), 131.25, 130.51, 128.93, 128.47, 122.24, 118.22 (t, JC—F=264.9 Hz), 115.49 (t, JC—F=248.5 Hz), 76.67 (t, J=23.6 Hz); 19F NMR (471 MHz, DMSO-d6) δ: -104.93~-107.10 (m), -127.74~-130.51 (m). ESI-HRMS calcd for C16H10Br2F4O2Na [M+Na]+: 490.8876, found 490.8880.
2, 2, 4, 4-Tetrafluoro-1, 3-bis(3-methoxy)-3-hydroxybutan-1-one (2h): 53% yield, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 7.58 (d, J=8.0 Hz, 1H), 7.43 (t, J=2.0 Hz 1H), 7.36 (t, J=8.0 Hz, 1H), 7.32 (t, J=8.0 Hz, 1H), 7.22~7.19 (m, 2H), 7.17~7.15 (m, 1H), 6.93~6, 91 (m, 1H), 6.49 (t, JH—F=54.5 Hz), 4.31 (s, 1H) 3.81 (s, 3H), 3.78 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 190.08 (t, J=29.5 Hz), 159.45 (d, J=13.7 Hz), 133.88, 133.57 (t, J=2.4 Hz), 129.44, 129.33, 122.84 (t, J=4.3 Hz), 121.28, 118.83, 117.10 (t, JC—F=267.6 Hz), 115.70 (t, JC—F=250.6 Hz), 114.51, 114.15 (t, J=2.9 Hz), 112.85, 77.97 (t, J=23.2 Hz), 55.28, 55.1; 19F NMR (471 MHz, CDCl3) δ: -105.12~-106.70 (m), -128.58~-130.57 (m). ESI-HRMS calcd for C18H16F4NaO4 [M+Na]+ 395.0877, found 395.0880.
1, 3-Bis(3-chlorophenyl)-2, 2, 4, 4-tetrafluoro-3-hydroxy-butan-1-one (2i): 52% yield, yellow oil. 1H NMR (500 MHz, DMSO-d6) δ: 7.99 (d, J=8.5 Hz, 2H), 7.90 (s, 1H), 7.64~7.62 (m, 2H), 7.60 (d, J=8.5 Hz, 2H), 7.51~7.48 (m, 2H), 6.94 (t, JH—F=53.5 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ: 188.18 (t, J=28.1 Hz), 139.59, 134.01, 132.45, 132.16, 131.85, 129.16, 128.78, 128.07, 118.78 (t, JC—F=265.2 Hz), 116.04 (t, JC—F=248.3 Hz), 77.12 (t, J=22.7 Hz); 19F NMR (471 MHz, DMSO-d6) δ: -105.73~-108.14 (m), -127.35~-130.34 (m). ESI-HRMS calcd for C16H10Cl2F4O2Na [M+Na]+ 402.9886, found 402.9885.
2, 2, 4, 4-Tetrafluoro-3-hydroxy-1, 3-di(thiophen-2-yl)-butan-1-one (2j): 60% yield, yellow oil. 1H NMR (500 MHz, DMSO-d6) δ: 8.20 (d, J=3.5 Hz, 1H), 8.01 (s, 1H), 7.91 (s, 1H), 7.60 (d, J=4.0 Hz, 1H), 7.29 (t, J=4.5 Hz, 1H), 7.26 (d, J=3.5 Hz, 1H), 7.09 (dd, J=5.0, 3.5 Hz, 1H), 6.72 (t, JH—F=54.0 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ: 181.40 (t, J=29.0 Hz), 139.49, 138.82, 137.88 (t, J=5.5 Hz), 136.63, 129.82, 127.94, 127.77, 127.60, 118.62 (t, JC—F=264.5 Hz), 115.78 (t, JC—F=250.5 Hz), 77.21 (t, J=24.4 Hz); 19F NMR (471 MHz, DMSO-d6) δ: -108.14~-109.72 (m), -127.08~-130.29 (m). ESI-HRMS calcd for C12H8F4O2S2Na [M+Na]+ 346.9794, found 346.9798.
To a solution of 1, 3-diphenylpropane-1, 3-dione (3a) (0.5 mmol, 1.0 equiv.) in 4.0 mL of CH3CN/H2O (V:V=9:1) was added Selectfluor® (0.53 mmol, 1.05 equiv.), the resulting solution was refluxed for 2.5 h. After that, cesium carbonate (0.75 mmol, 1.5 equiv.) was added to the solution and refluxed for 5 h. Then the reaction system was concentrated under reduced pressure and extracted with ethyl acetate (20 mL×3). The combined organic layer was dried over anhydrous sodium sulphate, filtered and con- centrated under reduced pressure. The residue was subjected to the column chromatography using ethyl acetate and petroleum ether as eluent to give product 2-fluoro-1-phenylethan-1-one (4a) in 68% yield. Other α-fluoroacetophenones 4b~4l were prepared in similar manner.
2-Fluoro-1-phenylethanone (4a):[22] 68% yield, yellow oil. 1H NMR (500 MHz, DMSO-d6) δ: 7.90~7.88 (m, 2H), 7.70 (tt, J=7.5, 1.5 Hz, 1H), 7.57~7.54 (m, 2H), 5.87 (d, JH—F=46.5 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ: 193.90 (d, J=14.5 Hz), 133.99, 133.50, 128.90, 127.58 (d, J=1.4 Hz), 84.68 (d, JC—F=176.4 Hz); 19F NMR (471 MHz, DMSO-d6) δ: -232.92 (t, J=46.6 Hz).
2-Fluoro-1-(4-fluorophenyl)ethanone (4b):[24] 65% yield, colorless solid. m.p. 49~51 ℃ (lit.[23] m.p. 47~49 ℃); 1H NMR (500 MHz, DMSO-d6) δ: 7.99~7.97 (m, 2H), 7.41~7.37 (m, 2H), 5.85 (d, JH—F=46.0 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ: 193.06 (d, J=14.5 Hz), 166.86 (d, JC—F=253.0 Hz), 131.21 (d, J=1.4 Hz), 131.14 (d, J=1.3 Hz), 130.77 (d, J=2.8 Hz), 116.58, 116.40, 85.05 (d, JC—F=176.5 Hz); 19F NMR (471 MHz, DMSO-d6) δ: -104.49~-104.55 (m), -232.58 (t, J=46.6 Hz).
1-(4-Chlorophenyl)-2-fluoroethanone (4c):[24] 75% yield, yellow solid, m.p. 50~51 ℃ (lit.[23] m.p. 51~52 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.86 (d, J=9.0 Hz, 2H), 7.48 (d, J=8.5 Hz, 2H), 5.52 (d, JH—F=47.0 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ: 192.96 (d, J=15.6 Hz), 141.08, 132.47, 129.83 (d, J=3.5 Hz), 129.69, 84.72 (d, JC—F=183.8 Hz); 19F NMR (471 MHz, CDCl3) δ: -229.60.
2-Fluoro-1-(p-tolyl) ethanone (4d):[22] 69% yield, colorless oil. 1H NMR (500 MHz, CDCl3) δ: 7.80 (d, J=8.0 Hz, 2H), 7.29 (d, J=8.0 Hz, 2H), 5.55 (d, JH—F=47.0 Hz, 2H), 2.42 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 193.21 (d, J=15.2 Hz), 145.30, 131.36, 129.72, 128.08 (d, J=2.6 Hz), 84.35 (d, JC—F=182.3 Hz), 21.89; 19F NMR (471 MHz, CDCl3) δ: -230.74.
2-Fluoro-1-(4-(trifluoromethyl)phenyl)ethenone (4e):[20] 68% yield, colorless solid. m.p. 36~37 ℃ (lit.[25] m.p. 36~37 ℃); 1H NMR (500 MHz, DMSO-d6) δ: 8.09 (d, J=8.0 Hz, 2H), 7.95 (d, J=8.5 Hz, 2H), 5.93 (d, JH—F=46.0 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ: 193.14 (d, J=14.6 Hz), 136.31, 132.82, 132.56, 128.10 (d, J=1.9 Hz), 125.47 (m), 122.14, 84.47 (d, JC—F=177.2 Hz); 19F NMR (471 MHz, DMSO-d6) δ: -61.71, -232.93 (t, J=45.7 Hz).
1-(4-Cyano)-2-fluoroethanone (4f):[26] 70% yield, colorless solid. m.p. 103~105 ℃ (lit.[23] m.p.104~105 ℃); 1H NMR (500 MHz, DMSO-d6) δ: 8.04 (s, 4H), 5.91 (d, JH—F=46.0 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ: 193.50 (d, J=14.6 Hz), 136.70, 132.87, 128.29, 117.99, 115.79, 84.91 (d, JC—F=177.4 Hz); 19F NMR (471 MHz, DMSO-d6) δ: -233.00 (t, J=45.7 Hz).
1-(4-Bromophenyl)-2-fluoroethanone (4g):[20] 66% yield, colorless solid. m.p. 72~73 ℃ (lit.[25] m.p. 72~73 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.78 (d, J=8.0 Hz, 2H), 7.66 (d, J=8.5 Hz, 2H), 5.52 (d, JH—F=47.0 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ: 192.67 (d, J=16.3 Hz), 132.36, 132.19, 129.38 (d, J=2.9 Hz), 84.19 (d, JC—F=183.8 Hz); 19F NMR (471 MHz, CDCl3) δ: -229.65.
2-Fluoro-1-(3-methoxyphenyl)ethanone (4h):[22] 61% yield, colorless solid. m.p. 52~54 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.47~7.46 (m, 2H), 7.39~7.38 (m, 1H), 7.27~7.24 (m, 1H), 5.86 (d, JH—F=46.5 Hz, 2H), 3.82 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ: 194.08 (d, J=14.4 Hz), 159.83, 135.13, 130.49, 120.30, 112.48, 85.07 (d, JC—F=175.8 Hz), 55.75. 19F NMR (471 MHz, DMSO-d6) δ: -232.72 (t, J=46.2 Hz).
1-(3-Chlorophenyl)-2-fluoroethan-1-one (4i):[24] 61% yield, yellow oil. 1H NMR (500 MHz, DMSO-d6) δ: 7.91 (t, J=2.0 Hz, 1H), 7.86~7.84 (m, 1H), 7.76~7.74 (m, 1H), 7.61 (t, J=8.0 Hz, 1H), 5.89 (d, JH—F=46.0 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ: 192.52 (d, J=14.5 Hz), 134.78, 133.32, 133.13, 130.39, 126.83, 125.74, 84.26 (d, JC—F=176.5 Hz). 19F NMR (471 MHz, DMSO-d6) δ:-232.96 (t, J=47.1 Hz).
2-Fluoro-1-(thiophen-2-yl)ethanone (4j):[22] 65% yield, colorless solid, m.p. 63~64 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 8.14 (d, J=4.5 Hz, 1H), 7.97 (dd, J=3.5Hz, 1Hz, 2H), 7.32~7.30 (m, 1H), 5.75 (d, JH—F=46.5 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ: 187.67 (d, J=16.5 Hz), 139.85, 136.07, 134.09 (d, J=1.9 Hz), 129.45, 84.62 (d, JC—F=177.5 Hz); 19F NMR (471 MHz, DMSO-d6) δ: -230.75 (t, J=46.2 Hz).
2-Fluoro-1-(2-methoxyphenyl)ethanone (4k):[22] 53% yield, colorless solid. m.p. 90~91 ℃; 1H NMR (500 MHz, DMSO-d6) δ: 7.83 (dd, J=8 Hz, 2 Hz, 1H), 7.65~7.62 (m, 1H), 7.23 (d, J=8.5 Hz, 1H), 7.12 (t, J=7 Hz, 1H), 5.54 (d, JH—F=48 Hz, 2H), 3.92 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ: 194.20 (d, J=14.4 Hz), 159.97, 135.81, 130.16 (d, J=1.9 Hz), 123.78, 121.31, 113.16, 87.38 (d, JC—F=176.5 Hz), 56.47; 19F NMR (471 MHz, DMSO-d6) δ: -223.01 (t, J=52.3 Hz).
2-Fluoro-1-(4-methoxyphenyl)ethanone (4l):[22] 62% yield, colorless solid. m.p. 81~82 ℃ (lit.[23] m.p. 78~79 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.90 (t, J=8.0 Hz, 2H), 6.97 (t, J=8.5 Hz, 2H), δ: 5.53 (d, JH—F=47.0 Hz, 2H) 3.89 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 192.38 (d, J=15.4 Hz), 164.61, 130.65 (d, J=2.6 Hz), 127.13, 114.50, 84.57 (d, JC—F=182.3 Hz), 55.92; 19F NMR (471 MHz, CDCl3) δ: -232.50 (t, J=44.7 Hz). Data matched those previously reported.
Supporting Information Copies of 1H NMR, 13C NMR and 19F NMR spectra for 2a~2j and 4a~4l. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Fujiwara, T.; O'Hagan D. J. Fluorine Chem. 2014, 167, 16.
(b) Ghosh, K.-G.; Chandu, P.; Mondal, S.; Sureshkumar, D. Tetrahedron 2019, 75, 4471.
(c) Wei, J.; Gu, D.-Y.; Wang, S.-D.; Hu, J.-B.; Dong, X.-W.; Sheng, R. Org. Chem. Front. 2018, 5, 2568.
(d) Cloutier, M.; Roudias, M.; Paquin, J.-F. Org. Lett. 2019, 21, 3866.
(a) Gillis, E.-P.; Eastman, K.-J.; Hill, M.-D.; Donnelly, D.-J.; Meanwell, N.-A. J. Med. Chem. 2015, 58, 8315.
(b) Vogt, D.-B.; Seath, C.-P.; Wang, H.; Jui, N.-T. J. Am. Chem. Soc. 2019, 141, 13203.
(c) Guan, Z.-P.; Wang, H.-M.; Huang, Y.-G.; Wang, Y.-K.; Wang, S.-C.; Lei, A.-W. Org. Lett. 2019, 21, 4619.
St-Gelais, J.; Bouchard, M.; Denavit, V.; Giguere, D. J. Org. Chem. 2019, 84, 8509. doi: 10.1021/acs.joc.9b00795
(a) Liu, C.-Z.; Wang, H.; Zhang, D.-W.; Zhao, X.; Li, Z.-T. Chin. J. Org. Chem. 2019, 39, 28(in Chinese). (刘传志, 王辉, 张丹维, 赵新, 黎占亭, 有机化学, 2019, 39, 28.)
(b) Hu, J. B.; Ding, K.-L. Acta Chim. Sinica 2018, 76, 905(in Chinese). (胡金波, 丁奎岭, 化学学报, 2018, 76, 905.)
(a) D'Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; D'Alonzo, D.; Piccialli, V.; Mayol, L.; Piccialli, G. Molecules 2012, 17, 13036.
(b) D'Errico, S.; Oliviero, G.; Piccialli, V.; Amato, J.; Borbone, N.; D'Atri, V.; D'Alessio, F.; Di Noto, R.; Ruffo, F.; Salvatore, F.; Piccialli, G. Bioorg. Med. Chem. Lett. 2011, 21, 5835.
(c) Naveen, N.; Balamurugan, R. Org. Biomol. Chem. 2017, 15, 2063.
(a) Yang, Q.; Mao, L.-L.; Yang, B.; Yang, S.-D. Org. Lett. 2014, 16, 3460.
(b) Purser, S.; Moore, P.-R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320.
(c) Zhang, P.; Wolf, C. J. Org. Chem. 2012, 77, 8840.
(a) He, Z.; Qi, X.-T.; She, Z.-J.; Zhao, Y.-S.; Li, S.-Q.; Tang, J.-B.; Gao, G.; Lan, Y.; You, J.-S. J. Org. Chem. 2017, 82, 1403.
(b) Bartlett, S.-L.; Beaudry, C.-M. J. Org. Chem. 2011, 76, 9852.
Kuang, J.-Q.; Zhou, T.; You, T.-J.; Chen, J.-H.; Su, C.-L.; Xia, Y.-Z. Org. Biomol. Chem. 2019, 17, 3940. doi: 10.1039/C9OB00494G
Chen, Y.-Z.; Zhu, J.; Wu, J.-J.; Wu, L. Org. Biomol. Chem. 2018, 16, 6675. doi: 10.1039/C8OB01640B
Han, J.-L.; Liao, Y.-T. Chang, C.-H. Eur. J. Org. Chem. 2019, 33, 5815.
Crosignani, S.; Linclau, B. J. Gen. Virol. 2006, 2, 129.
Tang, L.; Yang, Z.; Jiao, J.-C.; Cui, Y.; Zou, G.-D.; Zhou, Y.-Q.; Rao, W.-H.; Ma, X.-T. J. Org. Chem. 2019, 84, 10449. doi: 10.1021/acs.joc.9b01808
Wang, R.; Han, J.; Li, C.-C.; Zhang, J.; Liang, Y.; Wang, T.; Zhang, Z.-T. Org. Biomol. Chem. 2018, 16, 2479. doi: 10.1039/C8OB00135A
(a) Lin, Y.-M.; Yi, W.-B.; Shen, W.-Z.; Lu, G.-P. Org. Lett. 2016, 18, 592.
(b) Qian, J.-L.; Yi, W.-B.; Huang, X.; Jasinski, J.-P.; Zhang, W. Adv. Synth. Catal. 2016, 358, 2811.
(c) Yi, W.-B.; Qian, J.-L.; Lv, M.-F.; Cai, C. Synlett 2014, 26, 127.
Howard, J.-L.; Brand, M.-C.; Browne, D.-L. Angew. Chem., Int. Ed. 2018, 57, 16104. doi: 10.1002/anie.201810141
(a) He, Y.; Zhang, X.-Y.; Shen, N.-N.; Fan, X.-S. J. Fluorine Chem. 2013, 156, 9.
(b) Li, J.; Li, Y.-L.; Jin, N.; Ma, A.-L.; Huang, Y.-N.; Deng, J. Adv. Synth. Catal. 2015, 357, 2474.
(c) Wu, S.-W.; Liu, F. Org. Lett. 2016, 18, 3642.
(a) He, Y.; Zhang, X.-Y.; Shen, N.-N.; Fan, X.-S. J. Fluorine Chem. 2013, 156, 9.
(b) Singh, R.-P.; Martin, J.-L. J. Fluorine Chem. 2016, 181, 7.
(a) Barnette, W.-E. J. Am. Chem. Soc. 1984, 106, 452.
(b) Fuglseth, E.; Thvedt, T.-H.-K.; Moll, M.-F.; Hoff, B.-H. Tetrahedron 2008, 64, 7318.
(c) Kwiatkowski, P.; Beeson, T.-D.; Conrad, J.-C.; MacMillan, D.-W.-C. J. Am. Chem. Soc. 2011, 133, 1738.
(a) Wagner, P.-J.; Thomas, M.-J.; Puchalski, A.-E. J. Am. Chem. Soc. 1986, 108, 7739.
(b) Makosza, M.; Bujok, R. Tetrahedron Lett. 2004, 45, 1385.
Zhang, R.; Ni, C.-F.; He, Z.-B.; Hu, J.-B. J. Fluorine Chem. 2017, 203, 166. doi: 10.1016/j.jfluchem.2017.08.010
(a) Martinez-Haya, R.; Marzo, L.; Konig, B. Chem. Commun (Camb) 2018, 54, 11602.
(b) Leng, D.-J.; Black, C.-M.; Pattison, G. Org. Biomol. Chem. 2016, 14, 1531.
Li, J.; Li, Y.-L.; Jin, N.; Ma, A.-L.; Huang, Y.-N.; Deng, J. Adv. Synth. Catal. 2015, 357, 2474. doi: 10.1002/adsc.201500282
Chen, Z.; Zhu, W.; Zheng, Z.-B.; Zou, X.-Z. J. Fluorine Chem. 2010, 131, 340. doi: 10.1016/j.jfluchem.2009.11.008
He, Y.; Zhang, X.-Y.; Shen, N.-N.; Fan, X.-S. J. Fluorine Chem. 2013, 156, 9. doi: 10.1016/j.jfluchem.2013.08.006
Krane Thvedt, T.-H.; Fuglseth, E.; Sundby, E.; Hoff, B.-H. Tetrahedron 2009, 65, 9550. doi: 10.1016/j.tet.2009.09.070
Fuglseth, E.; Thvedt, T.-H.-K.; Møll, M.-F.; Hoff, B.-H. Tetrahedron 2008, 64, 7318. doi: 10.1016/j.tet.2008.05.060
表 1 Optimization of reaction conditionsa
|
|||
| Entry | Solvent | Base | Yieldb/% |
| 1 | CH3CN | Cs2CO3 | 31 |
| 2 | CH2Cl2 | Cs2CO3 | 12 |
| 3 | DMF | Cs2CO3 | 39 |
| 4 | THF | Cs2CO3 | 51 |
| 5 | 1, 4-Dioxane | Cs2CO3 | 49 |
| 6 | DMSO | Cs2CO3 | 56 |
| 7 | DMSO | K2CO3 | 18 |
| 8 | DMSO | KOH | 0 |
| 9 | DMSO | EtONa | 26 |
| 10 | DMSO | NaOAc | 49 |
| 11 | DMSO | DABCO | 13 |
| 12 | Solvent-free | Cs2CO3 | 82 |
| a Reaction condition: 1a (0.5 mmol) and base (1.5 mmol) in 2 mL of solvent stirred at room temperature for 10 h; b isolated yields. | |||
 下载: 导出CSV
下载: 导出CSV
表 2 Scope of the 2, 2-difluoro-1, 3-diketonesa
|
||
| Entry | R | Yieldb/% |
| 1 | Ph (1a) | 82 |
| 2 | 4-FC6H4 (1b) | 76 |
| 3 | 4-ClC6H4 (1c) | 72 |
| 4 | 4-MeC6H4 (1d) | 50 |
| 5 | 4-CF3C6H4 (1e) | 71 |
| 6 | 4-CNC6H4 (1f) | 68 |
| 7 | 4-BrC6H4 (1g) | 65 |
| 8 | 3-MeOC6H4 (1h) | 53 |
| 9 | 3-ClC6H4 (1i) | 52 |
| 10 | Thiophene (1j) | 60 |
| 11 | 2-MeOC6H4 (1k) | Trace |
| a Reaction condition: 1 (0.5 mmol), Cs2CO3 (1.5 mmol), solvent-free, r.t. for 10 h. b isolated yields. | ||
下载: 导出CSV
表 3 Optimization of reaction conditionsa
|
||||
| Entry | Solvent (volume ratio) | t/℃ | Base (equiv.) | Yieldb/% |
| 1 | CH3CN/H2O (20:1) | r.t. | Cs2CO3 (3.0) | 11 |
| 2 | CH3CN | r.t. | Cs2CO3 (3.0) | Trace |
| 3 | CH3CN/H2O (9:1) | r.t. | Cs2CO3 (3.0) | 25 |
| 4 | CH3CN/H2O (5:1) | r.t. | Cs2CO3 (3.0) | 20 |
| 5 | CH3CN/H2O (9:1) | 40 | Cs2CO3 (3.0) | 29 |
| 6 | CH3CN/H2O (9:1) | 60 | Cs2CO3 (3.0) | 43 |
| 7 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (3.0) | 53 |
| 8 | CH3CN/H2O (9:1) | Reflux | KOH (3.0) | 45 |
| 9 | CH3CN/H2O (9:1) | Reflux | K2CO3 (3.0) | 43 |
| 10 | CH3CN/H2O (9:1) | Reflux | NaOAc (3.0) | 10 |
| 11 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (4.0) | 41 |
| 12 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (2.0) | 56 |
| 13 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (1.5) | 69 |
| 14 | CH3CN/H2O (9:1) | Reflux | Cs2CO3 (1.0) | 52 |
| a Reaction condition: The mixture of 3a (0.2 mmol) and Selectfluor® (0.21 mmol) was stirred with various solvents (2.0 mL) for 2.5 h at different temperature, followed by the addition of various bases. The resulting mixture was continuously stirred for 5 h; byields were determined by 19F NMR using PhCF3 as an internal standard. | ||||
下载: 导出CSV
表 4 Scope of the 1, 3-diaryl-1, 3-diketonesa
|
||
| Entry | R | Yieldb/% |
| 1 | Ph (3a) | 68 |
| 2 | 4-FC6H4 (3b) | 65 |
| 3 | 4-ClC6H4 (3c) | 75 |
| 4 | 4-MeC6H4 (3d) | 69 |
| 5 | 4-CF3C6H4 (3e) | 68 |
| 6 | 4-CNC6H4 (3f) | 70 |
| 7 | 4-BrC6H4 (3g) | 66 |
| 8 | 3-MeOC6H4 (3h) | 61 |
| 9 | 3-ClC6H4 (3i) | 61 |
| 10 | Thiophene (3j) | 65 |
| 11 | 2- MeOC6H4 (3k) | 53 |
| 12 | 4-MeOC6H4 (3l) | 62 |
| a Reaction condition: 3 (0.50 mmol), Selectfluor® (0.53 mmol) was dissolved in 4.0 mL CH3CN/H2O (9:1) and refluxed for 2.5 h, after which Cs2CO3 (0.75 mmol) was added. The resulting mixture was refluxed for 5 h. b isolated yields. | ||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们






 下载:
下载: