





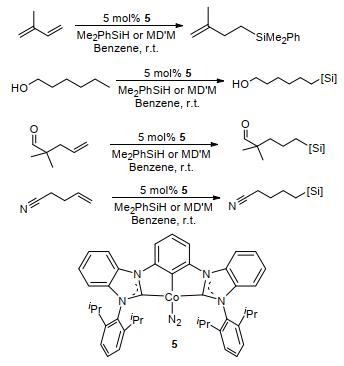
图 1.
双卡宾钴配合物催化烯烃硅氢加成反应
Figure 1.
Hydrosilylation of olefins catalyzes by bis(carbene) cobalt(I)-dinitrogen complex

硅氢加成反应是不饱和键(如C=C键或C=O键)中插入Si—H键形成C—Si键化合物的反应, 是合成有机硅化合物非常重要的反应之一[1~3].早期使用的硅氢加成催化剂主要是贵金属催化剂, 如铂[4, 5]、铑[6~8]、钯和铱[9]等, 应用最广泛的是Speier催化剂[4]和Karsted催化剂[5].但由于其价格昂贵且大多数情况下不可回收等缺点, 人们逐渐把研究方向转为对非贵金属如Fe、Co、Ni和Mn[10]等金属催化剂的研究.根据软硬酸碱理论, 钴的电负性较小, 比铁更易形成低价络合物, 比镍更易形成高价络合物[12].钴外层电子排布为3d74s2, 常见化合价为+2和+3, 容易与有机化合物配位.自从Chalk和Harrod等[11]首次报道了Co2(CO)8催化烯烃的硅氢加成反应以来, 钴配合物用于该反应的研究引起了越来越多科研工作者的关注和重视.已有文献对钴配合物催化硅氢加成反应进展进行了总结, 邓亮课题组[12]总结了从20世纪60年代到2016年钴配合物催化烯烃和炔烃硅氢加成反应的进展, 并且提出了该领域未解决的问题及其可能的解决方案.黄正[13]和Chirik课题组[14]在邓亮课题组综述的基础上对新发展的钴配合物催化烯烃硅氢加成反应进展进行了补充, 并且提出了该领域尚存的挑战和机遇.陆展课题组总结了2017年以前的钴配合物催化功能化烯烃和炔烃以及二烯、丙二烯、炔烃等硅氢加成反应的研究进展[15], 还对钴不对称催化官能化烯烃硅氢加成反应做了相应的总结[16]. Pawluc课题组[17]对钴催化末端烯烃硅氢加成反应进行总结.到目前为止, 没有相应的综述同时对钴催化剂催化烯烃、炔烃、醛酮等不饱和化合物硅氢加成反应做过全面的总结, 最近两年又报道了大量钴催化硅氢加成反应的研究工作.本文较全面地介绍了钴催化剂在硅氢加成反应的应用, 特别是近五年不同新型钴配合物催化剂用于催化烯烃、炔烃、醛酮等不饱和化合物硅氢加成反应的研究进展, 涉及马氏加成与反马氏加成反应, 还对一些副反应如脱氢反应也进行比较和总结.
1965年, Chalk和Harrod等[11]首次报道了钴催化烯烃硅氢加成反应.发现零价钴配合物Co2(CO)8是催化末端烯烃与硅烷硅氢加成的有效催化剂(Eq. 1).反应能在较温和的条件(0~60 ℃)下进行, 且没有脱氢产物和加氢产物等副产物生成, 但产物中有异构化的不活泼内烯烃生成.因此反应需要加入过量的烯烃, 这是Co2(CO)8催化烯烃硅氢加成反应的一个缺点.后来, Kalinin等[18]考察了Co2(CO)8催化硅氢加成反应的底物适应性.发现苯乙烯、烯丙基二(三乙氧基甲硅烷基)胺和乙烯基邻碳硼烷与(EtO)3SiH进行硅氢加成反应有良好的加成产物产率.
|
|
(1) |
Brookhart和Grant[19]报道了1 mol%的五甲基环戊二烯基钴(III)烷基络合物催化1-己烯与HSiEt3硅氢加成反应(Eq. 2), 得到反马氏加成产物, 分离产率为75%.
|
|
(2) |
2013年邓亮课题组[20]通过对钴进行连续C—H活化和硅烷功能化路线合成了一系列给电子硅基功能的N-杂环卡宾钴配合物1 (Eq. 3), 其中1a催化1-辛烯与PhSiH3反应生成反马氏加成产物的产率为70%, 仅有少量马氏加成产物和加氢产物生成.
|
|
(3) |
该课题组[21]又合成了含不同取代基的硅基酰胺钴配合物[Co(N(SiMe3)2)2]及其NHC卡宾配合物2 [(NHC)-Co(N(SiMe3)2)2], 是烯烃硅氢加成的有效催化剂(Eq. 4). 2a (5 mol%)作催化剂, 辛烯与(EtO)3SiH硅氢加成反应0.5和24 h分别以26%和79%的产率得到线性辛基三乙氧基硅烷.研究表明, 反应初期会产生烯烃异构体和脱氢副产物, 反应后期这些副产物完全转化为n-C8H17- Si(OEt)3和正辛烷.这一结果表明2a不仅能催化烯烃硅氢加成, 而且对烯烃异构化和氢化也有很好的催化效果. 2b和2c的催化性能与2a相似, 催化反应24和36 h分别得到68%和76%的产率. 2d-NHC配合物的空间体积更小, 硅氢加成产物n-C8H17Si(OEt)3的产率提升至86%. 0.1 mol%配合物2e可以有效地催化单取代脂族烯烃与(EtO)3SiH硅氢加成反应, 在0.5 h内几乎完全转化, 具有良好的反马氏加成产物选择性.烯烃结构对催化体系性能影响较大, 4-乙烯基-环己烯硅氢加成反应仅发生在乙烯基上, 与叔丁基取代烯烃硅氢加成产物产率较低(42%).与1-辛烯相比, 催化降冰片烯反应需在60 ℃条件下才能得到较高产率(87%), 而辛烯与三乙基硅烷不发生反应.
|
|
(4) |
不同类型的N-杂环卡宾钴配合物可以催化烯烃硅氢加成反应(Eq. 5)[22].以甲苯作溶剂, 3a催化苯乙烯硅氢加成得到反马氏硅氢加成产物, 分离产率为92%;双NHC钴络合物3b和3c作为催化剂时, 硅氢加成反应产物产率分别为66%和58%; 5 mol% 3b催化反应马氏加成产物产率可达到93%;相同反应条件下, 3d与3b催化性能相似.具有三咪唑环的NHC卡宾钴络合物3f和3e作为催化剂分别得到反马氏加成产物(23%和15%)、加氢产物(44%和48%)和脱氢偶联产物(30%和22%)的混合物.四咪唑环NHC卡宾配位的配合物3i和3h作为催化剂得到的产物也为混合物, 但3g催化该反应, 几乎只得到加氢产物.当3g用量提高到5 mol%, 减少反应时间, 加氢产物产率与选择性都会明显提高.
|
|
(5) |
2015年, Holland课题组[23]合成了β-二酮亚胺-钴(I)-芳烃配合物应用于PhSiH3和(EtO)3SiH与烯烃硅氢加成反应(Eq. 6).含有N-均三甲苯基取代的催化剂4活性最高.可有效催化含硅烷、甲硅烷基醚、卤化物、芳烃、酯、叔胺和酰胺取代基的烯烃硅氢加成反应, 都具有高的选择性, 产率为67%~94%.
|
|
(6) |
双卡宾钴配合物5可在室温条件下催化辛烯与Me2PhSiH, Ph2SiH2和(MeO)3SiH的硅氢加成反应(Scheme 1), 均可得到82%~94%产率[24].催化MD'M (1, 1, 1, 3, 5, 5, 5-七甲基三硅氧烷)可得到41%的加成产率; 催化Et3SiH反应转化率较低, 反应21 h后加成产物分离产率仅为35%;催化Ph3SiH反应1 h后基本无产物生成.相同条件下, 可催化Me2PhSiH和MD'M与带有官能团的末端烯烃硅氢加成反应.催化含多个烯键的底物时, 小位阻的双键会选择性地发生加成反应.含有酮、腈和胺等基团的烯烃均能高收率地反应, 生成相应加成产物, 但环己烯和1, 1-二取代烯烃(如柠檬烯)无硅氢加成产物生成.
Pawluć课题组[25]利用1-甲基-2-咪唑甲醛与2-(1-甲基肼基)吡啶缩合形成的三齿Schiff碱配体, 与CoCl2配位得到的配合物6催化烯烃硅氢加成反应(Eq. 7).苯基硅烷仅生成加氢产物; Et3SiH作为底物时不发生反应; 乙烯基硅烷与苯基硅烷进行硅氢加成反应, 转化率为41%~99%, 但主要为加氢产物; 1-己烯或1-辛烯等直链烯烃与硅烷反应, 没有检测到加成产物, 烯烃仅发生异构化反应; 环己烯与苯基硅烷硅氢加成反应会生成脱氢产物.
|
|
(7) |
2, 6-二(芳基亚氨基)吡啶(PDI)配体7a~7d与CoCl2形成的配合物, 可在NaBHEt3的存在下催化烯烃与硅烷硅氢加成反应[26]. 7a与CoCl2的配合物催化效果最好, 催化4-甲基-苯乙烯与(EtO)2MeSiH, PhSiH3, Et2SiH2和PhMeSiH2的加成反应, 反马氏加成产物分离产率在78%以上; Ph2SiH2为底物仅得到50%产率; 以(EtO)3Si- H, (TMSO)2MeSiH为底物产率更低; Ph3SiH, EtSiH3, Cl3SiH作为底物则没有加成产物生成.催化1, 3-二烯和1, 4-二烯分别与PhSiH3, Ph2SiH2和PhMeSiH2发生硅氢加成反应[27], 可高选择性地得到单加成反马氏产物, 不受另一双键的影响.当反应底物为芳环共轭的二烯时, 形成马氏和反马氏加成产物.催化含芳基取代基烯烃与苯基硅烷反应的加成产率为86%~97%, 催化醚基烯烃硅氢加成反应, 产率89%~92%.
|
|
具有高自旋性的吡啶二亚胺双(羧酸盐)钴络合物可用于催化(EtO)3SiH与1-辛烯硅氢加成反应(Eq. 8)[28]. 1 mol%钴催化剂, 23 ℃下反应1 h, 除配合物8b外, 其他配合物催化该反应的转化率均在98%以上, 分离产率超过95%, 配合物8e的催化性能最佳, 烯烃转化率与加成产物选择性均在98%以上.催化剂具有良好的底物适应性, 含酰胺、叔胺和仲胺基团的烯烃与(EtO)3SiH在0.25 mol%催化剂作用下均可获得74%以上的产率.与苯乙烯加成反应可得到98%的分离产率; 该配合物还适用于催化含溴烯烃和长链烯烃的硅氢加成反应; 对含氧官能团如酯、醚、环氧化物和酮等不饱和化合物的硅氢加成反应同样适用.值得注意的是, 催化烯丙基缩水甘油醚与(EtO)3SiH硅氢加成反应具有较高的选择性, 未发现环氧化物开环副反应, 性能优于铂催化剂, 在工业上具有很高的实用价值.该产物已被广泛应用于表面涂料, 也应用于玻璃纤维行业使用的填缝剂, 其产品优于含胺基和硫基的产品, 因为胺基和硫基会导致变色和产生有气味的蒸汽, 而铂催化剂会导致烯烃的异构化和马氏加成产物的生成.
|
|
(8) |
Ge课题组[29]报道了由乙酰丙酮钴和膦或氮配体9a~9d得到的配合物催化的烯烃硅氢加成反应. 9a和9b与Co(acac)2形成的催化剂在室温下催化苯乙烯与PhSiH3加成仅能得到15%以下的转化率; 而9c与Co(acac)2形成的催化剂在室温下能得到98%以上的转化率, 并且具有更好的反马氏加成产物选择性.含氮配体9d与Co(acac)2可在50 ℃下催化苯乙烯得到98%以上的转化率和69%选择性.同样, 9c与Co(acac)2形成的催化剂可在50 ℃条件下催化苯乙烯与Ph2SiH2反应, 得到98%以上的转化率, 但选择性仅为36%.相同条件下, 9b仅能得到12%的转化率, 而9a可得到98%以上的转化率和98%的选择性. 9d作为配体时, 无催化活性.
|
|
利用氨甲基吡啶10a~10e和CoBr2作为催化剂可以催化乙烯基五甲基二硅氧烷和二甲氧基甲基硅烷硅氢加成反应(Eq. 9)[30]. 10a-CoBr2作催化剂可得到98%的反马氏加成产物; 10b作配体时可得到反马氏加成产物、脱氢偶联产物和加氢产物的混合物(三种产物的物质的量之比为10:27:15); 10c作配体没有催化活性, 含吸电子基团的10d作为配体时加成产率较低(45%), 而含给电子基团的10e作为配体时可得到98%的产率. 10f和10g作为配体时, 分别在22和80 ℃温度下均能得到大于98%收率.
|
|
(9) |
陆展课题组[31]合成的手性噁唑啉亚氨基(OIP)钴配合物11a~11h可催化苯乙烯硅氢加成反应, 以马氏加成产物为主(Eq. 10).以叔丁醇钠作为活化剂, 11a (5 mol%)催化苯乙烯和PhSiH3加成反应产物收率为73%, ee值为72%. 11c作催化剂时, 对映选择性增加至98.6%; 11f作催化剂时得到最高的产率90%与区域选择性98% ee, 乙醚作溶剂时催化效果最好.使用11f催化苯基硅烷与多种烯烃(含苯基、醚基、硫醚、三氟甲基、卤化物、缩酮、酯基、酰胺、胺基、酮、硅基和自由基醇), 对芳香族烯烃而言, 对位取代基底物的硅氢加成产率较高(83%~94%), 间位取代基底物加成产率为74%~84%, 邻位最低(62%), ee值为81%~99%.
|
|
|
|
(10) |
|
|
一系列亚胺吡啶咪唑啉钴络合物可催化芳香族烯烃的脱氢硅烷化反应得到具有高附加值的烯基硅烷, 反应具有高化学选择性、区域选择性和立体选择性的(Eq. 11)[32]. 3 mol%配合物12和9 mol% NaBHEt3在室温下催化苯乙烯与Ph2SiH2硅氢加成反应, 10 min后得到烯基硅烷的产率为77%.
|
|
(11) |
黄正课题组[33]报道了喹啉噁唑啉钴配合物13催化共轭二烯与硅烷的不对称1, 2-马氏硅氢加成反应(Eq. 12).加成产物具有高区域选择性和对映选择性, 并能够催化多种共轭二烯, 包括具有芳基和烷基取代的单烯或1, 2-二取代二烯, 该催化剂催化呋喃环基芳香共轭二烯得到最高产率(96%), ee值为88%;催化甲硫基芳香共轭二烯可以获得最好的产物ee值(96%), 收率为88%.
|
|
(12) |
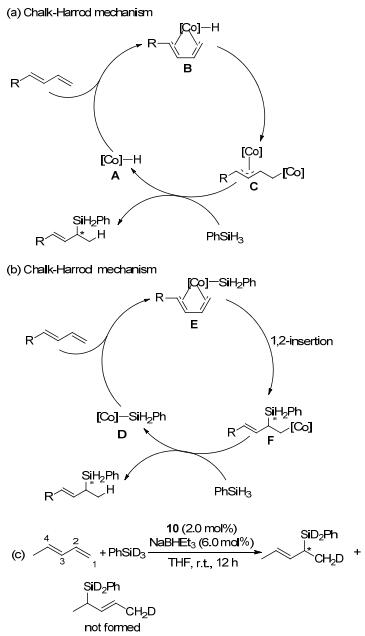
经过实验和计算确定反应历程是按改进型Chalk- Harrod机理进行(Scheme 2), 传统的Chalk-Harrod机理是烯烃插入Co—H键, 必须通过加氢金属化形成的π-烯丙基钴中间体C进行, 而改进型Chalk-Harrod机理是烯烃插入Co—Si键, 通过由1, 2-插入1, 3-二烯末端的C=C键变成Co—Si键(F)进行.例如, 1, 3-戊二烯与PhSiD3反应生成单一产物CH3CH=CHCH(SiPhD2)- CH2D, 其中D和Si原子分别添加到1, 3-戊二烯的C(1)和C(2)位置(Scheme 2c).该反应途径与传统的Chalk- Harrod机理相对立, 否则烯丙基中间体[Co]-(CH3CH- CHCHCH2D) (C)会按1:1的比例生成CH3CH=CH- CH(SiPhD2)CH2D与CH3CH(SiPhD2)CH=CHCH2D.因此, 改进型Chalk Harrod机理加成反应速度决定步骤就是将1, 3-二烯的末端双键1, 2-插入Co—Si键(E→F)[33].
2014年, 邓亮课题组[34]报道了钴配合物14可催化1-辛炔与Ph2SiH2 (1.2 equiv.)的反应, 在室温下反应3 h可得到96%的β-(E)加成产物(Eq. 13).当催化剂量降至0.1 mol%时, 催化反应12 h仍可得到95%的产率; 催化剂量为0.01 mol%时, 反应48 h仅得到12%产率.同时, 还测试了Ph2SiH2与其他末端炔烃反应.结果表明, 9种末端炔烃都能高选择性地生成β-(E)加合物.在苯基和烷基取代的炔烃硅氢加成中, E-乙烯基硅烷产率可以达到88%.具有氯、胺、烯烃和酯官能基团的炔烃也能高效地生成E-乙烯基硅烷(产率70%~82%); 催化1, 6-二炔硅氢加成得到1, 2-二亚烷基环戊烷, 产率为56%~62%.
|
|
(13) |
吡啶(双噁唑啉)钴络合物15a和15b能催化Ph2SiH2与苯乙炔反应(Eq. 14)[35]. 15b具有更好的催化活性和选择性, 可得到94%的产率.相同条件下, 15b能够催化多种末端芳基炔烃与Ph2SiH2进行硅氢加成反应, 得到具有高区域选择性的α-乙烯基硅烷, 选择性均高于92%.带有给电子基团或吸电子基团的芳基炔烃作为底物时能得到较高产率.具有邻位取代基的底物得到的产率不如对位或间位取代的同类炔烃卤素取代芳基炔烃, 对溴、对氯苯乙炔加成产物选择性高达99%, 且未观察到脱卤产物.催化叔胺、醚、酯和酮等官能团取代的杂化芳烃如3-乙炔基噻吩和3-乙炔基吡啶与Ph2SiH2反应, 产率分别为83%和55%, 区域选择性分别为97%和94%.
|
|
(14) |
该课题组[36]将乙酰丙酮钴与双噁唑啉基吡啶生成的催化剂16a~16f用于催化苯乙炔与手性二氢硅烷Ph(Ar)- SiH2 (Ar=2, 6-Me2C6H3)反应(Eq. 15).在25 ℃条件下反应, 16a催化苯乙炔硅氢加成可得到支链乙烯基硅烷, 分离产率为97%, 选择性为98%, 但对映选择性只有22%. 16b作配体时, 加成产物产率为82%, 选择性为97%.使用16c为配体时, 对映选择性有所增强, 但产率降低为58%.使用配体16d时, 产率更低(<10%).配体16e的催化选择性最强, 可达到99%, 同时还获得82% ee.甲基叔丁基醚作为溶剂催化反应效果最佳.降低反应温度可以进一步改善对映选择性和区域选择性, 但催化反应活性降低.
|
|
(15yj) |
陆展课题组[37]报道了亚氨基吡啶二氯化钴配合物17催化Ph2SiH2与1-(3-(烯丙氧基)丙-1-炔基)苯硅氢加成反应, 得到环化加成产物, 产率为71% (Eq. 16). NaBHEt3作为还原剂催化效果最佳, 溶剂对反应影响不大.将反应物浓度降低至0.125 mol/L可使加成产物产率增加至81%.其催化18种功能化炔烃的产率均在51%以上.以Me2PhSiH为底物, 加成产物产率降低.
|
|
(16) |
在前期工作基础上, 他们[38]又合成了一系列亚氨基吡啶类钴配合物18a~18e, 其中噁唑啉基亚氨基吡啶类配体18c与溴化钴形成的配合物催化活性最高. 6 mol% NaHBEt3, 2 mol%钴配合物, 以四氢呋喃(THF)为溶剂催化苯乙炔硅氢加成, 室温下反应5 min, 加成产物产率为90%, ee值为97%. 18c还能催化非端位芳香烷基炔烃的硅氢加成反应, 加成产率为79%~90%.转化速率(TOF)最高为65520 h-1.
|
|
CoCl2-19a和15 mol% NaBHEt3催化苯乙炔与Ph2SiH2反应得到硅氢加成产物, 产率为44%, 支链加成产物和线性加氢产物的混合产率为37% (Eq. 17)[39].当噁唑啉上的基团为异丙基时(19b), 硅氢加成产物和加氢产物总产率为32%, 二者物质的量之比为93:7.使用2, 6-二甲基亚胺(19c)时, 选择性得到明显改善, 支链加成产物产率为83%, rr值为99:1, ee值为80%.使用2, 4, 6-三甲基亚胺时(19d), 反应生成支链加氢产物的对映选择性增加至88%, 未检测到乙烯基硅烷.噁唑啉基团上具有较小的甲基配体(19e)时反应仅得到支链加氢产物, 产率为91%, ee值为88%.
|
|
(17) |
具有咪唑基团的NNN钴配合物20催化末端炔烃与硅烷硅氢加成反应, 产物以马氏加成为主.反应也需要加入NaBHEt3作为活化剂, 该催化体系可催化支链炔烃、芳环炔烃、含卤化物、酯、酰胺、醚、含甲硅烷基醇、游离醇和二草酸酯等炔烃与硅烷在室温下进行加成反应, 加成产物分离产率为51%~96%[40].利用该催化剂催化炔烃的选择性双硅氢加成反应[41], 以炔为氢源, 催化一种或两种不同的硅烷双硅氢加成反应, 该催化系统可催化各种功能化的底物, 得到相应的包含4个可变Si-H键的双二氢硅基烷烃, 产率51%~96%.
用CoBr2•Xantphos和CoBr2•OIP(噁唑啉-亚氨基吡啶)催化脂族炔进行双硅烷硅氢加成反应, 具有高对映选择性(Eq. 18)[42].催化Ph2SiH2和PhSiH3与4-苯基-1-丁炔硅氢加成反应, 48 h后分离产率为84%, ee值为99.9%.
|
|
(18) |
Ge课题组[43]用2, 6-二亚胺吡啶类配体与Co(OAc)2或Co(acac)2生成的催化剂21在室温下THF中催化苯乙炔与PhSiH的硅氢加成反应(Eq. 19). 21a与Co(acac)2作催化剂, 催化反应24 h, 苯乙炔的转化率为74%. 21a和Co(OAc)2作催化剂, 苯乙炔在12 h内完全转化, 得到比例为65:34的Z/E异构体混合物; 反应4.5 h, 苯乙炔完全转化, 但几乎全部得到Z-异构体.这表明该钴配合物催化炔烃硅氢加成反应是Z-选择性的, 并通过Z-乙烯基硅烷的异构化产生E-乙烯基硅烷.相同条件下, 以21a/Co(OAc)2作为催化剂, 加入10 mol%苯酚作为助剂, 可得到98%的对映选择性, 苯酚的加入使炔1, 2-插入Co—Si键更有利, 并且优先形成α-乙烯基硅烷, 抑制了Z-乙烯基硅烷的Z/E-异构化. 21b~21g/Co(OAc)2作为催化剂时, 除21g转化率较低, 其他配体均能催化底物全部转化, 21a的选择性最高(98%).
|
|
(19) |
Co(acac)2与膦配体催化末端炔烃与苯基硅烷加成反应, 能高效生成相应的(E)-乙烯基硅烷(Eq. 20)[44]. 7b作为配体时, 单加成反马氏产物的选择性最高, 能达到83%. PhSiH3用量为1.5 equiv.时, 单加成产物选择性为98%; PhSiH3用量为0.5 equiv.时, 双加成产物选择性为98%, 转化率为93%.在优化反应条件下, 考察了催化剂的底物适应性, 取代基为含有吸电子基团的芳香族末端炔烃和脂肪族取代基炔烃具有较高的反应活性, 得到相应的E-乙烯基硅烷, 产率在59%以上. Ph2SiH2作底物时, Co(acac)2/22e催化这些末端炔烃没有反应.而用Co(acac)2/22f作为催化剂时, 反应在室温下快速进行.
|
|
(20) |
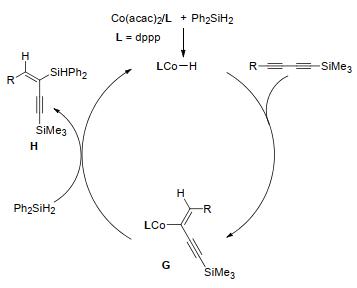
Co(acac)2与dppp共催化对称与不对称1, 3-二炔和Ph2SiH2硅氢加成反应, 生成1, 3-烯炔[45].多数甲硅烷基取代的1, 3-二炔在2 mol%催化剂存在下可与Ph2SiH2反应得到相应的双(甲硅烷基)取代的1, 3-烯炔, 分离产率为70%~90%, 区域选择性与立体选择性>99%.对称的1, 3-二炔、一系列1, 4-双(芳基)或1, 4-双(烷基)取代的1, 3-二炔也能高效反应生成相应的1, 3-烯炔.经计算和实验得出Co(acac)2/dppp催化1, 3-二炔的反应机理(Scheme 3):在dppp存在下, 用Ph2SiH2活化Co(acac)2会生成氢化钴配合物(L)Co-H, 然后将1, 3-二炔迁移插入Co—H键会生成乙烯基钴G, 随后G与Ph2SiH2反应生成产物H, 并生成活性中间体(L)Co—H, 从而进入下一催化循环.
2018年, 杨勇课题组以新型NNN(胺-吡啶-亚胺)API钳形配体与CoX2 (X=Br, Cl)配位生成的配合物为催化剂, 催化苯乙炔与硅烷反应[46].以NaBHEt3为活化剂, 2 mol%钴催化剂23a~23f, 室温条件下反应可得到99%以上转化率, 以α-乙烯基硅烷为主, 其中23d得到最高的α-乙烯基硅烷产率(90%), 23a活性最低, 仅得到63%的加成产物产率.该配合物催化其他炔类硅氢加成反应时, 由于苯乙炔空间位阻效应, 苯环对位或间位有取代基的底物转化率和α选择性比邻位取代基底物要高.卤素取代的苯基炔烃也能高选择性地得到马氏加成产物, 无脱卤产物生成, 间氯苯乙炔作底物加成产率为82%, 选择性为93%.杂芳族炔烃, 如3-乙炔基吡啶和2-乙炔基噻吩作为底物, 得到的马氏加成产物选择性也较高, 分别为94%和92%. 1, 4-二乙炔苯与PhSiH3反应得到二乙烯基硅烷产物产率为58%, 具有良好的区域选择性(93%).
|
|
亚胺基吡啶类配体24与Co(OAc)2于30 ℃条件下催化炔烃硅氢加成反应可高选择性得到马氏加成产物(Eq. 21)[47].苯环上含有不同官能团的苯乙炔与PhSiH3硅氢加成反应得到马氏加成产物分离产率为74%~96%.催化含有胺、腈基、羰基、卤素、氮和硫类炔烃、烯炔、内部炔烃得到马氏加成产物产率较高; 除具有吸电子取代基的芳族炔烃产率较低外, 催化杂芳族炔烃和内炔烃与Ph2SiH2也能得到中等以上产率的马氏加成产物.
|
|
(21) |
陈大发课题组[48]用三联吡啶配体钴配合物25催化Ph2SiH2与炔烃硅氢加成反应. 2 mol%配合物, 6 mol% NaBHEt3, THF作溶剂, 催化苯乙炔硅氢加成反应室温下反应5 min可得到86%分离产率.催化具有给电子基团的苯乙炔衍生物可得到89%~96%的分离产率.其他吸电子基团, 如氯、溴和三氟甲基则得到较低的产率, 分离产率为66%~83%.其他杂芳族炔烃, 例如2-噻吩或3-吡啶炔烃也能得到高产率与高区域选择性.链烯基和烷基取代的末端炔烃虽然转化率较高, 但产物区域选择性较低.内二烷基炔硅氢加成可得到良好的产率和产物立体选择性(80%~86%).
|
|
Nishiyama课题组[49]报道了用双(噁唑啉基苯基)胺配体与乙酸钴作催化剂还原各种酮生成相应醇的研究结果(Eq. 22).催化联苯乙酮与二乙氧基甲基硅烷硅氢加成反应, 发现用26a~26d作配体, 底物转化率均可达到99%, 26d作配体选择性最高(94%).
|
|
(22) |
Gade课题组[50]基于1, 3-双(2-吡啶基亚氨基)异吲哚啉(BPI)骨架, 从非手性廉价原料出发设计了具有不同空间结构的配体, 合成了新型手性三齿单阴离子NNN-钳形配体(Eq. 23).使用吡啶烷基钴前体络合物与之反应得到相应钴烷基络合物27.该配合物催化手性烷基芳基酮的不对称硅氢加成反应得到手性醇, 产率高达100%, ee值达91% (Eq.23).最优反应条件为: THF作溶剂, 催化剂用量为2.5 mol%, 温度为15 ℃, 最低反应时间为8 h, Me(EtO)2SiH与苯乙酮反应得到产物, 产率>99%, 91% ee.
|
|
(23) |
Flork课题组[51]通过硫代水杨醛与CoMe(PMe3)4反应合成硫配位酰基(氢)钴(III)配合物28, 能够在温和条件下有效催化醛和酮的硅氢加成反应(Eq. 24).催化苯甲醛、2-糠醛以及溴、羟基和甲氧基取代的苯甲醛在40 ℃条件下反应2 h, 均可完全转化生成加成产物.在相同条件下, 具有吸电子基团的底物反应活性依次为: 4-氟苯甲醛>2-氯苯甲醛>2, 6-二氯苯甲醛.相同反应条件下, 催化酮的转化率较低, 均在37%以下; 脂族酮如环己酮作底物则无加成产物生成.杂环芳族酮比苯基和脂肪族酮具有更高的反应活性.
|
|
(24) |
2015年, Li课题组[52]合成的N-亚苄基-萘胺(CNC钳形配体)氢化钴配合物29可催化醛和酮硅氢加成反应(Eq. 25). 29用量为1 mol%, 苯乙醛转化率为96%.催化苯甲醛、对氟苯甲醛和1-萘甲醛硅氢化反应得到相应的醇, 产率分别为93%、93%和91%.催化糠醛也可获得88%的分离产率.由于空间位阻效应, 邻位取代的醛如2-氯苯甲醛、2-氟苯甲醛和2-溴苯甲醛反应活性较低, 需增加催化剂量和延长反应时间才能还原得到相应的醇.催化苯乙醛则需催化剂量为5 mol%, 反应16 h, 苯乙醇产率为78%.若以酮为底物, 反应速率明显降低, 即使再提高催化剂量或延长反应时间, 转化率也无明显提高.
|
|
(25) |
陆展课题组[53]合成的氨基苯基噁唑啉基苯胺(IPOPA) (Eq. 26)与CoCl2作催化剂, 催化酮硅氢加成反应, 获得具有高对映选择性的手性醇. 30a催化效果最好, 2.5 mol% CoCl2, 4 mol%配体和2.5 mol% NaHBEt3, 催化苯乙酮硅氢加成反应得到手性醇产率高达99%, 同时具有97% ee值.催化剂量降至0.5 mol%, 产率也可达到96%, ee值为97%.在邻位, 间位和对位上有卤化物、醚、酯、硫醚、甲硅烷基醚和三氟甲基等给电子和吸电子取代基的乙酰基苯, 也可以顺利地进行不对称催化硅氢加成反应, 得到65%~98%的分离产率和93%~99% ee值.使用长链烷基代替甲基, 具有96%~98% ee值.空间位阻较大的酮, 如异丁基苯和叔丁基苯酮作为底物时反应活性较低.杂环底物, 如2-萘基、苯并噻吩、苯并呋喃和2-乙酰基吡啶作为底物可有效地反应得到相应的醇, 分离产率为80%~97%.
|
|
(26) |
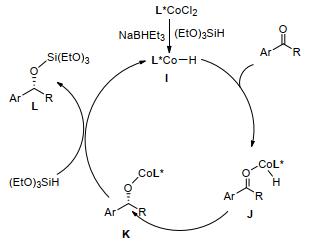
该反应的催化机理(Scheme 4)为[53]: NaBHEt3加速IPOPA-CoCl2去质子化, 形成IPOPA-CoCl2中间体, IPOPA-CoCl2中间体与(EtO)3SiH进行氢化物-氯化物交换生成氢化钴物质I, I再与酮配位形成中间体J.然后, 将酮迁移插入Co—H键中生成配合物K, (EtO)3SiH插入配合物K反应生成活性钴物质, 同时生成的氢硅烷化产物L从催化剂上离去, 该产物在碱性条件下进行了硅烷化反应, 得到相应的醇.
Fenske课题组[54]合成了钴配合物31 (Eq. 27), 并将其用于催化醛酮硅氢加成反应, 具有吸电子基团或给电子基团的芳香族醛都可以发生硅氢加成反应. THF作溶剂, 2 mol%催化剂, 80 ℃条件下反应4 h, 分离产率可达到77%~92%.以1-萘甲醛为底物, 转化率93%, 产物分离产率为88%.芳族酮作为反应底物的转化率和产物分离产率都低于醛; 脂族酮的转化率明显低于芳族酮的转化率, α, β不饱和酮比一般的酮化合物更具有反应活性.
|
|
(27) |
综上所述, 用于硅氢加成反应的钴配合物由最初的羰基类钴配合物逐渐发展成含氮、膦元素的配合物, 尤其是近10年内主要形成了NNN型钴配合物、NPN型钴配合物和POP型钴配合物三大类催化剂.这些钴配合物催化烯烃、炔烃和醛酮等不饱和化合物硅氢加成反应表现出优异的催化性能, 可以得到较高的转化率、立体选择性或区域选择性, 取得了长足的进步.
但是, 已报道的钴配合物种类还不够丰富, 作为硅氢加成催化剂的应用范围还十分有限, 仍然面临很大的挑战.大多数钴催化剂对空气较为敏感, 不能长期稳定保存.钴催化剂整体的催化活性不够高, 大部分钴配合物作为催化剂需要借助强还原剂才能发挥较好的催化作用, 这样的反应条件很难应用到实际工业生产中.催化硅氢加成反应选择性问题也是目前需要解决的一个重要问题, 例如炔烃硅氢加成会产生马氏产物、Z-式反马氏产物和E-式马氏产物.目前, 为了提高加成产物的区域选择性, 通常会提高催化剂用量或加入助剂.
因此, 简化该类催化剂的配体结构以及合成步骤, 合成稳定的催化活性高的催化剂将是该领域研究的总方向.另外, 含氮配体具有更好的助催化活性, 相对于于膦配体而言更环保, 将是以后的重要发展方向之一.提高钴催化剂的活性、构建不加还原剂的催化剂体系和实现无溶剂催化也是今后主要发展研究方向之一.将钴催化剂从均相到多相催化剂发展, 合成可回收重复使用的钴催化剂, 实现钴催化硅氢加成反应的绿色化反应将是该领域研究的重要内容和长远目标.
Marciniec, B.; Gulinski, J.; Urbaniak, W.; Kornetka, Z. W. In Comprehensive Handbook on Hydrosilylation, Pergamon, Oxford, U. K., 1992.
Ojima, I. In the Chemistry of Organic Silicon Compounds, Vol. 1, Wiley, Chichester, U. K., 1989, Chapter 25.
Roy, A. K. Adv. Organomet. Chem. 2007, 55, 1. doi: 10.1016/S0065-3055(07)55001-X
Speier, J. L.; Webster, J. A.; Barnes, G. H. J. Am. Chem. Soc. 1957, 79, 974. doi: 10.1021/ja01561a054
Karstedt, B. D. US 3775452, 1973. https://www.researchgate.net/publication/291785390_Platinum_complexes_of_unsaturated_siloxane_and_platinum_containing_organopolysiloxanes
Dong, H.; Berke, H. Adv. Synth. Catal. 2009, 351, 1783. doi: 10.1002/adsc.200900246
Ojima, I.; Kogure, T.; Nagai, Y. Tetrahedron Lett. 1974, 15, 1889. doi: 10.1016/S0040-4039(01)82586-7
Brunner, H.; Becker, R.; Riepl, G. Organometallics 1984, 3, 1354. doi: 10.1021/om00087a006
张琪, 刘奥, 于海珠, 傅尧, 化学学报, 2018, 76, 113. doi: 10.3866/PKU.WHXB201707101Zhang, Q.; Liu, A.; Yu, H.; Fu, Y. Acta Chim. Sinica 2018, 76, 113(in Chinese). doi: 10.3866/PKU.WHXB201707101
Yang, X.; Wang, C. Chin. J. Chem. 2018, 36, 1047. doi: 10.1002/cjoc.201800367
Harrod, J. F.; Chalk, A. J. J. Am. Chem. Soc. 1965, 87, 1133. https://www.researchgate.net/publication/231482881_Reactions_between_Dicobalt_Octacarbonyl_and_Silicon_Hydrides
Sun, J.; Deng, L. ACS Catal. 2016, 6, 290. doi: 10.1021/acscatal.5b02308
Du, X.; Huang, Z. ACS Catal. 2017, 7, 1227. doi: 10.1021/acscatal.6b02990
Obligacion, J. V.; Chirik, P. J. Nat. Rev. Chem. 2018, 2, 15. doi: 10.1038/s41570-018-0001-2
Chen, J.; Guo, J.; Lu, Z. Chin. J. Chem. 2018, 36, 1075. doi: 10.1002/cjoc.201800314
Chen, J.; Lu, Z. Org. Chem. Front. 2018, 5, 260. https://www.researchgate.net/publication/324160421_Lithocarpins_A-D_Four_Tenellone-Macrolide_Conjugated_42_Hetero-adducts_from_the_Deep-sea_Derived_Fungus_Phomopsis_lithocarpus_FS508
Zaranek, M.; Pawluc, P. ACS Catal. 2018, 8, 9865. doi: 10.1021/acscatal.8b03104
Magomedov, G. K. I.; Andrianov, K. A.; Shkolnik, O. V.; Izmailov, B. A.; Kalinin, V. N. J. Organomet. Chem. 1978, 149, 29. doi: 10.1016/S0022-328X(00)90374-7
Brookhart, M.; Grant, B. E. J. Am. Chem. Soc. 1993, 115, 2151. doi: 10.1021/ja00059a008
Mo, Z.; Liu, Y.; Deng, L. Angew. Chem., Int. Ed. 2013, 52, 10845. doi: 10.1002/anie.201304596
Liu, Y.; Deng, L. J. Am. Chem. Soc. 2017, 139, 1798. doi: 10.1021/jacs.6b12938
Gao, Y.; Wang, L.; Deng, L. ACS Catal. 2018, 8, 9637. doi: 10.1021/acscatal.8b02513
Chen, C.; Hecht, M. B.; Kavara, A.; Brennessel, W. W.; Mercado, B. Q.; Weix, D. J.; Holland, P. L. J. Am. Chem. Soc. 2015, 137, 13244. doi: 10.1021/jacs.5b08611
Ibrahim, A. D.; Entsminger, S. W.; Zhu, L.; Fout, A. R. ACS Catal. 2016, 6, 3589. doi: 10.1021/acscatal.6b01091
Gorczyński, A.; Zaranek, M.; Witomska, S.; Bocian, A.; Stefankiewicz, A. R.; Kubicki, M.; Patroniak, V.; Pawluć, P. Catal. Commun. 2016, 78, 71. doi: 10.1016/j.catcom.2016.02.009
Raya, B.; Biswas, S.; Rajanbabu, T. V. ACS Catal. 2016, 6, 6318. doi: 10.1021/acscatal.6b02272
Raya, B.; Jing, S.; Balasanthiran, V.; RajanBabu, T. V. ACS Catal. 2017, 7, 2275. doi: 10.1021/acscatal.6b03373
Schuster, C. H.; Diao, T.; Pappas, I.; Chirik, P. J. ACS Catal. 2016, 6, 2632. doi: 10.1021/acscatal.6b00304
Wang, C.; Teo, W. J.; Ge, S. ACS Catal. 2017, 7, 855. doi: 10.1021/acscatal.6b02518
Lee, K. L. Angew. Chem., Int. Ed. 2017, 56, 3665. doi: 10.1002/anie.201612460
Cheng, B.; Lu, P.; Zhang, H.; Cheng, X.; Lu, Z. J. Am. Chem. Soc. 2017, 139, 9439. doi: 10.1021/jacs.7b04137
程彪, 陆鹏, 赵家金, 陆展, 有机化学, 2019, 39, 1704. doi: 10.6023/cjoc201903018Cheng, B.; Lu, P.; Zhao, J.; Lu, Z. Chin. J. Org. Chem. 2019, 39, 1704(in Chinese). doi: 10.6023/cjoc201903018
Wen, H.; Wang, K.; Zhang, Y.; Liu, G.; Huang, Z. ACS Catal. 2019, 9, 1612. doi: 10.1021/acscatal.8b04481
Mo, Z.; Xiao, J.; Gao, Y.; Deng, L. J. Am. Chem. Soc. 2014, 136, 17414. doi: 10.1021/ja510924v
Zuo, Z.; Yang, J.; Huang, Z. Angew. Chem. 2016, 128, 10997. doi: 10.1002/ange.201605615
Wen, H.; Wan, X.; Huang, Z. Angew. Chem., Int. Ed. 2018, 57, 6319. doi: 10.1002/anie.201802806
Xi, T.; Lu, Z. J. Org. Chem. 2016, 81, 8858. doi: 10.1021/acs.joc.6b01555
Guo, J.; Lu, Z. Angew. Chem. 2016, 128, 10993. doi: 10.1002/ange.201605501
Guo, J.; Shen, X.; Lu, Z. Angew. Chem. 2017, 129, 630. doi: 10.1002/ange.201610121
Cheng, Z.; Xing, S.; Guo, J.; Cheng, B.; Hu, L.; Zhang, X.; Lu, Z. Chin. J. Chem. 2019, 37, 457. doi: 10.1002/cjoc.201900079
Cheng, Z. Chin. J. Chem. 2019, 37, 632. doi: 10.1002/cjoc.201900129
Guo, J.; Wang, H.; Xing, S.; Hong, X.; Lu, Z. Chem 2019, 5, 881. doi: 10.1016/j.chempr.2019.02.001
Teo, W. J.; Wang, C.; Tan, Y. W.; Ge, S. Angew. Chem., Int. Ed. 2017, 56, 4328. doi: 10.1002/anie.201700868
Wu, C.; Teo, W. J.; Ge, S. ACS Catal. 2018, 8, 5896. doi: 10.1021/acscatal.8b01410
Sang, H.; Hu, Y.; Ge, S. Org. Lett. 2019, 21, 5234. doi: 10.1021/acs.orglett.9b01836
Zhang, S.; Ibrahim, J. J.; Yang, Y. Org. Lett. 2018, 20, 6265. doi: 10.1021/acs.orglett.8b02746
Zong, Z.; Yu, Q.; Sun, N.; Hu, B.; Shen, Z.; Hu, X.; Jin, L. Org. Lett. 2019, 21, 5767. doi: 10.1021/acs.orglett.9b02254
Kong, D.; Hu, B.; Chen, D. Chem.-Asian J. 2019, 14, 2694. doi: 10.1002/asia.201900577
Inagaki, T.; Phong, L.T.; Furuta, A.; Ito, J.; Nishiyama, H. Chem.-Eur. J. 2010, 16, 3090. doi: 10.1002/chem.200903118
Sauer, D. C.; Wadepohl, H.; Gade, L. H. Inorg. Chem. 2012, 51, 12948. doi: 10.1021/ic3020749
Niu, Q.; Sun, H.; Li, X.; Klein, H. F.; Flork, U. Organometallics 2013, 32, 5235. doi: 10.1021/om4005687
Zhou, H.; Sun, H.; Zhang, S.; Li, X. Organometallics 2015, 34, 1479. doi: 10.1021/om5011929
Chen, X.; Lu, Z. Org. Lett. 2016, 18, 4658. doi: 10.1021/acs.orglett.6b02260
Yang, F.; Wang, Y.; Lu, F.; Xie, S.; Qi, X.; Sun, H.; Li, X.; Fuhr, O.; Fenske, D. New J. Chem. 2018, 42, 15578. doi: 10.1039/C8NJ02979B

图 1 双卡宾钴配合物催化烯烃硅氢加成反应
Figure 1 Hydrosilylation of olefins catalyzes by bis(carbene) cobalt(I)-dinitrogen complex

图 2 Chalk-Harrod机理与改进型Chalk-Harrod机理
Figure 2 Simplified Chalk-Harrod and modified Chalk-Harrod mechanisms


 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




























 下载:
下载:

 下载:
下载: