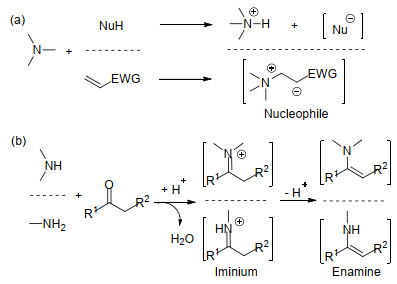
图式 1.
胺作为有机催化剂的活化模式
Scheme 1.
Activation modes of amine organocatalysts

由于能实现手性增值, 通过不对称催化反应是获得手性化合物的最为高效的一种方法, 其中, 发展高效的手性催化剂是各种不对称反应获得高活性和高对映选择性的关键[1].在人工设计化学催化剂方面, 继金属催化之后, 不含金属的有机小分子催化剂由于操作简单、对空气和水不敏感, 并且得到的产物无重金属残留等优点, 已经迅速发展并成为有机化学中最活跃和最有吸引力的研究领域之一[2~5].
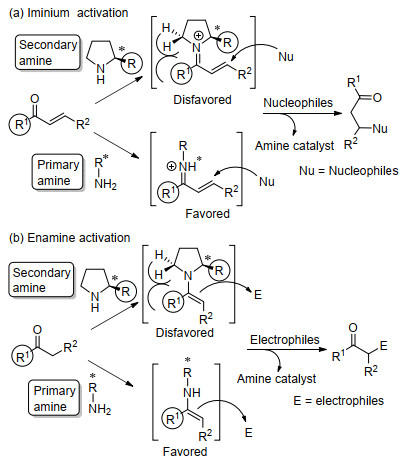
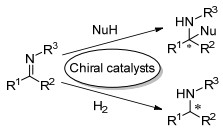
手性有机胺广泛分布在自然界中, 手性来源广泛.用手性有机胺化合物直接作为催化剂, 不仅原料易得, 并且手性骨架结构多样, 能有效进行改造和修饰, 可以得到结构丰富的手性催化剂.因而, 近年来在有机催化发展中, 对手性有机胺催化剂进行了大量研究, 已经证实手性有机胺催化剂对大量的不对称反应取得了好的活性和优秀的立体选择性.有机胺类化合物按氮上的取代基不同可以分为叔胺(Tertiary Amine)、仲胺(Secondary Amine)和伯胺(Primary Amine)三类, 因它们具有不同的化学性质, 导致对底物的活化模式各有特点.普遍情况下如Scheme 1所示, 手性叔胺催化剂一般起有机碱的作用, 既能作为Brønsted碱与质子结合, 又能作为Lewis碱进入空轨道, 活化底物形成具有活性的亲核给体(Scheme 1a)[6, 7]; 而手性伯胺和仲胺催化剂能与羰基化合物作用, 形成具有更低LUMO轨道能量的亚胺盐活性中间体[8]或具有更高HOMO轨道能量的烯胺中间体[9, 10](Scheme 1b).
伯胺催化剂和仲胺催化剂在不对称反应中对底物的活化模式有很多相似点, 一般情况下, 如Scheme 2所示, 仲胺和伯胺作为催化剂与羰基化合物形成亚胺离子中间体或烯胺中间体.通过亚胺离子活化能降低亲电试剂的LUMO能级, 使其更易受亲核试剂的进攻(Scheme 2a)[8]; 而通过烯胺中间体增加了亲核试剂的HOMO能级, 促进其与亲电试剂的反应(Scheme 2b)[9, 10].约二十年前, List等[11]将脯氨酸用于催化未修饰的丙酮和各种醛的直接不对称Aldol反应后, 标志着有机催化研究领域的复兴.在这阶段前十多年, 以脯氨酸及其衍生物[12]和手性联芳基骨架[13]为代表的仲胺有机催化剂蓬勃发展, 实现了催化大量高对映选择性的反应[8~10, 11, 12].最近十多年, 手性伯胺催化剂的研究也取得了许多令人振奋的发现, 在多种类型的不对称催化反应中表现出高效的催化活性和立体选择性[14~19].其中罗三中[17, 18, 20]和陈应春[15, 21]等不仅在手性伯胺催化剂早期发展中参与了开创性的工作, 而且在最近十年来也发展了一些用手性伯胺催化剂高对映选择性的化学反应.同样是通过烯胺和亚胺离子活化, 伯胺和仲胺催化各有特点, 在实现高对映选择性的反应中互为补充.相比于仲胺催化剂, 伯胺催化剂主要特色如下:一是在形成的亚胺盐或烯胺中间体结构中, 伯胺催化剂氮原子上存在的氢, 既可以促进活性催化中间体的有效形成, 也可能稳定过渡态的立体构型, 从而得到高立体选择性产物; 二是伯胺相对具有较小的位阻, 可以克服含有较大取代基羰基化合物作为底物存在的位阻问题, 例如α, β-不饱和酮和α取代的α, β-不饱和醛等[18].在自然界中, 天然酶如Ⅰ型醛缩酶和脱羧酶通常是使用赖氨酸部分的伯胺形成烯胺中间体进行催化的[22].
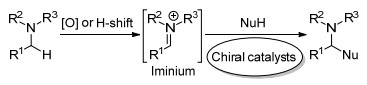
手性含氮化合物广泛分布在天然产物中, 其含氮结构单元对它们的生物活性起着很重要的作用, 并且由于氮的孤对电子能作为一个碱, 使得它们自己本身就能作为一个手性配体起到不对称催化作用, 它们也是一类重要的有机中间体和手性助剂.因此, 实现高效、高选择性的手性含氮化合物的合成有着重要的意义.从原子经济性的角度来说, 对于手性含氮化合物的合成最有效的策略是对含有C=N潜手性的亚胺不对称加成[23, 24], 也包括酮亚胺的不对称氢化[25, 26], 这样所得到的产物手性中心是产生在离氮最近的α位, 通过在这类反应中加入手性催化剂进行手性增值的催化不对称合成方法无疑是获得这类手性含氮化合物最高效途径(Scheme 3).然而, 相比于对含潜手性C=C键和C=O键化合物的不对称反应, 伯胺催化含潜手性C=N键的亚胺化合物的不对称反应这一具有挑战的领域发展相对缓慢.在研究环状亚胺的一些不对称反应时, 我们发现伯胺有机催化剂能适用于这类反应, 从而获得高对映选择性.因此, 本文对伯胺作为有机催化剂对含有C=N键的亚胺的不对称反应进行综述, 其中所定义的亚胺的不对称反应特指对潜手性C=N键的对映选择性加成得到含有α位手性中心的含氮化合物结构.
另外, 本文的亚胺针对一个较广的范围, 不仅为上面提到的直接用亚胺作为底物, 而且也包括在反应过程中涉及到对含有潜手性碳氮双键中间体的对映选择性加成反应.因此, 对于α位含有亚甲基的胺类化合物, 通过氧化或者氢迁移反应能原位产生C=N键中间体(亚胺盐), 然后在手性伯胺催化剂存在下, 对碳氮双键进行对映选择性加成得到产物, 即总反应为胺的α位C(sp3)—H催化不对称官能团化(Scheme 4)[27, 28], 该类型反应也在本文综述范围内.
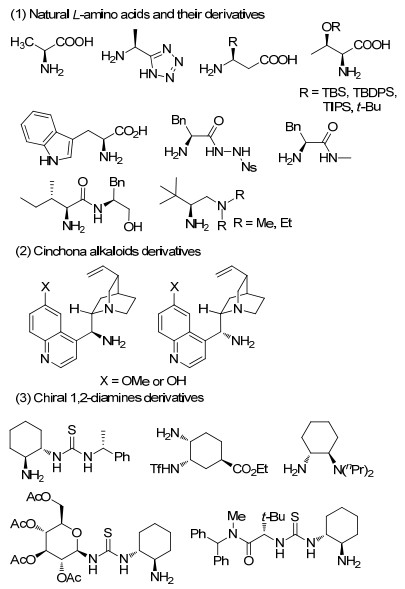
用于亚胺的不对称反应的手性伯胺催化剂, 结构如图 1所示.按伯胺官能团所在的手性骨架来源, 可以将它们分为以下三类: (1)天然L型氨基酸及其衍生的手性伯胺催化剂(Natural L-amino acids and their derivatives); (2)金鸡纳生物碱衍生的手性伯胺催化剂(Cinchona alkaloids derivatives); (3)手性1, 2二胺衍生的伯胺催化剂(Chiral 1, 2-diamines derivatives).下面将按所用到的这三类催化剂的类型进行分类, 对其在亚胺中的不对称反应中的应用进行讨论.
氨基酸由于其结构简单、廉价易得、可控位点较多等优点, 已成为最具价值的有机催化剂来源之一.直接利用非环状天然L型氨基酸就能作为伯胺催化剂, 并且氨基酸种类较多且易于修饰, 在对含C=N键化合物的不对称反应表现出好的活性和高的立体选择性.
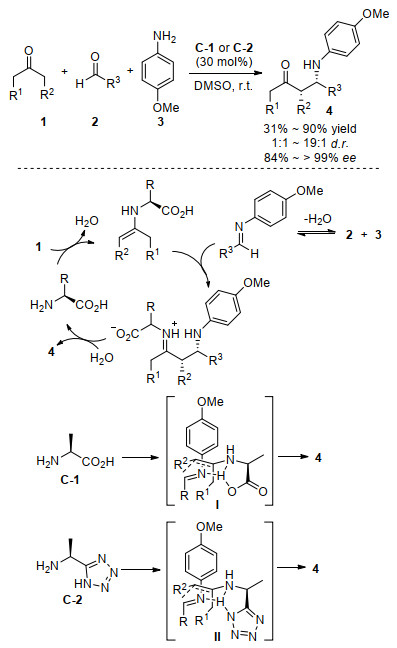
2005年, Córdova研究组[29]报道了第一个由伯胺氨基酸催化的不对称Mannich反应.如Scheme 5所示, 最优的催化剂为简单的丙氨酸C-1和丙氨酸-四唑衍生物C-2, 对酮1、醛2和对茴香胺3的三组分反应, 以很高的化学选择性和立体选择性得到相应的顺式为主的Mannich产物4, 其ee可高达99%以上.对反应条件的筛选表明, 简单非环状手性天然L型氨基酸, 如丙氨酸、丝氨酸、缬氨酸、苯丙氨酸、天冬氨酸和异亮氨酸等催化该反应都具有很好的化学选择性, 丙氨酸C-1和丙氨酸-四唑衍生物C-2给出了最高的不对称诱导.当供体为链状或环状酮时, 该反应均表现出很好的非对映选择性和对映选择性.作者提出的反应机理为经过伯胺催化剂与羰基给体形成烯胺中间体完成, 原位生成的亚胺与该手性烯胺中间体反应, 得到的亚胺离子产物水解后得到相应的Mannich产物.通过对产物绝对构型的确认, 推出丙氨酸C-1和丙氨酸-四唑衍生物C-2催化的不对称Mannich反应分别是通过六元椅式过渡态Ⅰ和Ⅱ进行的, 其中催化生成的手性烯胺的Si面与原位生成的受体亚胺的Si面接触.
2007年, Barbas等[30]用手性伯氨基酸作为催化剂实现了反式Mannich反应和顺式Aldol反应, 如Scheme 6所示, 对羟基酮5的三组分反式Mannich反应能得到具有光学活性的反式-1, 2-氨基醇6 (Scheme 6). O-叔丁基- L-苏氨酸C-3和L-色氨酸C-4被证明是最好的催化剂, 用N-甲基吡咯烷酮(NMP)作溶剂时, 适合用O-叔丁基- L-苏氨酸C-3作为催化剂, N, N-二甲基甲酰胺(DMF)作为溶剂时, 适合用L-色氨酸C-4作为催化剂.在标准反应条件下, 羟基丙酮5还可以与对硝基苯甲醛发生顺式-Aldol反应从而得到顺式-1, 2-二醇, 获得了>95%的收率、18/1顺式dr值和98% ee值.
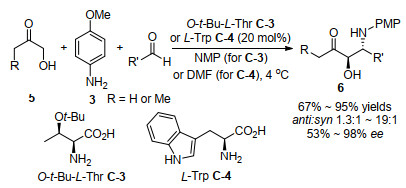
该项研究工作[30]的亮点在于用伯胺催化剂实现了反式选择性的Mannich反应, 而仲胺催化剂得到的是顺式选择性的Mannich反应.如Scheme 7所示, 对仲胺和伯胺催化的中间体进行比较, 对这种不同选择性进行了合理分析.仲胺催化剂(S)-脯氨酸与α-羟基酮形成两种烯胺中间体, 由于空间位阻作用, 发生Mannich反应时(E)-烯胺中间体A优于(Z)-烯胺中间体B, 从而占主导地位(Scheme 7a), (E)-烯胺A与亚胺发生C—C键加成形成过渡态C, 最后得到顺式-Mannich产物.当伯胺催化剂与α-羟基酮形成烯胺中间体时, 由于(Z)-烯胺中的羟基可以和伯胺N原子形成分子内氢键, 所以伯胺催化剂所形成的(Z)-烯胺中间体D优于(E)-烯胺中间体E (Scheme 7b), (Z)-烯胺D与亚胺发生C—C键加成形成过渡态F, 最后反应得到反式-Mannich产物.
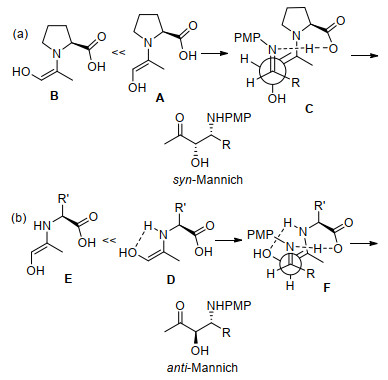
紧随其后, Lu课题组[31]也报道了L-苏氨酸衍生物C-5能有效催化对苄基羟基丙酮7、茴香胺3和芳香醛或脂肪醛在水相中的直接反式Mannich反应, 得到具有高对映选择性(高达97% ee)的反式1, 2-氨基醇8 (Scheme 8).该工作首次实现了可以用伯胺氨基酸在水溶剂中催化三组分的直接Mannich反应, 在没有水的情况下, 对映选择性较低.对催化剂的筛选表明苏氨酸衍生的疏水性有机催化剂比丝氨酸衍生的有机催化剂更有效.优化的催化反应条件对各种芳香醛和脂肪醛都能得到相应的具有很好对映体选择性的Mannich产物.将氨基醇转化为相应的Boc保护的噁唑烷酮, 并与单一构型的已知化合物进行比较, 确定了产物的相对构型和绝对构型.为了解释立体化学结果, 作者提出Scheme 8所示的含多个氢键的过渡态, 由O-苄基羟基丙酮和O-(叔丁基二苯基硅烷)苏氨酸催化剂生成的烯胺结构的几何构型为Z, 这可能是由于疏水基团的疏水相互作用或芳香组分的π-π堆积所致.
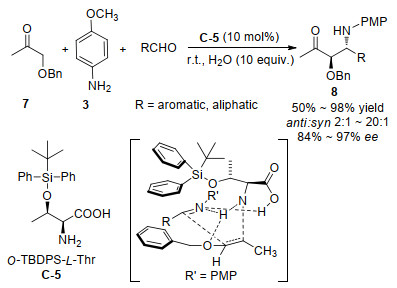
同一年, Lu等[32]用L-苏氨酸衍生C-6作为催化剂, 继续实现了对O-TBS-羟基丙酮9与芳香醛衍生的N-对甲苯磺酰亚胺10发生反式选择性的Mannich反应, 并以良好的收率和几乎99%的对映选择性以及最高达102:1的非对映选择性得到了产物11 (Scheme 9).不考虑芳醛的电子性质, 该方法几乎适用于任何芳香醛, 但是当使用脂肪醛作为底物时, Mannich反应不能够进行.亚胺的氮取代基对于反应的进行至关重要, 使用甲磺酰基取代的亚胺也能得到98%的ee和14:1的dr, 而对N上带有PMP、Bn或Boc的亚胺只能得到非常差的对映选择性.天然色氨酸或苏氨酸对该反应是完全无效的, 说明苏氨酸上羟基位置带有空间位阻的硅氧取代基对反应顺利进行是关键的.为了解释反应的立体选择性, 作者也进行了相应的计算研究, 发现砜的两个氧原子分别与来自NH和羧酸上的活泼氢形成氢键而相互作用.
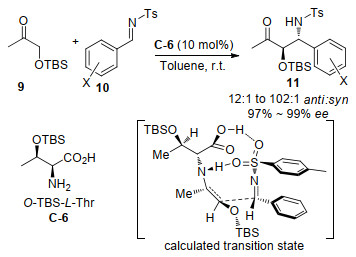
继上面Barbas和Lu发表伯氨基酸催化反式Mannich反应论文后同一年, Córdova课题组[33]也报道了用非环状的β-氨基酸催化环己酮和甘氨酸酯衍生的亚胺12的反式选择性Mannich反应.如Scheme 10所示, 对不同催化剂研究, 非环状β-氨基酸C-7、C-8和C-9催化的反应具有很好的反式选择性, 相比之下, α-氨基酸如丙氨酸C-1则以较高的顺式选择性催化反应.有趣的是, 添加少量从Aldrich购买的合成海水ASTM D665 (含0.5~0.9 mol/L NaCl)提高了对映选择性, 并加速了β-氨基酸催化的反应.当合成海水被0.8 mol/L的NaCl溶液替换时, 也能得到相似的结果.反应扩展到其它环状酮, 也获得了高的非对映选择性和对映选择性, 以高达19:1的dr和99% ee得到相应的手性氨基酸衍生物, 而使用支链酮如3-戊酮作为供体则反应时间慢, 收率低(17%), 但是有较高的对映选择性(88%)及非对映选择性(>19:1).
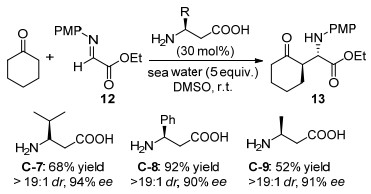
2008年, Barbas等[34]继续用O-叔丁基-L-苏氨酸C-3作为催化剂, 实现了二羟基丙酮衍生物14和对甲氧基苯基(PMP)取代的亚胺15不对称反式Mannich反应, 产物16为多羟基化合物的氨基糖结构(Scheme 11).通过简单的重结晶, 所得Mannich产物的非对映选择性和对映选择性可以提高到99:1的dr和99% ee.并且发现添加5-甲基四氮唑, 反应的速率和对映选择性均能提高.之前有报道[35]使用仲胺L-脯氨酸催化二羟基丙酮衍生物不对称Mannich反应, 但是只能得到顺式的Mannich产物, 并且二羟基丙酮衍生物的底物范围只能限制为2, 2-二甲基-1, 3-二噁烷-5-酮.该方法得到的反式Mannich产物能应用于合成各类含胺基的碳水化合物, 对基于脯氨酸催化策略合成该产物体系是进一步补充.
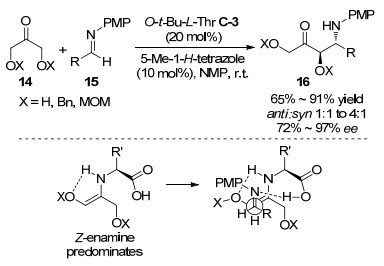
2013年王锐等[36]通过手性离子对催化策略实现了无金属催化的不对称氧化脱氢偶联反应, 如Scheme 12所示, 在苯丙氨酸C-10作为催化剂和2, 3-二氯-5, 6-二氰对苯醌(DDQ)作为氧化剂条件下, 不同取代的四氢异喹啉17和各种酮能发生脱氢偶联反应生成旋光活性的碳1位烷基化的四氢异喹啉衍生物18.底物扩展实验显示对环状的酮(环己酮和带杂原子的环状酮)能取得3:1~13:1 dr和61%~90% ee的立体选择性, 对于非环状的丁酮得到了较差的30% ee的对映选择性.作者认为反应过程包含有单电子转移(SET)阳离子机理过程, 即四氢异喹啉在DDQ存在下发生单电子转移得到自由基阳离子, 实验推测随后快速的发生不可逆的氢转移过程形成了含有环内碳氮双键的亚胺盐中间体, 该亚胺盐中间体与催化剂C-10和酮形成的烯胺羧基阴离子形成离子对过渡态, 最终通过手性伯胺催化酮对碳氮双键的加成完成反应.
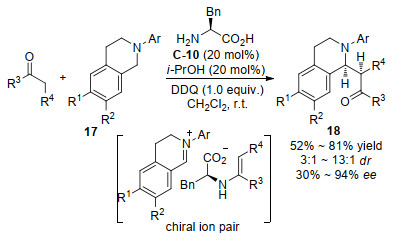
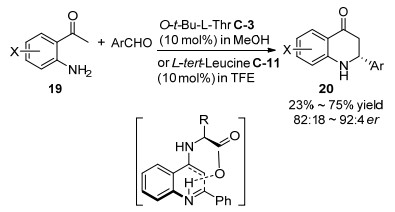
2014年Pan等[37]发现L-苏氨酸衍生物C-3或L-叔亮氨酸C-11催化2-氨基苯乙酮19与醛的不对称分子内的Mannich反应, 以23%~75%收率和好的对映选择性获得2-芳基-2, 3-二氢-4-奎诺酮20 (Scheme 13).对一些手性伯胺氨基酸作为催化剂进行了筛选, 发现用OtBu-L-苏氨酸C-3催化剂在甲醇中能得到最好的对映选择性, 而用L-叔亮氨酸C-11作催化剂在三氟乙醇(TFE)能得到最好的对映选择性, 作者最终选用OtBu-L-苏氨酸作催化剂进行了底物拓展.研究结果证实手性伯胺在该反应的催化效果要优于手性仲胺如脯氨酸.机理被认为是伯胺催化剂与酮形成烯胺中间体, 然后从新形成的碳氮双键的Si面进攻, 并且催化剂的羧基同时活化了碳氮双键.
2015年汪志勇和罗德平等[38]报道了L-苯丙氨酸衍生的含有多个氢键的双官能团手性伯胺磺酰肼C-12催化的五元环状酮亚胺21与酮的非对映选择性和对映选择性Mannich反应, 有效地构建了四取代的α-氨基酯衍生物(Scheme 14).在这一有效的转化过程中, 链状的酮和环状酮都可以用作底物, 分别得到α-氨基酯22和23.用活性较低的苯乙酮代替丙酮作为亲核试剂时, Mannich产物仍具有好的80% ee和90%的收率.另外, 用乙醇洗涤产物, 浓缩滤液后得到98% ee的白色固体, 可进一步提高产物的对映体选择性.以α-氨基酯产物22为原料, 分三步可以合成具有生物活性的螺四氢呋喃, 生理活性测试显示α-氨基酯产物23的其中一个具有潜在的HIV-1抑制剂活性.
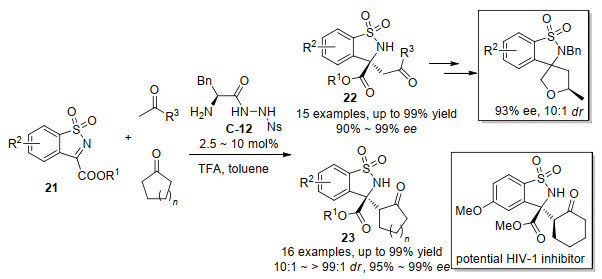
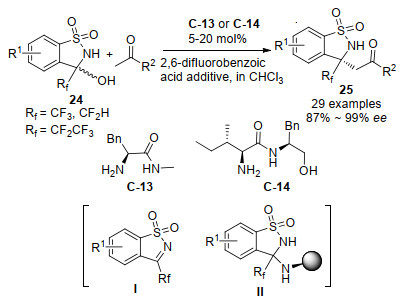
随后, 汪志勇等[39]用L-苯丙氨酸衍生的C-13或L-异亮氨酸衍生的C-14作为催化剂, 报道了氟烷基取代的醇胺24与酮的不对称脱水Mannich反应, 直接获得了一系列具有潜在生物活性的β-氨基酮25 (Scheme 15).使用两种不同的氨基酸伯胺催化剂对于烷基酮或者芳基酮的底物都具有非常好的对映选择性(高达99%), 在该Mannich反应中, 以前较少探索的芳基酮显示出很好的反应性.作者通过19F NMR检测到了参与脱水Mannich反应的两个中间产物Ⅰ和Ⅱ, 并通过HRMS进一步确定了中间体的结构.考虑到反应中存在原位生成的酮亚胺, 在标准反应条件下, 作者对脱水的酮亚胺底物进行了尝试, 得到了92%ee和99%的收率, 这一发现证实了该脱水Mannich反应经过原位产生的酮亚胺中间体完成.
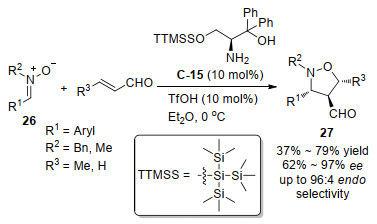
硝酮(Nitrone)是亚胺的N-氧化物, 对用其作为底物涉及到碳氮双键参与反应产生α位立体手性中心含氮化合物的反应也进行介绍.硝酮的碳氮双键除了具有亚胺作为亲电受体的性质外, 其与氮相邻的氧还能作为亲核给体, 因此能作为1, 3-偶极体发生1, 3-偶极环加成反应. 2015年, Kwon和Nakano[40]设计合成了一类含有超大体积硅基伯氨基醇C-15作为有机催化剂, 能成功应用于硝酮26和α, β不饱和醛(巴豆醛和丙烯醛)的1, 3-偶极环加成反应(Scheme 16).在对不同硝酮的底物实验中均得到endo构型选择性的五元环异恶唑啉27为产物, 并取得了67%~97% ee高的对映选择性, 用巴豆醛作为底物取得了好的非对映选择性(89:11~96:4 endo/exo), 优于丙烯醛作为底物的非对映选择性(61:39 endo/ exo).得到的异恶唑啉产物能通过还原开环转化为含有三个手性中心的γ氨基二醇.作者推测的反应过程为伯胺催化剂、三氟甲磺酸和α, β不饱和醛现成具有更低LUMO轨道能量的亚胺盐活性中间体, 然后该中间体与硝酮发生环加成反应, 过渡态的理论计算显示超大体积硅基TTMSS (tris(trimethylsilyl)silyl)作为位阻基团对反应的选择性起到重要的作用, 进一步理论计算显示中间体亚胺盐活性中间体的LUMO和硝酮的HOMO轨道相互作用是匹配的, 允许环加成反应发生.
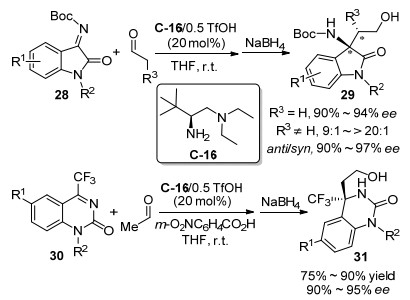
2017年, 邵志会等[41]报道了L-叔亮氨酸衍生的手性伯胺C-16催化的醛与酮亚胺28和30的不对称Mannich反应(Scheme 17). N-Boc靛红酮亚胺28和醛在有机溶剂或水性介质中能发生对映选择性Mannich反应, 对于乙醛能得到90%~94% ee值, 对其它醛得到高非对映选择性和对映选择性的反式Mannich产物29 (90%~97%ee).值得注意的是, 当使用脯氨酸衍生的仲胺催化该反应, 则得到与之相反的顺式Mannich产物, 也具有优异的对映选择性(92%~99% ee).该伯胺催化体系对环状的三氟甲基取代的酮亚胺30和乙醛也能实现高对映选择性的Mannich反应, 以90%~95% ee值获得产物31, 而使用手性仲胺作催化剂则不能催化该反应的进行.随后, Houk等[42]通过密度泛函理论(density functional theory)计算和扭曲相互作用(distortion-intera- ction)分析, 从理论上证明了该Mannich反应的手性产生过程.计算结果表明, 优势过渡态虽然为含有氢键的九元环状结构, 但是在能量上非常稳定, 该优势过渡态为含有八个重原子的冠状(椅式−椅式)构象, 亚胺底物的氮上Boc基团和伯胺催化剂C-16的叔丁基之间相互引起位阻作用.
2017年罗三中等[43]开发了亚胺前体试剂N, O-乙缩醛32的不对称Mannich反应, 在手性伯胺C-17催化作用下, 以近乎光学纯的立体选择性地高效合成了一系列β-氨基羰基以及α-氨基酸酯化合物33 (Scheme 18).甲醛的不对称Mannich反应(也称为氨甲基化反应)以及乙醛酸酯的不对称Mannich反应是合成手性β-氨基酸以及α-氨基酸类化合物的有效手段, 但是由于其不稳定容易自身聚合, 实现其不对称Mannich反应具有一定挑战.在该研究中, 设计了用甲醛与乙醛酸酯衍生的稳定亚胺前体试剂32, 并将其成功应用于链状和环状的β酮羰基化合物α位的不对称Mannich反应, 高效、立体专一地合成了手性的α-和β-氨基的羰基类化合物33, 该反应对其它非官能团酮底物, 如丙酮、环戊酮和环己酮, 也能获得高的对映选择性(94%~98% ee).基于实验结果以及该课题组以往的研究, 提出了如下的反应过渡态:首先, 亚胺前体试剂32在伯胺催化剂的作用下原位产生亚胺中间体, 烯胺从Re面与亚胺发生加成, 质子化叔胺的N—H键与亚胺(Ⅰ)或亚胺酯(Ⅱ)之间的氢键作用决定了反应具有很高的对映选择性以及非对映选择性.随后, 该研究团队[44]将同样的Mannich反应继续拓展到了含氟或含氯基团取代的N, O-乙缩醛, 即Scheme 18中N, O-乙缩醛32的R为CF3, CF2H, C2F5和CCl3等基团, 用同样的手性伯胺C-17和三氟甲基磺酸盐作为催化剂.主要进行了两类底物的拓展实验, 对含CF3的N, O-乙缩醛进行底物实验时N上保护基为Cbz时立体选择性最佳, 取得了98%~>99% ee优秀对映选择性, 不同酮底物导致了差别比较大的非对映选择性; 对含CF2H的N, O-乙缩醛进行底物实验时N上保护基为Fmoc时立体选择性最佳, 取得了95%~>99% ee优秀对映选择性.
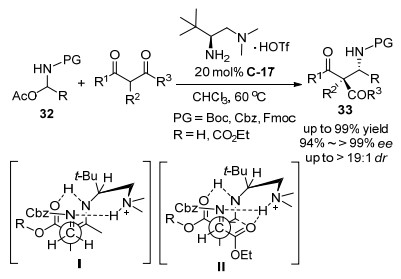
2018年, Rueping等[45]用可见光氧化还原催化剂和手性伯胺有机催化剂合并的催化体系, 实现了四氢异喹啉17和环状酮的氧化交叉脱氢偶联的不对称α-烷基化反应(Scheme 19).在这一双催化体系中, 四氢异喹啉17被可见光氧化还原催化剂活化, 酮被手性伯胺催化剂活化, 在对一系列伯氨基酸作为手性伯胺催化剂进行筛选中, 发现最优的手性伯胺催化剂为苯甘氨酸C-18.并且该反应的加料顺序能明显提高对映选择性, 通过对可见光催化剂和手性伯胺催化剂进行分段试验操作的方式, 在优化条件下, 能以好的收率、高的对映和非对映选择性得到目标产物34.
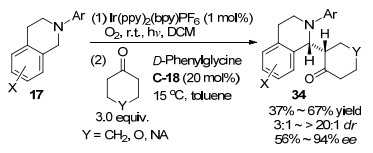
2019年, 罗三中等[46]用其研究团队开发的手性伯胺C-16和三氟甲磺酸成盐作为伯胺有机催化剂, 也能实现硝酮26和环状烯酮35的不对称1, 3-二偶极环加成反应, 以好到优秀的对映和非对映选择性得到含有多个立体中心的稠合双环异恶唑啉结构化合物36 (Scheme 20).作者也给出了该反应的机理过程, 伯胺催化剂与烯酮形成具有更低LUMO轨道能量的亚胺盐中间体作为亲偶极体, 随后通过如Scheme 20所示的过渡态发生环加成反应, 硝酮可以认为既作为氧亲核给体又作为含有碳氮双键的亲电受体.
金鸡纳碱是存在于金鸡纳类植物树皮中的一类天然产物, 目前分离出来的最受关注的是奎宁、奎尼丁、辛可宁、辛可尼丁四种.金鸡纳生物碱及其衍生物一直以来都以医学药用为主, 近30年来人们才将其用作手性试剂、手性配体以及优先考虑的手性催化剂用于不对称合成中.其中, 用于不对称反应的伯胺催化作用的金鸡纳生物碱催化剂均为9-位含有伯胺官能团的金鸡纳生物碱衍生物[15, 16, 47].
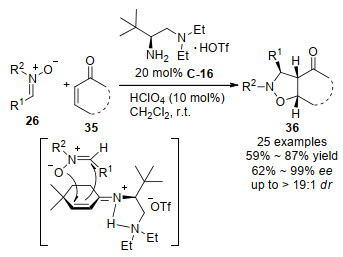
2007年, 四川大学的陈应春等[48]用金鸡纳生物碱衍生的具有多重官能团的伯胺催化剂C-19, 实现了对映选择性的偶氮次甲基亚胺37和环己烯酮35a的1, 3-二偶极[3+2]环加成, 以优秀的立体选择性(dr>99:1, 86%~95% ee)构建了一类新颖的三环产物38 (Scheme 21).反应条件优化显示了不同的酸对反应活性和对映选择性影响很大, 最后筛选最优的酸为2, 4, 6-三异丙基苯磺酸(TIPBA), 分子筛的加入虽然提高了反应的对映选择性, 但降低了反应活性.在催化模型中, 催化剂喹啉环上的酚羟基与1, 3-二偶极体形成的氢键, 对反应所得的高立体选择性起到了必要的控制作用.
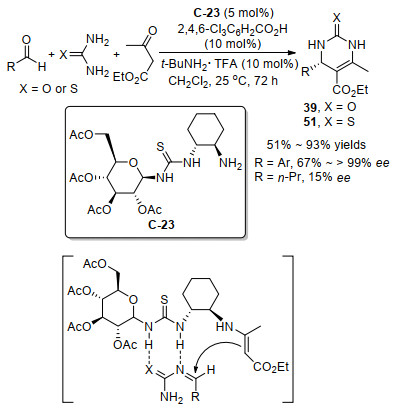
在2010年, Zhao课题组[49]和Xu课题组[50]先后独立报道了奎宁衍生的伯胺C-20作为催化剂, 实现了同样类型的不对称Biginelli反应, 金鸡纳生物碱衍生的伯胺催化剂也能实现对映选择性的Biginelli反应, Biginelli反应虽然没有直接用到亚胺作为底物进行反应, 但是在该反应过程中, 尿素与醛原位产生N-酰基亚胺中间体, 随后其碳氮双键接受伯胺与乙酰乙酸乙酯生成的手性烯胺中间体作为碳亲核试剂的进攻, 从而产生新的手性中心, 最后得到手性的二氢吡啶二酮衍生物39作为产物(Scheme 22). Zhao等[49]用伯胺C-20和氯化氢作为酸共催化可以获得最高78%的ee值.紧随其后, Xu等[50]发现金属Lewis酸作为共催化剂能得到更好的反应活性和更高的对映选择性, 相比更低负载量伯胺催化剂情况下, 获得更好的收率和最高84% ee值. Xu课题组进一步试验发现, 对于模型底物得到的产物(65% ee), 通过简单的一次乙醇重结晶, 即可获得优秀的>99% ee值.随后2012年, 周伟等[51]也用同样的手性伯胺与金属盐的复合催化体系, 研究了这个Biginelli反应, 对大量的手性伯胺催化剂和金属盐进行筛选, 筛选的伯胺催化剂除了有金鸡纳生物碱衍生的伯胺, 还有手性1, 2二胺衍生的伯胺以及手性1, 2二胺和手性氨基酸组合起来的伯胺, 最后仍然是用奎宁衍生的伯胺C-20和NbC15共催化取得最好的结果(74%收率和69% ee).
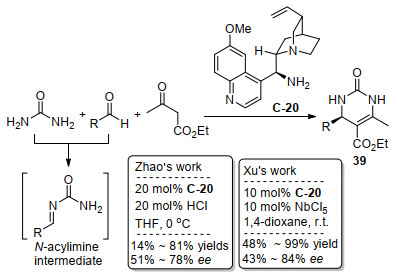
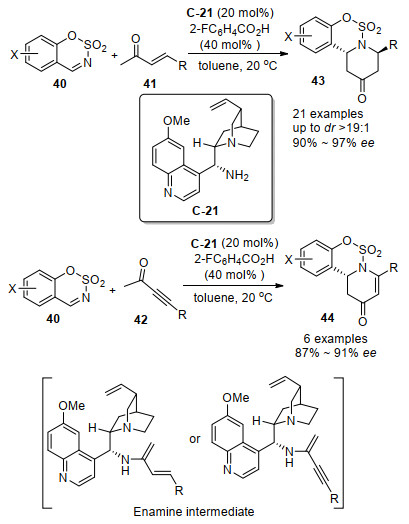
2013年, 康泰然、刘全忠和何龙等[52]用奎尼丁衍生的伯胺C-21和2-氟苯甲酸合并作为催化剂, 实现了烯酮41或者炔酮42对氮磺酰基环状亚胺40的[4+2]环加成反应合成了2, 6-二取代的哌啶-4-酮43或44, 对烯酮底物41取得了优秀的90%~97% ee值, 而对炔酮底物32取得了略微降低的87%~91% ee值(Scheme 23).作者提到, 对于烯酮41和氮磺酰基环状亚胺40的反应, 有少量的Mannich反应产物被观察到.对于该环加成反应, 烯酮41或者炔酮42与伯胺催化剂形成烯胺活性中间体, 接下来烯胺与亚胺底物完成反应存在两种可能, 尽管作者倾向于经过分步的Mannich-Michael加成反应机理, 但是也不能排除另一种的协同反应机理.
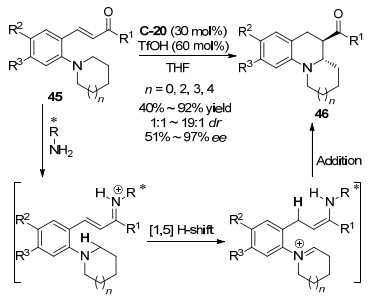
2013年, Kim等[53]报道了用奎宁衍生的伯胺C-20实现了四氢异喹啉45分子内的不对称C—H官能团化(Scheme 24).该反应的亚胺盐形成过程和氧化脱氢偶联反应不同, 手性伯胺催化剂与底物作用后, 经过1, 5-氢迁移得到了烯胺-亚胺盐的中间体, 该中间体的环化包含了伯胺催化活化的分子内烯胺对亚胺盐的碳氮双键的对映选择性的加成过程.底物实验显示, 该催化体系对各种含有环状胺和非环状不饱和羰基官能团的底物都能顺利反应, 以51%~97% ee的对映选择性得到四氢喹啉衍生物46, 但是对于含有环状胺和环己酮官能团的底物只得到7% ee, 对含有非环状胺和非环状不饱和羰基官能团的底物不能得到目标产物, 这说明该催化反应体系只对邻位环状含氮官能团取代的芳基不饱和酮具有活性.
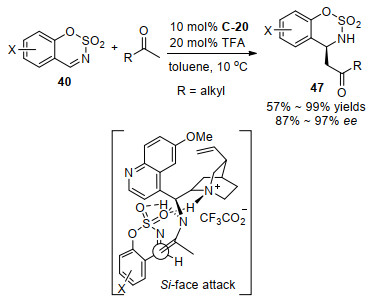
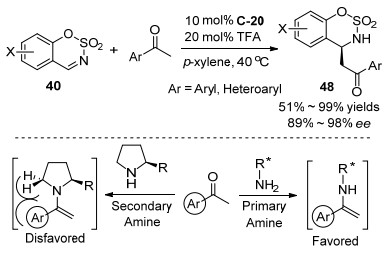
2014年, 我们课题组[54]报道了由奎宁衍生的伯胺C-20和TFA共催化的甲基烷基酮与环状亚胺苯并[e][1, 2, 3]恶噻嗪2, 2-二氧化物40的高对映选择性直接Mannich反应(Scheme 25).这是首次实现N-磺酰基环状亚胺作为受体的不对称Mannich反应, 用常见的仲胺催化剂脯氨酸的标准条件对该反应没有活性.对各种取代的环状亚胺40的底物实验, 都能取得高的对映选择性(93%~97%ee).在用不同的非对称的烷基甲基酮进行底物实验中, 该催化体系对酮作为给体表现出了高的区域选择性, 即完全专一选择在酮的α位位阻较小的甲基位置加成, 然而, 对于甲基叔丁基甲酮底物没有反应活性, 可能因为叔丁基较大位阻的原因.用X射线单晶衍射确定了产物的绝对构型, 推测了由丙酮和伯胺催化剂形成的烯胺中间体的碳亲核基团从Si面进攻N-磺酰基环状亚胺的碳氮双键.
随后, 我们课题组[55]用相似的伯胺催化体系实现了低反应活性的芳基酮的不对称Mannich反应.如Scheme 26所示, 在伯胺C-20和TFA共催化条件下能高对映选择性地构建一系列含有氮氧硫的杂环氨基磺酸酯类化合物48.反应对含不同取代基的亚胺底物40和各种芳基甲基酮都能给出高的对映选择性(89%~98% ee), 苯环上带有给电子基的取代芳基甲基酮在较短的反应时间内比带吸电子取代基的取代芳基甲基酮具有更高的产率, 由于空间位阻作用, 邻位取代芳基甲基酮的产率和对映体选择性均低于对位取代或间位取代的芳基甲基酮.进一步, 含有氮和硫的杂环芳基甲基酮也能用于该反应, 得到高的对映选择性.在该研究中, 用伯胺催化剂能实现活性较低的芳基酮的不对称Mannich反应, 伯胺催化剂与酮形成烯胺中间体, 相比仲胺催化剂, 其较小的位阻对具有α芳基的底物显示出相应的优势.
光学纯的手性1, 2-二胺如环己二胺、二苯二胺, 这类化合物作为手性源, 其原料廉价易得, 对其中一个氨基能方便地衍生为叔胺或其它含有氢键给体官能团, 能得到含有伯胺的双官能类催化剂, 近来对亚胺的不对称反应有少量成功的例子报道.
2008年, Tsogoeva研究小组[56]首次将手性伯胺-硫脲有机催化剂成功应用于未修饰酮的形式上的不对称Mannich反应.如Scheme 27所示, 用手性环己二胺衍生的伯胺-硫脲C-22作为催化剂, 对含有亚胺基甘氨酸乙酯49和各种链状与环状酮能顺利的反应, 以45%~89%产率和82%~99% ee得到产物50, 但是非对映选择性不是太理想, 非环状的酮得到反式产物, 而环状酮则得到过量的顺式产物.底物扩展实验显示含有支链或具有空间位阻的酮, 其ee值高达99%以上, 当底物49的R为苯甲酰基时, 苯环上的吸电子取代基提高了反应的速率, 而给电子取代基则降低了反应速率.不同于常规伯胺活化羰基化合物烯胺的机理, 该反应机理通过计算和实验证明以烯醇式的机理进行.这是第一个伯胺-硫脲催化的C—C键形成Mannich型反应的烯醇机理的证据, 进一步发现, 酮的烯醇式与催化剂的结合优先于烯胺的形成, 胺-硫脲可以通过氢键稳定酮的烯醇互变异构.
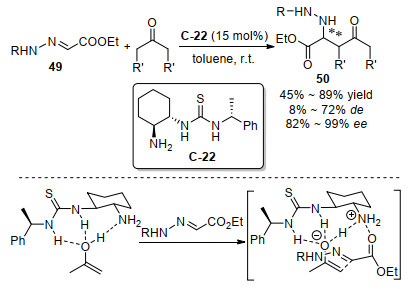
2009年, 苗志伟和陈茹玉等[57]用含有手性糖和手性1, 2-环己二胺结构单元的双官能团伯胺-硫脲催化剂C-23, 实现了高对映选择性的Biginelli反应, 合成了二氢嘧啶类产物39和51 (Scheme 28).底物实验发现, 该催化体系对芳基醛表现出好到高的对映选择性, 但是对脂肪族醛虽然也能得到目标产物, 但是只获得了很低的对映选择性.三氟乙酸的叔丁基胺盐加入能将ee值从63%提高到94%, 显著提高了反应的对映选择性.三元化合物的Biginelli反应虽然没有直接用亚胺作为底物, 但是在反应过程中经过醛和尿素或者硫脲原位产生的N-酰基亚胺中间体, 在这里伯胺催化剂与乙酰乙酸乙酯产生的烯胺中间体对N-酰基亚胺中间体的C=N立体选择性加成, 作为关键步骤产生新的手性中心.随后2011年, 他们研究团队[58]用同样的伯胺催化剂C-23在食盐水中也实现了Biginelli反应, 不同的是用三氟甲磺酸替代了2, 4, 6-三氯苯甲酸, 在研究不同浓度食盐水对反应影响时, 发现随着食盐浓度增加反应对映选择性增加, 最后在饱和食盐水中对各类不同醛底物获得了44%~>99% ee值.随后2013年, 王立新等[59]以基于手性1, 2-二苯基乙二胺骨架的伯胺-硫脲为催化剂(20 mol%), 加入樟脑磺酸(20 mol%), 实现了靛红、脲和乙酰乙酸乙酯的一个不对称Biginelli反应的例子, 虽然获得了79%的收率, 但是对映选择性非常低, 仅为22% ee值.
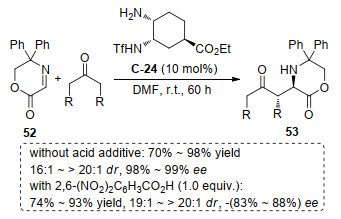
2012年, Maruoka小组[60]报道了用手性顺式环己二胺衍生的伯胺催化剂C-24催化环状亚胺52与酮的不对称Mannich反应, 在同样催化剂作用下, 仅仅在有或没有非手性酸作为添加物的情况下, 均能高对映选择性的获得不同绝对构型的产物53(Scheme 29).通过研究Brønsted酸作为添加物的过程中, 发现酸作为添加物对烯胺催化过程的立体选择性有很大的影响, 在反应中加入10 mol%的2, 6-二硝基苯甲酸可以有效地逆转相应产物的对映选择性, 同时保持良好的催化活性和立体选择性.在同样手性催化剂条件下, 能通过简单的改变条件同时获得两种不同对映体产物的目标, 这对手性合成是非常有用处的.进一步, 该催化体系也能用于酮酯作为受体的Aldol反应.有趣的是, 水的加入使Aldol反应对映选择性翻转, 但是水的加入对Mannich反应并没有使对映选择性翻转.
2017年, 罗三中和吴骊珠等[61]合作报道了可见光促进的四氢异喹啉17和酮的不对称脱氢偶联反应, 如Scheme 30所示, 该反应催化体系通过协同多重催化作用, 手性伯胺C-25和三氟甲磺酸盐的有机催化剂与酮作用形成烯胺中间体, 而通过Ru/Co催化作用让四氢异喹啉17原位产生亚胺盐中间体.该反应催化体系能应用于广泛的底物范围, 对多种不同类型的底物都能以合理的收率得到目标产物, 其中, 对不同取代的四氢异喹啉底物能取得3:1~12:1 dr和95%~99% ee的立体选择性, 对各种环状的酮和两个链状的酮能取得2:1~>19:1 dr和25%~97% ee的立体选择性, 对简单的丙酮取得了最差的25%的对映选择性, 对各种不同取代的4-取代环己酮的去对称化反应也能取得4:1~11:1 dr和87%~97% ee的立体选择性.值得注意的是, 尽管得到的产物54也为C1位烷基化的四氢异喹啉结构, 但是和上面提到的在手性伯氨基酸催化得到的产物18 (Scheme 12)或34 (Scheme 19)相对构型相反, 即互为非对映选择性异构体, 说明手性伯胺催化剂的骨架决定了最终产物的立体选择性.
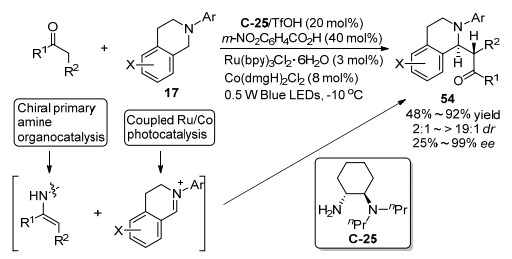
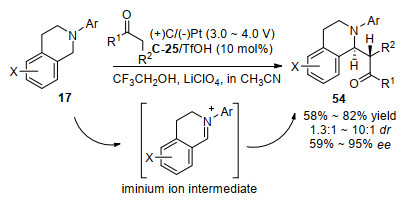
同一年, 罗三中小组[62]结合电化学氧化和手性伯胺催化的策略继续实现了四氢异喹啉17和酮的氧化偶联反应(Scheme 31).该催化体系主要拓展到了各种环状酮底物, 对各种大小的环状酮底物能得到好到高的对映选择性(74%~95% ee), 但是对大环的七元环状环庚酮取得了差的非对映选择性(1.5:1~1.3:1 dr).另外, 作者也报道了一个非环状亚胺作为底物的例子, 对α, β不饱和酮4-苯基-3-丁烯-2-酮, 改变伯胺催化剂为苯丙氨基酸衍生的伯-叔二胺为催化剂, 得到了中等的59% ee值.进一步用廉价的铅笔芯作为电极, 该电化学氧化偶联反应也能放大到毫摩尔规模, 对模型反应能获得63%的收率、9:1 dr和93% ee的立体选择性.通过进一步对照实验, 该反应机理过程认为先通过电化学氧化得到亚胺盐中间体, 然后与手性伯胺催化剂和酮形成的烯胺反应产生最后的产品.
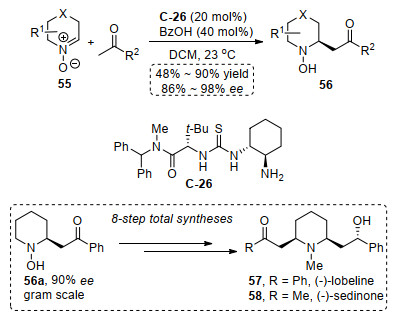
最近, Snyder小组[63]报道了含有环内碳氮双键的环状硝酮55作为亚胺底物与甲基酮的不对称Mannich型反应构建2-取代哌啶衍生物56, 用手性环己二胺衍生的双官能的伯胺硫脲衍生物C-26作为催化剂, 能得到高达98%的对映选择性(Scheme 32).作为底物的环状硝酮55可通过二级胺或羟胺的氧化合成得到, 并且作为稳定的环状亚胺等价物具有反应活性较强的E式碳氮双键结构.此外, Mannich产物中的羟胺部分可防止脂肪族胺α位外消旋的可能性, 且易被转化为其它含氮化合物.在没有催化剂的情况下, 环状硝酮和β-酮酸之间的脱羧Mannich反应可以在室温下进行, 因此不能用手性催化剂进行不对称诱导.最后, 以产物中的一个56a为起始原料, 经过8步全合成, 其中包含一步的β-酮酸的脱羧Mannich反应, 最后完成了天然产物(-)-lobeline (57)和(-)-sedinone (58)的全合成.这一开创性的工作可能激发更多有趣的通过有机催化向环状硝基化合物进行亲核加成的方法, 而且为制备手性氮杂环提供了一种潜在的更普适性的方法.
最近十几年, 手性伯胺化合物作为有机小分子催化剂的不对称加成反应已经取得了很大的发展, 尽管对含有潜手性的C=N键的亚胺的不对称加成反应是构建光学活性的含氮化合物最有效、最直接的方法, 但是应用手性伯胺催化剂对C=N键的高对映选择性的加成反应报道仍然较少.作为仲胺催化剂的补充, 伯胺催化剂通常也是通过烯胺和亚胺离子活化, 但是其在催化过程中含有独特的活泼质子和位阻小的特点, 使其也能在部分仲胺催化剂表现欠佳的反应中获得优秀的结果.并且, 用手性伯胺催化剂, 对部分亚胺的不对称加成反应被证明采用了不同寻常的烯醇式反应机理, 这也拓展了催化反应的新思路.基于伯胺催化剂在亚胺的不对称加成反应中表现的独特优良性能, 进一步研究拓展其它类型的伯胺催化剂, 用于新的含有C=N键的亚胺底物的不对称加成反应将具有重要的学术和应用价值.
李月明, 范青华, 陈新滋, 不对称有机反应, 北京化学工业出版社, 北京, 2005.Li, Y. M.; Fan, Q. H.; Chen, X. Z. Asymmetric Organic Reactions, Beijing Chemical Industry Press, Beijing, 2005 (in Chinese).
Dalko, P. I. Comprehensive Enantioselective Organocatalysis:Catalysts, Reactions, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013.
Jacobsen, E. N.; MacMillan, D. W. C. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20618. doi: 10.1073/pnas.1016087107
Maruoka, K.; List, B.; Yamamoto, H.; Gong, L.-Z. Chem. Commun. 2012, 48, 10703. doi: 10.1039/c2cc90327j
Zhan, G.; Du, W; Chen, Y.-C. Chem. Soc. Rev. 2017, 46, 1675. doi: 10.1039/C6CS00247A
Wurz, R. P. Chem. Rev. 2007, 107, 5570. doi: 10.1021/cr068370e
Palomo, C.; Oiarbide, M.; López, R. Chem. Soc. Rev. 2009, 38, 632. doi: 10.1039/B708453F
Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416. doi: 10.1021/cr068388p
Notz, W.; Tanaka, F.; Barbas, C. F., Ⅲ Acc. Chem. Res. 2004, 37, 580. doi: 10.1021/ar0300468
Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471. doi: 10.1021/cr0684016
List, B.; Lerner, R. A.; Barbas, C. F., Ⅲ. J. Am. Chem. Soc. 2000, 122, 2395. doi: 10.1021/ja994280y
Donslund, B. S.; Johansen, T. K.; Poulsen, P. H.; Halskov, K. S.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2015, 54, 13860. doi: 10.1002/anie.201503920
Kano, T.; Maruoka, K. Chem. Sci. 2013, 4, 907 doi: 10.1039/C2SC21612D
Xu, L.-W.; Luo, J.; Lu, Y. Chem. Commun. 2009, 1807.
Jiang, L.; Chen, Y.-C. Catal. Sci. Technol. 2011, 1, 354. doi: 10.1039/c0cy00096e
Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748. doi: 10.1002/anie.201109036
Zhang, L.; Luo, S. Synlett 2012, 1575. doi: 10.1055/s-0031-1290405
Zhang, L.; Fu, N.; Luo, S. Acc. Chem. Res. 2015, 48, 986. doi: 10.1021/acs.accounts.5b00028
Reddy, U. V. S.; Chennapuram, M.; Seki, C.; Kwon, E.; Okuyama, Y.; Nakano, H. Eur. J. Org. Chem. 2016, 4124.
Luo, S.; Xu, H.; Li, J.; Zhang, L.; Cheng, J.-P. J. Am. Chem. Soc. 2007, 129, 3074. doi: 10.1021/ja069372j
Xie, J.-W.; Chen, W.; Li, R.; Zeng, M.; Du, W.; Yue, L.; Chen, Y.-C.; Wu, Y.; Zhu, J.; Deng, J.-G. Angew. Chem., Int. Ed. 2007, 46, 389. doi: 10.1002/anie.200603612
Gefflaut, T.; Blonksi, C.; Perie, J.; Willson, M. Prog. Biophys. Molec. Biol. 1995, 63, 301. doi: 10.1016/0079-6107(95)00008-9
Enders, D.; Reinhold, U. Tetrahedron:Asymmetry 1997, 1895. doi: 10.1002/chin.199745303
Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626. doi: 10.1021/cr100204f
王东, 侯传金, 陈丽凤, 刘小宁, 安庆大, 胡向平, 有机化学, 2013, 33, 1355. doi: 10.6023/cjoc201211036Wang, D.; Hou, C.; Chen, L.; Liu, X.; An, Q.; Hu, X. Chin. J. Org. Chem. 2013, 33, 1355(in Chinese). doi: 10.6023/cjoc201211036
Xie, J.-H.; Zhu, S.-F.; Zhou, Q.-L. Chem. Rev. 2011, 111, 1713. doi: 10.1021/cr100218m
Qin, Y.; Lv, J.; Luo, S. Tetrahedron Lett. 2014, 55, 2423; doi: 10.1016/j.tetlet.2014.02.126
Cheng, M.-X.; Yang, S.-D. Synlett 2017, 28, 159.
Ibrahem, I.; Zou, W.; Engqvist, M.; Xu Y.; Córdova, A. Chem.-Eur. J. 2005, 11, 7024. doi: 10.1002/chem.200500746
Ramasastry, S. S. V.; Zhang, H.; Tanaka, F.; Barbas Ⅲ, C. F. J. Am. Chem. Soc. 2007, 129, 288. doi: 10.1021/ja0677012
Cheng, L.; Wu, X.; Lu, Y. Org. Biomol. Chem. 2007, 5, 1018. doi: 10.1039/b701579h
Cheng, L.; Han, X.; Huang, H.; Wong, M. W.; Lu, Y. Chem. Commun. 2007, 4143.
Dziedzic, P.; Córdova, A. Tetrahedron:Asymmetry 2007, 18, 1033. doi: 10.1016/j.tetasy.2007.04.024
Zhang, H.; Ramasastry, S. S. V.; Tanaka, F.; Barbas Ⅲ, C. F. Adv. Synth. Catal. 2008, 350, 791. doi: 10.1002/adsc.200800069
Westermann, B.; Neuhaus, C. Angew. Chem., Int. Ed. 2005, 44, 4077. doi: 10.1002/anie.200500297
Zhang, G.; Ma, Y.; Wang, S.; Kong, W.; Wang, R. Chem. Sci. 2013, 4, 2645. doi: 10.1039/c3sc50604e
Mondal, B.; Pan, S. C. Org. Biomol. Chem. 2014, 12, 9789. doi: 10.1039/C4OB02146K
Zhang, S.; Li, L.; Hu, Y.; Zha, Z.; Wang, Z.; Loh, T.-P. Org. Lett, 2015, 17, 1050. doi: 10.1021/acs.orglett.5b00196
Zhang, S.; Li, L.; Hu, Y.; Li, Y.; Yang, Y.; Zha, Z.; Wang, Z. Org. Lett. 2015, 17, 5036. doi: 10.1021/acs.orglett.5b02514
Otsuki, T.; Kumagai, J.; Kohari, Y.; Okuyama, Y.; Kwon, E.; Seki, C.; Uwai, K.; Mawatari, Y.; Kobayashi, N.; Iwasa, T.; Tokiwa, M.; Takeshita, M.; Maeda, A.; Hashimoto, A.; Turuga, K.; Nakano, H. Eur. J. Org. Chem. 2015, 7292.
Dai, J.; Xiong, D.; Yuan, T.; Liu, J.; Chen, T.; Shao, Z. Angew. Chem., Int. Ed. 2017, 56, 12697. doi: 10.1002/anie.201706304
Chen, S.; Houk, K. N. J. Org. Chem. 2018, 83, 3171. doi: 10.1021/acs.joc.8b00037
You, Y.; Zhang, L.; Cui, L.; Mi, X.; Luo, S. Angew. Chem., Int. Ed. 2017, 56, 13814. doi: 10.1002/anie.201707005
You, Y.; Luo, S. Org. Lett. 2018, 20, 7137. doi: 10.1021/acs.orglett.8b03083
Hou, H.; Zhu, S.; Atodiresei, I.; Rueping, M. Eur. J. Org. Chem. 2018, 1277.
Yang, Q.; Zhang, J.; Jia, Z.; Yang, C.; Zhang, L.; Luo, S. Asian J. Org. Chem. 2019, 8, 1049. doi: 10.1002/ajoc.201900013
Duan, J.; Li, P. Catal. Sci. Technol. 2014, 4, 311. doi: 10.1039/C3CY00739A
Chen, W.; Du, W.; Duan, Y.-Z.; Wu, Y.; Yang, S.-Y.; Chen, Y.-C. Angew. Chem., Int. Ed. 2007, 46, 7667. doi: 10.1002/anie.200702618
Ding, D.; Zhao, C.-G. Eur. J. Org. Chem. 2010, 3802. doi: 10.1002/ejoc.201000448
Cai, Y.-F.; Yang, H.-M.; Li, L.; Jiang, K.-Z.; Lai, G.-Q.; Jiang, J.-X.; Xu, L.-W. Eur. J. Org. Chem. 2010, 4986. doi: 10.1002/chin.201105159
周伟, 赵金刚, 林晶, 徐燕霞, 刘琚, 赵淑娟, 谢恬, 杭州师范大学学报(自然科学版), 2012, 11, 352. doi: 10.3969/j.issn.1674-232X.2012.04.013Zhou, W.; Zhao, J.-G.; Lin, J.; Xu, Y.-X.; Liu, J.; Zhao, S.-J.; Xie, T. J. Hangzhou Normal Univ. (Nat. Sci. Ed.) 2012, 11, 352(in Chinese). doi: 10.3969/j.issn.1674-232X.2012.04.013
Liu, Y.; Kang, T.-R.; Liu, Q.-Z.; Chen, L.-M.; Wang, Y.-C.; Liu, J.; Xie, Y.-M.; Yang, J.-L.; He, L. Org. Lett. 2013, 15, 6090. doi: 10.1021/ol402977w
Kang, Y. K.; Kim, D. Y. Adv. Synth. Catal. 2013, 355, 3131. doi: 10.1002/adsc.201300398
Wang, Y.-Q.; Cui, X.-Y.; Ren, Y.-Y.; Zhang, Y. Org. Biomol. Chem. 2014, 12, 9101. doi: 10.1039/C4OB01902D
Cui, X.-Y.; Duan, H.-X.; Zhang, Y.; Wang, Y.-Q. Chem. Asian J. 2016, 11, 3118. doi: 10.1002/asia.201601149
Yalalov, D. A.; Tsogoeva, S. B.; Shubina, T. E.; Martynova, I. M.; Clark, T. Angew. Chem., Int. Ed. 2008, 47, 6624. doi: 10.1002/anie.200800849
Wang, Y.; Yang, H.; Yu, J.; Miao, Z.; Chen, R. Adv. Synth. Catal. 2009, 351, 3057. doi: 10.1002/adsc.200900597
Wang, Y.; Yu, J.; Miao, Z.; Chen, R. Org. Biomol. Chem. 2011, 9, 3050. doi: 10.1039/c0ob01268h
杨清川, 彭林, 王斐英, 徐小英, 王立新, 合成化学, 2013, 21, 237. doi: 10.3969/j.issn.1005-1511.2013.02.028Yang, Q.-C.; Peng, L.; Wang, F.-Y.; Xu, X.-Y.; Wang, L.-X. Chin. J. Synth. Chem. 2013, 21, 237(in Chinese). doi: 10.3969/j.issn.1005-1511.2013.02.028
Moteki, S. A.; Han, J.; Arimitsu, S.; Akakura, M.; Nakayama, K.; Maruoka, K. Angew. Chem., Int. Ed. 2012, 51, 1187. doi: 10.1002/anie.201107239
Yang, Q.; Zhang, L.; Ye, C.; Luo, S.; Wu, L.-Z.; Tung, C.-H. Angew. Chem., Int. Ed. 2017, 56, 3694. doi: 10.1002/anie.201700572
Fu, N.; Li, L.; Yang, Q.; Luo, S. Org. Lett. 2017, 19, 2122. doi: 10.1021/acs.orglett.7b00746
Lisnyak, V. G.; Lynch-Colameta, T.; Snyder, S. A. Angew. Chem., Int. Ed. 2018, 57, 15162. doi: 10.1002/anie.201809799

图式 2 伯胺和仲胺在烯胺和亚胺离子的活化过程催化作用比较
Scheme 2 Catalysis comparison between primary amine and secondary amine in iminium and enamine activation modes

图式 4 胺的α位C(sp3)—H催化不对称官能团化
Scheme 4 Catalytic asymmetric α-C(sp3)—H functionalization of amines

图式 5 伯胺催化的三组分不对称Mannich反应
Scheme 5 Amino acid and its derivatives catalyzed asymmetric three-component Mannich reaction

图式 6 有机催化的三组分反式Mannich反应合成反式-1, 2-氨基醇
Scheme 6 Asymmetric synthesis of anti-1, 2-amino alcohols through organocatalytic three-component anti-Mannich reactions

图式 7 仲胺和伯胺催化分别得到顺式和反式Mannich产物机理
Scheme 7 Mechanism of syn- and anti-Mannich products catalyzed by secondary amine and primary amine respectively

图式 8 在纯水相中的不对称三组分Mannich反应
Scheme 8 Asymmetric three-component Mannich reactions in a purely aqueous system

图式 9 苏氨酸衍生物C-6催化N-甲苯磺酰亚胺的不对称Mannich反应
Scheme 9 Asymmetric Mannich reactions employing N-tosyl- imines catalyzed by C-6

图式 10 β-氨基酸催化的反式选择性不对称Mannich反应
Scheme 10 β-Amino acid-catalyzed asymmetric anti-selective Mannich-type reaction

图式 11 手性伯胺催化Mannich反应合成氨基糖
Scheme 11 Chiral primary amine catalyzed anti-Mannich reactions to synthesize amino sugars

图式 12 对映选择性的叔胺和酮的氧化脱氢偶联
Scheme 12 Enantioselective oxidative cross-dehydrogenative coupling of tertiary amines with ketones

图式 13 伯胺氨基酸催化的分子内的不对称Mannich反应
Scheme 13 Primary amino acid-catalyzed asymmetric intramolecular Mannich reaction

图式 14 环状氮磺酰基酮亚胺与酮的直接不对称Mannich反应
Scheme 14 Asymmetric Mannich reaction of cyclic sulfonyl ketimines with ketones

图式 15 氟烷基醇胺与酮的不对称脱水Mannich反应
Scheme 15 Asymmetric dehydrated Mannich reaction of fluoroalkylated hemiaminals with ketones

图式 16 硅氧伯氨基醇C-15催化硝酮的不对称1, 3-二偶极环加成
Scheme 16 Silyloxy primary amino alcohol C-15 catalyzed asymmetric 1, 3-dipolar cycloaddition of nitrones

图式 17 手性伯胺C-16催化酮亚胺的不对称Mannich反应
Scheme 17 Chiral primary amine C-16 catalyzed asymmetric Mannich reactions of ketimines

图式 18 N, O-缩醛被用作为N-氨甲酰基亚胺等价体的催化不对称Mannich反应
Scheme 18 Catalytic asymmetric Mannich reaction using N, O- acetals as N-carbamoyl imine surrogates

图式 19 四氢异喹啉和环状酮的不对称α-烷基化
Scheme 19 Asymmetric α-alkylation of tetrahydroisoquinolines with cyclic ketones

图式 20 伯胺催化作用的硝酮和烯酮的不对称1, 3-二偶极环加成
Scheme 20 Asymmetric 1, 3-dipolar cycloadditions between nitrones and enones by primary amine catalysis

图式 21 偶氮次甲基亚胺的对映选择性的1, 3-二偶极[3+2]环加成
Scheme 21 Enantioselective 1, 3-dipolar [3+2] cycloaddition of azomethine imines

图式 22 奎宁衍生的伯胺催化经过N-酰基亚胺中间体的Biginelli反应
Scheme 22 Quinine-derived primary amine catalyzed Biginelli reaction involving N-acylimine intermediate

图式 23 奎尼丁衍生的伯胺催化氮磺酰基环状亚胺的对映选择性[4+2]环加成
Scheme 23 Quinidine-derived primary amine catalyzed enantioselective [4+2] cycloaddition of N-sulfonyl cyclic imines

图式 24 通过1, 5-氢迁移的手性伯胺催化的C-H官能化
Scheme 24 Chiral primary amine-catalyzed C-H functionalization via 1, 5-hydride transfer

图式 25 甲基烷基酮与氮磺酰基环状亚胺的不对称Mannich反应
Scheme 25 Asymmetric Mannich reaction of methyl alkyl ketones with N-sulfonyl cyclic imines

图式 26 芳基甲基酮与氮磺酰基环状亚胺的不对称Mannich反应
Scheme 26 Asymmetric Mannich reaction of aryl methyl ketones with N-sulfonyl cyclic imines

图式 27 伯胺硫脲催化通过烯醇式机理的对映选择性的反应
Scheme 27 Enol mechanism in enantioselective reaction catalyzed by primary amine-thiourea

图式 28 伯胺硫脲催化经过N-酰基亚胺中间体的Biginelli反应
Scheme 28 Primary amine-thiourea catalyzed Biginelli reaction involving N-acylimine intermediate

图式 29 非手性酸在不对称Mannich反应中诱导的对映选择性翻转
Scheme 29 Achiral-acid-induced switch in the enantioselectivity of asymmetric Mannich reactions

图式 30 可见光促进的四氢异喹啉的不对称交叉脱氢偶联
Scheme 30 Visible-light-promoted asymmetric cross-dehydrogenative coupling of tetrahydroisoquinolines

图式 31 电化学氧化和手性伯胺催化合并作用的叔胺氧化偶联
Scheme 31 Oxidative coupling of tertiary amines by the combination of electrochemical oxidation and chiral primary amine catalysis

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:



 下载:
下载: