
图 1.
含手性芳基醇骨架的生物活性分子
Figure 1.
Bioactive compounds containing chiral aryl alcohol

手性芳基醇结构广泛存在于许多生物活性分子及天然产物中, 如抗抑郁药物西酞普兰(4)[1a]、抗组胺药氯马斯汀(5)[1b]、止咳药氯苯达诺(6)[1c]等都含有手性芳基醇骨架.此外, 对手性醇类化合物进行简单的衍生化还可以得到其他类型的药物分子如抗过敏药西替利嗪等(图 1). “沙利度胺”事件之后, 单一对映体的手性药物研究受到学术界和工业界的重视[2].鉴于手性芳基醇类化合物及其衍生物展现出的广泛的生物活性, 以及它们作为重要手性合成砌块在不对称合成中的应用, 发展高效构建手性芳基醇骨架的方法对手性药物合成以及新药创制都具有十分重要的意义.
与烯烃的不对称双羟化[3a, 3b]、手性硼酸酯的氧化[3c]、环氧化合物的开环[3d, 3e]以及醇的去对称化[3f]等构建手性醇的方法相比, 有机金属试剂如格氏试剂、锂试剂、有机锌试剂及有机锡试剂等对羰基化合物的不对称加成, 是构建手性醇类特别是手性叔醇类化合物最简单直接的方法[4].然而这些有机金属试剂存在着制备较为繁琐, 对水和空气敏感, 反应需要在较低的温度下进行, 官能团耐受性差, 以及底物适应性差等特点, 很大程度上限制了该类方法的应用.
1998年Miyaura小组[5]首次报道了铑催化的芳基硼酸对醛的不对称1, 2-加成, 尽管反应的对映选择性并不理想(41% ee), 但芳基硼酸方便易得、低毒、对水和空气稳定、官能团耐受性好等优点引起化学家们的极大关注和兴趣[6].发展高效的催化体系, 解决反应的立体选择性控制困难的问题成为该领域研究的焦点.在过去的二十年里, 化学家们发展了各种不同类型的催化体系来实现该类反应的立体选择性控制, 本文详细归纳了这一领域的研究进展和取得的结果.
1998年Miyaura小组[5]报道了Rh(Ⅰ)催化的芳基硼酸对醛的加成, 使用膦配体反应可以取得非常好的收率, 之后他们利用(S)-MeO-MOP (L1)作为配体进行了不对称加成的尝试, 得到41% ee值的产物12, 首次实现了铑催化的芳基硼酸对醛的不对称加成, 开启了过渡金属催化的有机硼试剂对羰基化合物的不对称加成研究的先河, 他们认为在这个反应中水作为质子源可以促进反应的进行(Eq. 1).
|
|
(1) |
在接下来的几年里, 过渡金属催化的芳基硼酸对醛的不对称加成一直没有报道.直到2005年, Bolm等[7]将平面手性的咪唑鎓盐作为N杂环卡宾(NHC)的前体用于Rh(Ⅱ)催化的芳基硼酸对醛的不对称加成, 反应可以取得最高38% ee值的产物.研究发现, 配体中的二苯基氧膦对反应的立体控制很重要, 将二苯氧膦换成甲氧基时反应的立体选择性会出现下降, 在咪唑基团的氮原子上引入手性基团对部分底物的立体选择性的控制有一定的帮助(Eq. 2).
|
|
(2) |
2006年, Aoyama课题组[8]报道了Rh(Ⅰ)/(R, R)-iPr-Duphos催化的芳基硼酸对芳基醛的不对称加成, 在100 ℃的加热条件下, 反应可以以优异的收率得到最高66% ee值的产物.同年, 该课题组又利用手性双膦配体L5实现了三价铑催化的芳基硼酸对芳基醛的不对称加成, 反应能取得最高85% ee.有意思的是, ESI-MS显示一价铑/膦配体的钠盐可能是其催化的活性形式, 该催化剂的形成原因可能是膦配体在将三价铑还原成了一价铑的同时自身被部分氧化成氧膦(Eq. 3).
|
|
(3) |
2006年, 周其林课题组[9]将基于手性螺环骨架的单膦配体L6用于一价铑催化的芳基硼酸对醛的不对称加成反应之中, 取得最高87% ee值的结果.该催化体系表现出较好的底物适应性, 对邻位取代的芳基醛以及芳基硼酸都可以取得非常优异的收率, 而且对于硝基、酯基、卤素以及杂环都能够耐受.值得一提的是, 该催化体系对于兼具1, 2-和1, 4-加成反应位点的肉桂醛也有很好的化学选择性, 反应可以以96%收率和75% ee得到单一的1, 2-加成产物(Eq. 4).
|
|
(4) |
同年, Minnaard等[10]报道了Rh(Ⅰ)/手性双齿亚磷酰胺L7催化的芳基硼酸对醛的不对称加成反应, 可以取得最高75% ee (Eq. 5).与单齿亚磷酰胺配体相比, 乙二胺桥连的双齿亚磷酰胺配体能实现更好地立体选择性控制; 同时相比其他溶剂, 异丙醇能给出更好的转化率, 这可能与其作为溶剂可以更好地促进中间体硼酸酯18转变成产物有关(Scheme 1).
|
|
(5) |
2007年, Van der Eycken等[11a]利用Rh(Ⅰ)/[2.2.1]庚二烯骨架的手性双烯配体实现了芳基硼酸对醛的不对称加成, 他们发现在桥环庚二烯骨架上引入取代基有利于催化剂反应活性的提高, 而使用醇类溶剂能取得最优的立体选择性(Eq. 6).
|
|
(6) |
2009年, Hayashi课题组[11b]将他们发展的[2.2.2]辛三烯骨架的手性双烯配体L9用于醛的不对称芳基化反应中, 在温和的条件下反应就能顺利进行, 对各种取代的芳基硼酸以及芳基醛都表现出较好的适应性.他们认为桥环上四氟苯基的引入可以减小二面角的大小以及调整配体的电性, 从而可以提高配体与过渡金属配位的能力.此外该反应体系在间苯二醛的不对称双芳基化中也能取得不错的结果(Eq. 7).
|
|
(7) |
2010年, Amii小组[12]发展了一类基于手性联二萘酚(BINOL)骨架的含氟代醇的单膦配体, 该配体在Rh(Ⅰ)催化的芳基硼酸对醛的不对称加成反应中可以实现中等到良好的立体选择性控制, 给出最高92% ee值的结果.他们发现配体L10中醇羟基以及氟原子的存在对于催化剂的反应活性和立体选择性的控制都起到十分重要的作用:当配体中的醇变成酸时, 催化剂反应活性消失; 当将与醇连接的三氟甲基变为氢原子或者甲基时, 反应的收率和立体选择性都出现了明显的下降.而变成甲基时, 反应的立体选择性甚至出现了翻转, 一方面可能是由于三氟甲基的引入使得羟基具有弱酸性, 从而增强了与醛羰基之间的相互作用, 另一方面三氟甲基也参与了过渡态面选择性的调控(Eq. 8).
|
|
(8) |
马玉道课题组[13]利用面手性的NHC配体实现了铑催化的芳基硼酸对醛的不对称加成的立体选择性控制, 该催化体系表现出非常好的反应活性, 在0.01 mol%催化剂用量的条件下, 反应也能取得91%的收率, 但是立体选择性并不理想, 模板反应最高只能取得41% ee的结果.对配体进行改造, 将苯环上的溴原子换为OMe时, 反应的ee值可以提高到52%, 尝试制备好的NHC-Rh配合物作为催化剂, 反应的立体选择性和收率反而出现明显的下降, 通过降低温度超声的方法可以在一定程度上提高反应的收率和立体选择性.此外, 他们发现直接使用RhCl3作为催化剂反应可以取得58% ee值.最后他们还对配体分子进行构象限制, 设计了乙二醇桥连的配体, 收率提高但选择性没有得到改善(Eq. 9).
|
|
(9) |
2016年, Li等[14]报道了Rh(Ⅱ)/手性苯并咪唑盐L11催化的醛的不对称加成的例子, 在温和的反应条件下大部分底物都能给出比较高的收率, 但最高也仅能取得56% ee值.而当改用醋酸钯作为催化剂时, 反应的收率和立体选择性相比醋酸铑都出现了明显的下降(Eqs. 10, 11).
|
|
(10) |
|
|
(11) |
2017年, 日本的Kamikawa和Ogasawara课题组[15]在他们之前发展的基于面手性π-芳烃-铬络合物结构的膦烯配体的基础上发展了一类在空气中更加稳定的基于锰的平面手性配体L12, 该配体除了在铑催化的芳基硼酸对烯基酮的不对称1, 4-加成中能取得非常好的收率和立体选择性外, 在苯硼酸对1-萘醛10的不对称1, 2-加成反应中也能取得很好的结果, 结果表明与环戊二烯相连的磷上的取代基对反应的收率和立体选择性的控制起到了十分关键的作用(Eq. 12).这是目前铑催化的醛的不对称芳基化取得的最好选择性结果.
|
|
(12) |
在目前已报道的芳基硼酸对醛的不对称加成的工作中, 铑是最为常见的金属催化剂, 除此之外, 其他地表含量更为丰富以及更为廉价的金属也被尝试用于醛的不对称催化加成反应中, 也取得了一些不错的结果.
2009年, Miyaura教授课题组[16a]最先报道了钌催化的芳基硼酸对醛的不对称加成, 以Me-BIPAM (L13)为配体,反应可以实现很好的立体选择性控制, 取得最高99% ee值.该反应体系对于杂环芳醛如2-噻吩甲醛, 2-呋喃甲醛甚至是6-甲氧基-吡啶-3-甲醛都能取得不错的结果, 这在其他的催化体系中较少见到(Eq. 13).作者在文中提到钌与双齿亚磷酰胺的配位模式可能和铑的平面四边形的配位模式有所区别.值得一提的是, 该催化体系同样可以实现烷基醛以及烷基酮酸酯的高效不对称加成[16b, 16c].
|
|
(13) |
2016年, Paquin小组[17]将上述催化体系进一步拓展到五氟巯基取代的芳基醛21的不对称加成反应中, 高效地构建了一系列五氟巯基取代的手性二芳基醇类化合物22, 取得最高98% ee值的结果(Eq. 14).
|
|
(14) |
汤文军课题组[18]利用钌/手性单膦配体L14的催化体系也实现了芳基硼酸对醛的不对称加成(Eq. 15).在机理探讨过程中, 他们发现产物的ee值和配体的ee值呈线性关系, 这表明催化剂中钌和单膦配体是1:1配位的, 通过对催化剂活性物种进行单晶培养发现, 钌除了和配体中的磷配位外, 还和其中与氧连接的苯基配位(Eq. 14).基于此方法, 他们以醛23为底物, 实现了选择性磷酸二酯酶抑制剂CDP-840关键中间体的合成(Scheme 2).
|
|
(15) |
最近, 汪君课题组[19]通过氧杂苯并降冰片烯的不对称膦氢化构建了一类含氧桥的手性膦配体, 并成功将其用于钌催化的芳基硼酸对醛的不对称加成反应中, 取得了中等到优秀的收率和对映选择性.研究发现钌和配体L15的比例很重要,两者摩尔比为1:1时的结果要优于1:2的结果, 机理研究表明钌同时与配体中的磷和桥环上的氧配位(Eq. 16).
|
|
(16) |
2007年, Aoyama[20a]报道了Ni(cod)2/(R, R)-Et-Du- phos (L16)催化的芳基硼酐对芳基醛的不对称加成的例子, 在硼酐作为硼源, NaOtBu作为碱,100 ℃条件下, 反应可以以最高93%的收率以及78% ee得到手性的二芳基醇类化合物.随后, 他们用方便易得的芳基氟硼酸钾代替硼酐, 发现反应在无外加碱的条件下也能顺利进行, 并能给出最高81% ee值的结果[19b](Eq. 17).
|
|
(17) |
2010年, Cheng课题组[21]利用廉价的钴作为催化剂实现了芳基硼酸对醛的加成, 在以(R, R)-BDPP (L17)作为手性配体的反应条件下, 可以取得最高99% ee的结果, 该催化体系表现出很好的底物适应性, 无论是氰基、硝基取代的醛, 还是大位阻的1-萘醛亦或是脂肪醛都能取得不错的结果, 同时对于大位阻的2-位取代的芳基硼酸反应也能取得优异的收率和对映选择性(Eq. 18).
|
|
(18) |
此外钯作为催化剂也被用于芳基硼酸对醛的不对称加成反应之中[22], 但结果并不理想, 取得相对较好结果的是Shi课题组[22c]报道的基于联萘二胺(BINAM)骨架的氮杂卡宾钯络合物催化的例子, 反应可以取得最高65% ee值的结果(Eq. 19).
|
|
(19) |
手性的3-羟基吲哚酮是一类非常重要的结构单元, 这类骨架存在于许多具有生物活性的天然产物和药物分子中如:化合物26是一种非常有效的生长激素促分泌素[23], 化合物27是一类Maxi-K通道激活剂(图 2)[24], 因此, 此类手性分子骨架的高效合成一直是一个研究热点,受到有机和药物化学家们的广泛关注.过渡金属催化的芳基硼酸对靛红的不对称加成是构建此类结构最直接和高效的方法.

2006年Hayashi等[25]首次报道了Rh(Ⅰ)/单齿膦配体MeO-MOP (L1)催化的靛红的不对称芳基化的例子, 在温和的条件下能以中等到优秀的收率和对映选择性得到一系列光学活性的3-羟基吲哚酮29, 对于靛红氮上没有保护基的底物也能取得87%的ee值.值的一提的是, 该反应条件对于靛红的苯乙烯基化也可以取得不错的结果(Eq. 20).
|
|
(20) |
同年, Feringa和Minnaard等[26]利用手性亚磷酰胺配体也实现了Rh(Ⅰ)催化的苯硼酸对靛红的不对称芳基化立体选择性的调控, 反应可以以几乎定量的收率得到55% ee的产物31, 31的ee值可通过结晶提高到94% (Eq. 21).
|
|
(21) |
2012年, 廖建课题组[27]利用他们发展的手性磷-亚砜配体L19, 同样实现了靛红的不对称芳基化的立体选择性的控制, 取得最高92% ee值的结果.除了邻位取代的芳基硼酸可以取得较为优异的结果外, 该反应体系对于氮上没有保护的靛红也可以取得不错的结果, 这为产物的进一步衍生化提供了便利(Eq. 22).
|
|
(22) |
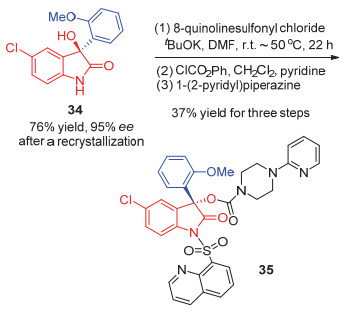
近年来, 杜海峰课题组[28a]发展了一系列简单的烯烃配体, 2014年, 他们将发展的硫烯配体L20用于铑催化的芳基硼酸对靛红的不对称加成之中, 对于膦配体以及NHC结果都不理想的大位阻的硼酸, 硫烯配体能取得中等的选择性[28b].此外, 对于靛红氮上没有保护的底物, 反应也表现出较好的适应性(Eq. 23).他们以此反应为依托, 将相应产物34重结晶, 再通过3步反应合成了对加压素或催产素依赖疾病具有预防作用的生物活性分子35 (Scheme 3).
|
|
(23) |
2016年, Burke等[29]利用(R)-BINAP作为配体实现了Rh(Ⅰ)催化的芳基硼酸对三氮唑取代靛红底物36的不对称加成, 反应可以取得最高95% ee值的产物.由于底物中的邻二羰基以及三氮唑都有可能干扰铑和配体的配位, 因此在反应之前需要先制备好金属络合物, 此外选择大位阻的有机胺作为碱对反应的收率也有一定的帮助作用(Eq. 24).
|
|
(24) |
2009年, 秦勇课题组[30]发展了一类手性四取代的联苯磷亚胺配体L22, 并将其用于靛红的不对称芳基化的尝试.当使用Rh(CH3CN)(cod)BF4或者Rh2(OAc)4作为催化剂时, 由于存在严重的背景反应, 得到的都是消旋的产物.而使用Pd(OAc)2作为催化剂时, 反应能以中等的收率以及ee值得到产物, 实验结果证明反应的立体选择性主要是通过联苯的轴手性而不是通过磺酰亚胺的硫手性来控制的.此外他们还发现在反应体系中加入BF3•Et2O有助于反应收率的提高(Eq. 25).
|
|
(25) |
2011年, Shi和Li等[31]报道了Pd-NHC物种催化的芳基硼酸对靛红的不对称加成, 反应可以获得79%~94%收率以及中等偏上的对映选择性(Eq. 26).在反应优化过程中他们发现碱的影响比较大, LiOH•H2O作为碱时, 无论是收率还是对映选择性都明显优于其他碱, 在反应机理考察的过程中, 他们认为加入的LiOH•H2O可以与芳基醇原位生成硬的Lewis酸LiOAr, 生成的LiOAr与底物中两个羰基氧作用, 从而可以活化底物提高反应收率(Scheme 4).
|
|
(26) |
2013年, Zhou课题组[32]利用基于H8-BINOL骨架的膦噁唑啉配体L23同样实现了钯催化的芳基硼酸对靛红的不对称加成, 取得最高88% ee值的产物, 其中H8-BINOL骨架和噁唑啉的手性匹配可以提高反应的收率以及立体选择性, 同时此骨架的大位阻有利于反应的立体选择性控制(Eq. 27).
|
|
(27) |
Yamamoto小组将发展的手性亚磷酰胺配体L13[33a], 相继用于醛[16a]、乙醛酸酯[16b]以及酮酸酯[16c]的不对称芳基化后, 又成功地将其用于钌催化的芳基硼酸对靛红的不对称加成中, 也可以取得不错的结果(Eq. 28).反应机理被认为与铑催化的1, 2-加成类似, 也是经历了转金属化、芳基钌物种对不饱和键插入, 然后再水解得到产物的过程.
|
|
(28) |
之后, 该小组[33b]又利用相同的催化剂实现了2, 3-二酮苯并呋喃底物42的不对称芳基化, 添加的乙腈对于反应收率非常重要, 被认为可以防止二酮的两个羰基毒化催化剂.从反应结果来看, 底物中的4位甲基的存在对反应收率和ee值都有非常重要的影响, 这是因为4位甲基位阻的存在可以防止底物的水解同时提高反应的立体选择性(Eq. 29).
|
|
(29) |
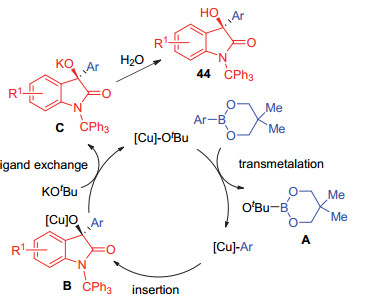
2010年, Hayashi和Shintani等[34]共同报道了Cu/ NHC催化的芳基硼酸酯对靛红的不对称加成, 反应能以中等到优异的收率得到ee值为67%~92%的产物(Eq. 30).他们通过实验证明, 反应可能经历了与铑催化的反应相似的过程:氯化铜与卡宾L24形成反应的活性成分叔丁醇氧N-杂环卡宾铜, 然后与硼酸酯发生转金属化后形成芳基铜物种, 该物种再与底物反应形成烷氧基铜中间体, 此中间体与叔丁醇钾生成活性的叔丁醇氧N-杂环卡宾铜进入下一个循环同时释放出产物, 这样一个反应循环也可以在一定程度上解释添加剂叔丁醇钾的重要性(Scheme 5).
2015年, Qiu等[35]报道了基于手性桥环联苯骨架的手性亚磷酰胺配体, 并将其用于铱催化的芳基硼酸对靛红的不对称加成反应之中, 以中等到优异的收率得到ee值为76%~95%的手性的3-芳基-3-羟基-2-吲哚酮产物.他们在实验中发现, 配体中比较拥挤的环境不仅有利于促进反应的进行从而提高反应收率, 而且也有助于反应的立体选择性控制(Eq. 31).
|
|
(31) |
手性2-羟基二芳基乙酸酯是合成毒蕈碱受体拮抗剂的关键中间体[36], 芳基硼酸对芳基酮酸酯进行1, 2-加成便可以直接构建此类分子骨架, 但是反应的立体选择性通常难以控制, 因此寻找合适的反应体系至关重要.近年来各种不同的反应体系也被成功发展并用于该类化合物的构建之中.
2008年, 周其林课题组[37]利用他们发展的螺环的亚磷酸酯配体, 首次实现了Rh(Ⅰ)催化的芳基硼酸对α-酮酸酯的不对称1, 2-加成, 反应可以取得中等到优异的对映选择性.他们认为加入的LiF不仅仅作为碱, 其中的氟离子也可以和芳基硼酸中的硼络合加速芳基铑物种的形成, 从而有利于反应收率的提高.利用该催化体系, 他们还选择性地实现了同时具有1, 2-和1, 4-加成反应位点的α, β-不饱和酮酸酯的不对称1, 2-加成(Eq. 32).
|
|
(32) |
2011年, Ready等[38]设计合成了手性联烯骨架的双膦配体, 并将其用于Rh(Ⅰ)催化的芳基硼酸对芳基酮酸酯的不对称加成反应之中, 在温和的条件下可以以52%~98%收率得到72%~95% ee的产物.络合物单晶结构表明, 配体中两个磷以及联烯中的一个双键以三齿配体的方式和铑配位.两个磷原子上连有强吸电子的3, 5-二三氟甲基苯基的配体的活性最好, 可能的原因是催化剂与底物络合后通过诱导效应, 可有效减弱反应底物的电负性, 使得反应更易进行(Eq. 33).
|
|
(33) |
几乎同时, 徐明华课题组[39a]报道了一类铑/手性硫烯配体催化的芳基硼酸对α-芳基酮酸酯的不对称1, 2-加成, 也取得了很好的结果, 其中手性硫烯配体L28结构极其简单, 合成简便.该催化体系被进一步拓展到3-吲哚-α-酮酸酯的不对称1, 2-加成之中, 反应也表现出了很好的底物适应性[39b], 此外,对于苯并呋喃以及苯并噻吩取代的酮酸酯, 都能给出非常好的收率和对映选择性, 与芳香杂环的Friedel-Crafts反应相比, 该方法受底物的电子效应影响较小, 为含芳香杂环季碳手性中心的α-羟基羰基化合物的合成提供了新方法(Eq. 34).
|
|
(34) |
通过在酮酸酯芳基邻位引入取代基团, 徐明华小组[40]又成功实现了以不同取代的直链手性硫烯L29和L30为配体的Rh(Ⅰ)催化的芳基硼酸对邻位官能团化的芳基酮酸酯51的不对称1, 2-加成, 并进一步通过后续分子内的环化反应高效构建了一系列含季碳手性中心的高光学活性的苯并二氢异呋喃52、3-芳基-3-羟基吲哚酮53和异色满-3-酮类化合物54 (Scheme 6).
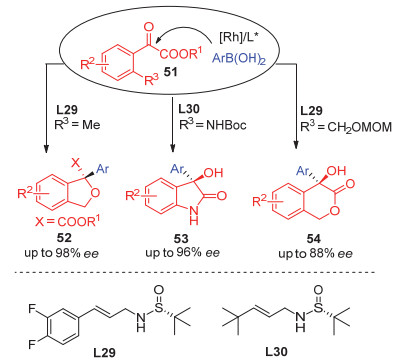
α-烷基取代的酮酸酯的不对称1, 2-加成非常挑战, 一直没有成功的报道. 2013年, 徐明华小组[41]研究发现, 以手性硫烯为配体, 反应可以取得中等选择性.有意思的是, 仅仅改变取代基在配体双键上的位置, 反应得到的产物的构型便发生了翻转, 直链配体L28得到(S)构型产物56, 而支链配体L31得到(R)构型产物57, 而以α, β-不饱和酮酸酯58为底物时, 发现支链配体主要得到1, 2-加成的(R)构型产物60, 而直链配体倾向于得到1, 4-加成产物59 (Scheme 7).
2018年, Strand等[42]发展了一类基于面手性的二苯并环辛二烯配体, 他们通过在该类化合物的5, 11位引入取代基, 可以在一定程度上阻止其构象翻转, 从而得到手性配体.这类配体在芳基硼酸对酮酸酯的不对称加成反应中表现出较好的立体选择性控制, 可以取得最高94% ee值的结果(Eq. 35).
|
|
(35) |
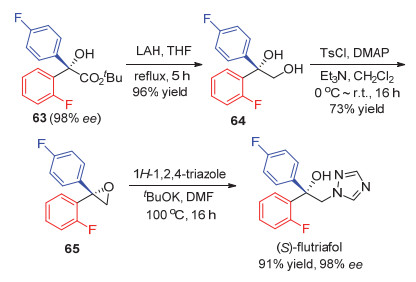
最近, Wu小组[43]以[2.2.1]庚二烯骨架的手性双烯L32为配体也实现了铑催化的芳基硼酸对酮酸酯的不对称加成, 取得最高99% ee值的结果(Eq. 36).该反应体系表现出较高的催化活性, 在催化剂的量为0.5 mol%时, 反应也能给出较高的收率.利用该方法, 以产物63为原料,经3步反应高效实现了具有抗真菌活性的脱甲基抑制剂(S)-flutriafol的对映选择性合成(Scheme 8).
|
|
(36) |
2017年, Johnson小组[44]以Rh(Ⅰ)/手性双烯L33和L34催化的芳基硼酸对酮酸酯的不对称加成为基础, 实现了β位取代的α-酮酸酯66的动态动力学拆分, 反应可以实现非常优异的非对映选择性以及对映选择性的控制.反应结果表明有机碱在反应的对映选择性的控制中起着非常关键的作用, 这是因为有机碱可以影响β取代基的消旋化速度(Eq. 37).
|
|
(37) |
光学活性的α-羟基酮是许多生物活性分子的重要结构单元, 如烟酸受体激动剂呋喃酸[45]与人体免疫缺损病毒(HIV)整合酶抑制剂Integrastatin[46].偶酰类二酮化合物的不对称加成是制备此类化合物的直接方法,但由于底物中同时存在两个羰基, 反应的选择性通常难以控制, 通过相应的傅克反应或有机锡试剂加成来制备, 又存在较大的底物局限性和锡试剂剧毒性的问题.
虽然芳基硼酸在对靛红、酮酸酯以及三氟甲基酮的不对称加成中都取得了不错的结果, 但对邻二酮底物的不对称芳基化却一直未见成功的例子报道.直到2012年, 徐明华课题组[39a]利用铑/手性硫烯配体的催化体系首次实现了邻二酮68的不对称芳基化, 对于大部分的二芳基邻二酮均可以取得大于95% ee值的结果, 而且对于更加挑战的甲基取代的烷基邻二酮, 反应也能取得中等的对映选择性(Eq. 38).
|
|
(38) |
同年, 杜海峰课题组[47]也报道了将硫烯配体L20用于铑催化的邻二酮的不对称加成的例子, 该反应体系对于环二芳基酮也能取得非常优异的收率和对映选择性, 但是对于含杂环的二酮底物, 反应的选择性并不理想(Eq. 39).
|
|
(39) |
2016年, 曾庆乐等[48]发展了一类环状亚磺酰胺结构的手性硫烯配体L35, 在铑催化的4-甲氧基苯硼酸对偶酰的不对称芳基加成反应中, 可以取得90%收率和90% ee的结果(Eq. 40).
|
|
(40) |
基于邻二芳基酮的不对称加成的优异结果, 徐明华小组[49]又设计了在一侧酮的芳基邻位引入卤素或酯基, 利用位阻以及反应活性的差异实现对其中一个酮羰基的选择性加成, 然后再串联分子内醚化环化或者酯化的策略.基于该策略, 成功地实现了一锅法合成高光学活性的异色满4-酮74以及异色满1, 4-二酮化合物76 (Eqs. 41, 42), 并进一步利用该方法, 以产物78为原料,通过两个线性步骤简单转化实现了Urotensin-Ⅱ抑制剂AC-7954 (80)的首次催化不对称合成, 该方法为手性异色酮类药物的合成提供了高效的不对称合成途径(Scheme 9).
|
|
(41) |
|
|
(42) |
最近, 徐明华小组[50]又设计了一类芳基邻位卤素取代的邻二芳基酮底物81, 将铑催化的不对称芳基化和钯催化的C—O偶联进行组合, 通过双金属协同催化的一锅法反应实现了偕二芳基取代的苯并呋喃酮82的高对映选择性合成, 反应能以高收率取得优秀的对映选择性(最高99% ee).值得一提的是, 该方法利用两种金属催化反应的温度差异来实现反应的调控; 需要指出的是, 底物中的卤素的选择也非常重要, 当为活性较高的溴时, 底物容易发生Suzuki偶联等副反应(Eq. 43).
|
|
(43) |
此外, 徐明华小组[51]还设计并合成了一类基于手性BINOL骨架的开链磷烯配体L36, 这类配体在铑催化的芳基硼酸对邻二酮的不对称1, 2-加成中也表现出非常优异的特性, 反应以非常优异的收率得到光学纯度高达96%的产物.当将配体中的双键还原或者移去后反应的立体选择性都出现大幅下降以及配体与铑配位的核磁谱图数据均证实了该类配体是通过双键和磷与同时铑配位而不是传统的只通过磷与铑配位的方式(Eq. 44).
|
|
(44) |
因为氟原子的引入不仅可以调节药物分子的理化特性, 还可以提高药物的生物利用度以及代谢稳定性[52], 近年来含氟药物在临床治疗药物中占的比例越来越大.手性的三氟甲基叔醇类骨架由于其存在于一些生物活性分子中而受到人们的关注, 酮的不对称三氟甲基化是构建此类骨架的传统方法, 但是反应的立体选择性通常难以控制.
2006年, Minnaard等[53]利用铑/磷酰亚胺L37的催化体系, 实现了三氟苯乙酮类化合物85的不对称芳基化, 虽然底物的适应性不广, 收率和对映选择性也还不够理想, 但是这是首次通过铑催化的芳基硼酸对三氟甲基酮的不对称加成来构建手性三氟甲基叔醇类化合物的报道(Eq. 45).
|
|
(45) |
2010年, Iuliano等[54]报道了以脱氧胆酸衍生物的亚磷酸盐L38作为手性配体诱导芳基硼酸对三氟苯乙酮的不对称加成的例子, 该体系具有较高的反应活性, 即使在常温条件下反应也能在短时间内给出70%~95%的收率, 但是遗憾的是反应的底物适应性较窄而且选择性较低(Eq. 46).
|
|
(46) |
2013年, 汤文军课题组[55]发展了一类新型手性双膦配体L39, 在铑催化的芳基硼酸对三氟苯乙酮的不对称加成反应中取得很好的结果, 该方法能够以最高93%的产率以及最高99% ee值得到加成产物, 并且该反应体系表现较为广泛的底物适应性, 无论是杂环的还是邻位取代的酮, 反应都能给出比较优异的结果.通过X-ray单晶衍射确定了产物的绝对构型, 并结合产物的立体化学对反应的过渡态进行了探讨(Eq. 47).
|
|
(47) |
相比活泼酮, 非活泼酮的不对称芳基化相对更加困难.传统的方法主要是使用芳基锌试剂对其进行加成来实现, 考虑到芳基锌试剂的制备与稳定性的问题, 芳基硼酸对非活泼酮的不对称加成更加引起人们的兴趣, 最近这方面的工作也取得了一定的进展.
2011年, Sakai等[56]在尝试了将双膦配体BINAP (L21)用于铑催化的苯硼酸对2-乙酰萘的不对称加成中, 虽然发现使用缺电子双膦配体与铑的络合物可以有效活化反应底物, 进而得到高收率的加成产物, 但是反应的对映选择性很不理想(Eq. 48).
|
|
(48) |
2012年, 胡桥生课题组[57]将[3.3.0]-辛二烯骨架的手性双烯配体L40用于铑催化的芳基硼试剂对非活泼酮的不对称芳基化中, 反应能以优异的收率得到最高为68% ee值的产物, 文中提到无水的反应体系对于催化剂的活性保持至关重要(Eq. 49).
|
|
(49) |
2016年, 汤文军课题组[58]在非活泼酮的不对称芳基化研究方面取得突破, 他们以大位阻的双膦配体WingPhos (L41)/铑作为催化剂, 硼酐作为硼源, 在体系中加入溴化镁帮助活化羰基, 实现了铑催化的芳基硼试剂对简单酮的高效不对称加成, 构建了一系列高光学活性的手性二芳基醇类化合物.该反应体系解决了长期困扰化学家们的简单酮的反应活性以及立体选择性不够理想的问题(Eq. 50).
|
|
(50) |
值得一提的是, 利用该方法可以实现两个手性药物分子氯马斯汀和西酞普兰的合成.以对氯苯乙酮96为底物, 通过一步加成反应即可构建氯马斯汀的关键中间体97 (Scheme 10).以4-氯-(2, 4-二氯苯基)-1-丁酮98为反应底物, 利用4-氟苯硼酐在铑催化条件下进行不对称加成即可构建相应的季碳手性中心, 然后将得到的叔醇化合物99与二甲胺盐酸盐反应引入N, N-二甲基, 再经钯催化双氰化引入氰基后, 其中一个氰基再内酯化关环, 最后将内酯的羰基还原得到药物分子西酞普兰101 (Scheme 11).
2009年, Itami课题组[59]报道了零价镍/卡宾配体L42催化的芳基硼酸酯对非活泼酮的分子间加成, 该方法对于芳基酮、二芳基酮以及脂肪酮底物都可以取得较好的收率, 但是基本都只能得到消旋产物, 仅有的一个最好的结果为36% ee值(Eq. 51).
|
|
(51) |
酮的反应活性相对较弱, 因此反应相对较难.而分子内的反应由于反应位点之间碰撞的概率更高, 在一定程度上可以弥补酮的反应活性较弱的问题, 并且分子内加成相对于分子间的加成反应的位阻也更大, 这一点对提高反应的立体控制是有利的.近年来也陆续有一些分子内加成反应的报道, 也取得了不错的成果.
2006年, 陆熙炎小组[60]首次报道了阳离子钯催化的分子内的芳基硼酸对酮的不对称加成(Eq. 52).在以离子交换树脂作为添加剂的条件下, 反应能以最高91%的收率得到最好为96% ee值的环化产物, 其中分子中的连接基团氧被认为具有导向以及活化作用, 当将氧换成碳时, 反应的收率和立体选择性都出现大幅降低.
|
|
(52) |
2012年, Sarpong课题组[61]报道了[Rh(cod)OH]2与手性二茂铁双膦配体催化的频哪醇硼酯对非活泼酮的不对称分子内加成的例子, 用于构建高光学活性的手性茚1-醇类化合物107, 与陆小组工作[60]不同的是, 该反应体系对于碳链连接的酮效果更好(Eq. 53),但当连接的碳链进一步增加1个碳时反应结果又不够理想.
|
|
(53) |
有意思的是, Lam小组[62]引入氮原子作为连接基团设计了底物108, 于同一时期报道了铑/硫烯配体L44催化的分子内芳基硼酸对酮的不对称加成, 高对映选择性地构建了一系列苯并氮杂六元环类化合物109, 该方法对于位阻较大的酮也能取得比较好的结果(Eq. 54).
|
|
(54) |
2016年, 徐明华课题组[63]利用课题组发展的手性硫烯配体L45, 也实现了铑催化的非活泼酮110的分子内的不对称芳基化.在温和的反应条件下成功地实现了一系列高光学活性的含季碳手性中心的3-羟基苯并二氢呋喃类化合物111的构建, 该反应体系不仅适合于芳基酮, 对于烷基酮也同时适用, 而且对于α位双取代的大位阻的酮也能取得不错的结果(Eq. 55).
|
|
(55) |
在过去的二十年里, 过渡金属催化的有机硼试剂对醛、酮的不对称加成研究取得了很大的进展, 特别是发展了一系列高效的催化体系, 在一些较为活泼的羰基化合物如α-芳基酮酸酯和1, 2-二芳基二酮类化合物的对映选择性加成中取得了非常优秀的结果; 对靛红的加成虽然也有一些不错的结果报道, 但反应的对映选择性受底物和芳基硼试剂上取代基的影响较大.值得关注的是, 简单醛、酮的高对映选择性加成仍是一个难点, 除了反应活性较高的2, 2, 2-三氟苯乙酮及其衍生物外, 大多数醛、非活泼酮的催化不对称加成都难以取得理想的收率和对映选择性, 或者存在明显的底物普适性问题, 仍有很大的提升空间; 一些催化体系在分子内酮的不对称加成反应中给出了不错的结果, 但同样受制于反应底物的设计, 分子结构的局限性较大, 因此还有待有机化学家们设计新的手性配体、发展高效的过渡金属催化体系来突破这些瓶颈, 取得更好的结果.另一方面, 我们也可以看到, 与有机硼试剂对亚胺的不对称加成类似[64], 有机硼试剂对醛、酮的不对称加成研究也是主要集中在贵金属铑和钯催化的反应中, 可喜的是, 其它过渡金属如铱、钌, 以及更为廉价的铜、钴、镍等催化的有机硼试剂对醛、酮的不对称加成研究已取得了一些成功, 虽然目前一些反应的选择性仍不尽理想, 但相信随着化学家们对反应机理和过渡态的理解和认识的深入, 通过进一步合理设计新型手性配体, 制备高效的金属络合物催化剂, 未来在这一领域取得突破值得期待.
(a) Dhillon, S.; Scott, L. J.; Plosker, G. L. CNS Drugs 2006, 20, 763.
(b) Fournier, A. M.; Brown, R. A.; Farnaby, W.; Miyatake-On- dozabal, H.; Clayden, J. Org. Lett. 2010, 12, 2222.
(c) Nathan, L. A. J. Appl. Ther. 1962, 4, 830.
白东鲁, 陈凯先, 李树坤, 冯松, 高等药物化学, 2011, p. 290.Bai, D. L.; Chen, K. X.; Li, S. K.; Feng, S. Adv. Med. Chem. 2011, p. 290 (in Chinese).
(a) Heravi, M. M.; Zadsirjan, V.; Esfandyari, M.; Lashaki, T. B. Tetrahedron: Asymmetry 2017, 28, 987.
(b) Zaitsev, A. B.; Adolfsson, H. Synthesis 2006, 1725.
(c) Scott, H. K.; Aggarwal, V. K. Chem. Eur. J. 2011, 17, 13124.
(d) Jacobsen, E. N.; Kakiuchi, F.; Konsler, R. G.; Larrow, J. F.; Tokunaga, F. Tetrahedron Lett. 1997, 38, 773.
(e) Wang, C.; Luo, L.; Yamamoto, H. Acc. Chem. Res. 2016, 49, 193.
(f) Sälinger, D.; Brückner, R. Chem.-Eur. J. 2009, 15, 6688.
(a) Hatano, M.; Ishihara, K. Synthesis 2008, 1647.
(b) Rong, J.; Pellegrini, T.; Harutyunyan, S. R. Chem. Eur. J. 2016, 22, 3558.
(c) Collados, J. F.; Sol, R.; Harutyunyan, S. R.; Macià, B. ACS Catal. 2016, 6, 1952.
(d) Pellissier, H. Tetrahedron 2015, 71, 2487.
(e) Liu, Y. L.; Lin, X. T. Adv. Synth. Catal. 2019, 361, 876.
Sakai, M.; Ueda, M.; Miyaura, N. Angew. Chem., Int. Ed. 1998, 37, 3279. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3279::AID-ANIE3279>3.0.CO;2-M
(a) Jiang, Z. T.; Wang, B. Q.; Shi, Z. J. Chin. J. Chem. 2018, 36, 954.
(b) Wang, G. N.; Gan, Y.; Liu, Y. H. Chin. J. Chem. 2018, 36, 916.
Focken, T.; Rudolph, J.; Bolm, C. Synthesis 2005, 429.
(a) Suzuki, K.; Ishii, S.; Kondo, K.; Aoyama, T. Synlett 2006, 648.
(b) Suzuki, K.; Kondo, K.; Aoyama, T. Synthesis 2006, 1360.
(c) Arao, T.; Suzuki, K.; Kondo, K.; Aoyama, T. Synthesis 2006, 3809.
Duan, H. F.; Xie, J. H.; Shi, W. J.; Zhang, Q.; Zhou, Q. L. Org. Lett. 2006, 8, 1479. doi: 10.1021/ol060360c
Jagt, R. B. C.; Toullec, P. Y.; de Vries, J. G.; Feringa, B. L.; Minnaard, A. J. Org. Biomol. Chem. 2006, 4, 773. doi: 10.1039/b518311a
(a) Noël, T.; Vandyck, K.; Van der Eycken, J. Tetrahedron 2007, 63, 12961.
(b) Nishimura, T.; Kumamoto, H.; Nagaosa, M.; Hayashi, T. Chem. Commun. 2009, 45, 5713.
Morikawa, S.; Michigami, K.; Amii, H. Org. Lett. 2010, 12, 2520. doi: 10.1021/ol100697a
(a) Ma, Q. S.; Ma, Y. D.; Liu, X.; Duan, W. Z.; Qu, B.; Song, C. Tetrahedron: Asymmetry 2010, 21, 292.
(b) Duan, W. Z.; Ma, Y. D.; Qu, B.; Zhao, L.; Chen, J. Q.; Song, C. Tetrahedron: Asymmetry 2012, 23, 1369.
(c) Duan, W. Z.; Ma, Y. D.; He, F. Y.; Zhao, L.; Chen, J. Q.; Song, C. Tetrahedron: Asymmetry 2013, 24, 241.
(d) Wang, D. X.; Ma, Y. D.; He, F. Y.; Duan, W. Z.; Zhao, L.; Song, C. Synth. Commun. 2013, 43, 810.
(e) Chen, J. Q.; Yang, S. B.; Chen, Z.; Song, C.; Ma, Y. D. Tetrahedron: Asymmetry 2015, 26, 288.
He, W. P.; Zhou, B. H.; Zhou, Y. L.; Li, X. L.; Fan, L. M.; Shou, H. W.; Li, J. Tetrahedron Lett. 2016, 57, 3152. doi: 10.1016/j.tetlet.2016.06.023
Kamikawa, K.; Tseng, Y.-Y.; Jian, J.-H.; Takahashi, T.; Ogasawara, M. J. Am. Chem. Soc. 2017, 139, 1545. doi: 10.1021/jacs.6b11243
(a) Yamamoto, Y.; Miyaura, N.; Kurihara, K. Angew. Chem., Int. Ed. 2009, 48, 4414.
(b) Shirai, T.; Watanabe, M.; Kurihara, K.; Miyaura, N.; Yamamoto, Y. Molecules 2011, 16, 5020.
(c) Yamamoto, Y.; Shirai, T.; Miyaura, N. Chem. Commun. 2012, 48, 2803.
Desroches, J.; Ariane Tremblay, A.; Paquin, J.-F. Org. Biomol. Chem. 2016, 14, 8764. doi: 10.1039/C6OB01663D
Li, K.; Hu, N. F.; Luo, R. S.; Yuan, W. C.; Tang, W. J. J. Org. Chem. 2013, 78, 6350. doi: 10.1021/jo400850m
Lu, Z. W.; Zhang, H. Y.; Yang, Z. P.; Ding, N.; Meng, L.; Wang. J. ACS Catal. 2019, 9, 1457. doi: 10.1021/acscatal.8b04787
(a) Arao, T.; Kondo, K.; Aoyama, T. Tetrahedron Lett. 2007, 48, 4115.
(b) Yamamoto, K.; Tsurumi, K.; Sakurai, F.; Kondo, K.; Aoyama, T. Synthesis 2008, 3585.
Karthikeyan, J.; Jeganmohan, M.; Cheng, C.-H. Chem.-Eur. J. 2010, 16, 8989. doi: 10.1002/chem.201001160
(a) Suzuma, Y.; Hayashi, S.; Yamamoto, T.; Oe, Y.; Ohta, T.; Ito, Y. Tetrahedron: Asymmetry 2009, 20, 2751.
(b) Loxq, P.; Debono, N.; Gülcemal, S.; Daran, J-C.; Manoury, E.; Poli, R.; Cetinkaya, B.; Labande, A. New J. Chem. 2014, 38, 338.
(c) Zhang, R.; Xu, Q.; Zhang, X.; Zhang, T.; Shi, M. Tetrahedron: Asymmetry 2010, 21, 1928.
(a) Tokunaga, T.; Hume, W. E.; Nagamine, J.; Kawamura, T.; Taiji, M.; Nagata, R. Bioorg. Med. Chem. Lett. 2005, 15, 1789.
(b) Tokunaga, T.; Hume, W. E.; Umezome, T.; Okazaki, K.; Ueki, Y.; Kumagai, K.; Hourai, S.; Nagamine, J.; Seki, H.; Taiji, M.; Noguchi, H.; Nagata, R. J. Med. Chem. 2001, 44, 4641.
Di Malta, A.; Garcia, G.; Roux, R.; Schoentjes, B.; Serradeil-le Gal, C.; Tonnerre, B.; Wagnon, J. WO 2003008407, 2003.
Shintani, R.; Inoue, M.; Hayashi, T. Angew. Chem., Int. Ed. 2006, 45, 3353. doi: 10.1002/anie.200600392
Toullec, P. Y.; Jagt, R. B. C.; de Vries, J. G.; Feringa, B. L.; Minnaard, A. J. Org. Lett. 2006, 8, 2715. doi: 10.1021/ol0608101
Gui, J.; Chen, G.; Cao, P.; Liao, J. Tetrahedron: Asymmetry 2012, 23, 554. doi: 10.1016/j.tetasy.2012.04.013
(a) Feng, X. Q.; Du, H. F. Chin. J. Org. Chem. 2015, 35, 259 (in Chinese). (冯向青, 杜海峰, 有机化学, 2015, 35, 259.)
(b) Feng, X. Q.; Nie, Y. Z.; Zhang, L. Q.; Yang, J.; Du, H. F. Tetrahedron Lett. 2014, 55, 4581.
Marques, C. S.; Burke, A. J. ChemCatChem 2016, 8, 3518. doi: 10.1002/cctc.201600901
Lai, H. S.; Huang, Z. Y.; Wu, Q.; Qin, Y. J. Org. Chem. 2009, 74, 283. doi: 10.1021/jo802036m
Liu, Z.; Gu, P.; Shi, M.; McDowell, P.; Li, G. G. Org. Lett. 2011, 13, 2314. doi: 10.1021/ol200566s
Li, Q.; Wan, P.; Wang, S.; Zhuang, Y.; Li, L.; Zhou, Y.; He, Y.; Cao, R.; Qiu, L.; Zhou, Z. Appl. Catal., A 2013, 458, 201. doi: 10.1016/j.apcata.2013.03.027
(a) Yamamoto, Y.; Yohd, M.; Shirai, T.; Ito, H.; Miyaura, N. Chem. Asian J. 2012, 7, 2446.
(b) Yohda, M.; Yamamoto, Y. Tetrahedron: Asymmetry 2015, 26, 1430.
Shintani, R.; Takatsu, K.; Hayashi, T. Chem. Commun. 2010, 46, 6822. doi: 10.1039/c0cc01635g
Zhuang, Y.; He, Y. W.; Zhou, Z. H.; Xia, W.; Cheng, C. Y.; Wang, M.; Chen, B.; Zhou, Z. Y.; Pang, J. Y.; Qiu, L.Y. J. Org. Chem. 2015, 80, 6968. doi: 10.1021/acs.joc.5b00595
(a) Skaddan, M. B.; Kilbourn, M. R.; Snyder, S. E.; Sherman, P. S.; Desmond, T. J.; Frey, K. A. J. Med. Chem. 2000, 43, 4552.
(b) Selent, J.; Brandt, W.; Pamperin, D.; Goeber, B. Bioorg. Med. Chem. 2006, 14, 1729.
Duan, H. F.; Xie, J. H. Qiao, X. C. Wang, L. X.; Zhou, Q. L. Angew. Chem., Int. Ed. 2008, 47, 4351. doi: 10.1002/anie.200800423
Cai, F.; Pu, X. T.; Qi, X. B.; Lynch, V.; Radha, A.; Ready, J. M. J. Am. Chem. Soc. 2011, 133, 18066. doi: 10.1021/ja207748r
(a) Zhu, T.-S.; Jin, S.-S.; Xu, M.-H. Angew. Chem., Int. Ed. 2012, 51, 780.
(b) Wang, H.; Zhu, T.-S.; Xu, M.-H. Org. Biomol. Chem. 2012, 10, 9158.
Li, Y.; Zhu, D.-X.; Xu, M.-H. Chem. Commun. 2013, 49, 11659. doi: 10.1039/c3cc47927g
Zhu, T.-S.; Xu, M.-H. Chin. J. Chem. 2013, 31, 321. doi: 10.1002/cjoc.201300063
Melcher, M.-C.; Ivšić, T.; Olagnon, C.; Tenten, C.; Letzen, A.; Strand, D. Chem.-Eur. J. 2018, 24, 2344. doi: 10.1002/chem.201704816
Chang, C.-A.; Uang, T.-Y.; Jian, J.-H.; Zhou, M.-Y.; Chen, M.-L.; Kuo, T.-S.; Wu, P.-Y.; Wu, H.-L. Adv. Synth. Catal. 2018, 360, 3381. doi: 10.1002/adsc.201800575
Bartlett, S. L.; Keiter, K. M.; Johnson, J. S. J. Am. Chem. Soc. 2017, 139, 3911. doi: 10.1021/jacs.7b00943
Jung, J. K.; Johnson, B. R.; Duong, T.; Decaire, M.; Uy, J.; Gharbaoui, T.; Boatman, P. D.; Sage, C. R.; Chen, R.; Richman, J. G.; Connolly, D. T.; Semple, G. J. Med. Chem. 2007, 50, 1445. doi: 10.1021/jm070022x
Singh, S. B.; Zink, D. L.; Quamina, D. S.; Pelaez, F.; Teran, A.; Felock, P.; Hazuda, D. J. Tetrahedron Lett. 2002, 43, 2351. doi: 10.1016/S0040-4039(02)00265-4
Feng, X.; Nie, Y.; Yang, J.; Du, H. Org. Lett. 2012, 14, 624. doi: 10.1021/ol203238j
Wen, Q.; Zhang, L.; Xiong, J.; Zeng, Q. L. Eur. J. Org. Chem. 2016, 5360.
Zhu, T.-S.; Chen, J.-P.; Xu, M.-H. Chem. Eur. J. 2013, 19, 865. doi: 10.1002/chem.201203701
Zhang, Z.-F.; Zhu, D.-X.; Chen, W.-W.; Xu, B.; Xu, M.-H. Org. Lett. 2017, 19, 2726. doi: 10.1021/acs.orglett.7b01070
Yu, Y.-N.; Xu, M.-H. Org. Chem. Front. 2014, 1, 738. doi: 10.1039/C4QO00135D
(a) Rowley, M.; Hallett, D. J.; Goodacre, S.; Moyes, C.; Crawforth, J.; Sparey, T. J.; Patel, S.; Marwood, R.; Thomas, S.; Hitzel, L.; O'Connor, D.; Szeto, N.; Castro, J. L.; Hutson, P. H.; MacLeod, A. M. J. Med. Chem. 2001, 44, 1603.
(b) van Niel, M. B.; Collins, I.; Beer, M. S.; Broughton, H. B.; Cheng, S. K.; Goodacre, S. C.; Heald, A.; Locker, K. L.; MacLeod, A. M.; Morrison, D.; Moyes, C. R.; O'Connor, D.; Pike, A.; Rowley, M.; Russell, M. G.; Sohal, B.; Stanton, J. A.; Thomas, S.; Verrier, H.; Watt, A. P.; Castro, J. L. J. Med. Chem. 1999, 42, 2087.
(c) Domagala, J. M.; Hanna, L. D.; Heifetz, C. L.; Hutt, M. P.; Mich, T. F.; Sanchez, J. P.; Solomon, M. J. Med. Chem. 1986, 29, 394.
(d) Rosenblum, S. B.; Huynh, T.; Afonso, A.; Davis, H. R. Jr.; Yumibe, N.; Clader, J. W.; Burnett, D. A. J. Med. Chem. 1998, 41, 973.
Martina, S. L. X.; Jagt, R. B. C.; Vries, J. G.; Feringa B. L.; Minnaard, A. J. Chem. Commun. 2006, 42, 4093.
Jumde, V. R.; Facchetti, S.; Iuliano, A. Tetrahedron: Asymmetry 2010, 21, 2775. doi: 10.1016/j.tetasy.2010.11.009
Luo, R. S.; Li, K.; Hu, Y. L.; Tang, W. J. Adv. Synth. Catal. 2013, 355, 1297. doi: 10.1002/adsc.201201125
Korenaga, T.; Ko, A.; Uotani, K.; Tanaka, Y.; Sakai, T. Angew. Chem., Int. Ed. 2011, 50, 10703. doi: 10.1002/anie.201104588
Liao, Y. X.; Xing, C. H.; Hu, Q. S. Org. Lett. 2012, 14, 1544. doi: 10.1021/ol300275s
Huang, L. W.; Zhu, J. B.; Jiao, G. J.; Wang, Z.; Yu, X. X.; Deng, W. P.; Tang, W. J. Angew. Chem., Int. Ed. 2016, 55, 4527. doi: 10.1002/anie.201600979
Bouffard, J.; Itami, K. Org. Lett. 2009, 11, 4410. doi: 10.1021/ol9017613
Liu, G. X.; Lu, X. Y. J. Am. Chem. Soc. 2006, 128, 16504. doi: 10.1021/ja0672425
Gallego, G. M.; Sarpong, R. Chem. Sci. 2012, 3, 1338. doi: 10.1039/c2sc01068b
Low, D. W.; Pattison, G.; Wieczysty, M. D.; Churchill, G. H.; Lam, H. W. Org. Lett. 2012, 14, 2548. doi: 10.1021/ol300845q
Zhu, D.-X.; Chen, W.-W.; Xu, M.-H. Tetrahedron 2016. 72, 2037.
陈雕, 徐明华, 有机化学, 2017, 37, 1589. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346089.shtmlChen, D.; Xu, M.-H. Chin. J. Org. Chem. 2017, 37, 1589 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346089.shtml

图 2 含3-羟基吲哚的活性分子
Figure 2 Biologically active molecules containing 3-hydroxy- 2-oxindoles

图式 6 不对称芳基加成-环化策略制备手性杂环
Scheme 6 Asymmetric arylation–cyclization strategy for the synthesis of chiral heterocycles

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:






























































 下载:
下载:










 下载:
下载: