
图式 1.
3-(噁唑-5-基)吲哚类天然产物
Scheme 1.
Natural products of 3-(5-oxazolyl)indole

天然产物因其骨架结构新颖、生物活性多样和作用机理独特等特点, 受到人们的广泛关注和研究.以天然产物为先导化合物进行新药研究, 是近年来新药创制的重点和前沿.吲哚类生物碱具有特殊的化学性质和生理活性, 关于吲哚类天然产物及其衍生物的化学合成及生物活性的研究和报道层出不穷, 如2015年杨光富等[1]发表了关于含吲哚类抗病毒药物的研究进展.具有3-(噁唑-5-基)吲哚结构的天然产物具有骨架新颖、生物活性广泛和类药性高等特点, 近年来是药物化学研究的热点方向之一.具有多种重要的医药和农业生物活性的3-(噁唑-5-基)吲哚结构的天然产物已被报道[2], 作为抗氧化、抗感染、抗癫痫、抗真菌和抗肿瘤等药物的先导结构具有很好的研究价值.鉴于此, 本文概述了具有代表意义的3-(噁唑-5-基)吲哚类天然产物的结构特点和生物活性, 系统总结了该类天然产物骨架的合成方法, 包括酰胺酮关环、TosMIC关环、金属催化偶联、α-氨基酸关环等.
最早被发现的3-(噁唑-5-基)吲哚类天然产物是Pimprinine I-1 (Scheme 1), 是1960年Bhate等[3]从链霉菌Streptomyces pimprina的培养液中提取分离得到, 但未确定化学结构. 1963年, Joshi等[4, 5]在前人的基础上对Pimprinine进行研究, 首次确认了Pimprinine的化学结构为3-(噁唑-5-基)吲哚. Koyama等[6~8]发现Pim- prinine具有一定的抗微生物活性和药理学活性, 既能作为单胺氧化酶抑制剂, 也具有一定的抗癫痫作用.此外, Pimprinine对小鼠还表现出了一定的抗痉挛活性, 同时也能抑制由震颤素(Tremorine)引起的颤抖和痛觉缺失.先正达公司(Syngenta)的生物活性测试结果表明, Pimprinine具有一定的农业杀菌活性, 其在100 mg/L浓度下, 对出芽短梗霉菌(Aureobasidium pullulans)、葡萄灰霉菌(Botrytis cinerea)、稻瘟病菌(Magnaporthe grisea)和小麦叶枯病(Septoria tritici)等多种农作物病菌具有广谱的抑制作用.
在Pimprinine的生物活性、分离鉴定以及合成方法被陆续报道之后, 具有3-(噁唑-5-基)吲哚结构的天然产物的研究已经成为一个热点课题.数目众多的类似天然产物也被相继报道出来, 如Scheme 1所示.其中的Pim- printhine I-2, Pimprinaphine I-3是从橄榄网状链轮丝菌(Streptoverticillium olivoreticuli)的代谢产物中被分离鉴定出来, 具有一定的抗生活性[6, 9]; WS-30581 A I-4和WS-30581 B I-5是从Streptoverticillium wasksmanni中分离得到的, 被报道具有抑制血小板聚集的作用[8, 10]; 2002年, Pettit课题组[11, 12]从假单胞菌(Pseudomonas syringae)中分离得到Labradorin 1 I-6和Labradorin 2 I-7这两个Pimprinine系列的新化合物都表现出很强的抗肿瘤活性, 其中Labradorin 1对人体癌细胞BXPC-3(人胰腺癌细胞)和NCI-H(人肺癌细胞)的GI50值分别为6.2和9.8 μg/mL. 1988年, Watanabe等[13]从海洋链霉菌Streptomyces sp.中分离得到一种小分子吲哚生物碱Streptochlorin I-8, 经X光单晶衍射表征其结构为4-氯- 3-(噁唑-5-基)吲哚, 与Pimprinine结构很相似. Streptochlorin被报道具有抗微生物、抗血管形成、抗肿瘤等活性, 该化合物在微摩尔浓度下, 能有效抑制内皮细胞入侵和血管内皮细胞生长因子(VEGF)受激的微管形成, 并且对正常细胞不产生毒害.此外, Streptochlorin能够抑制TNF-α诱导的NF-κB活化[14~16]; 2015年, Shim等[17]报道了一种Streptochlorin衍生物HIS I-9合成方法与活性, HIS能够抑制β干扰素TIR结构域衔接蛋白(TRIF)依赖性信号转导, 且能抑制急性肺损伤和大肠杆菌引起的感染性休克. 1996年, Takahashi等[18]从红藻(Martensia Fragilis Harvey)中分离得到次生代谢产物Martefragin A I-10, 该化合物是迄今发现唯一来自海洋的具有3-(噁唑-5-基)吲哚类结构的新型抗氧化剂, 具有两个手性中心, 噁唑环的2位和4位均被取代, 其抗氧化活性是维生素E的30多倍, 维生素C的70多倍[19, 20]. 1996年, Diaye等[21]从塞内加尔海岸的红藻(Red alga, Haralditvllum sp.)中分离得到Almazole C I-11, 该天然产物对革兰氏阴性的粘质沙雷菌和伤寒沙门氏菌LXD具有较强的抗菌活性, 且没有发现细胞毒性和溶血活性, 使它成为一个有开发潜力的先导物, 用于探索开发新的抑制剂分子. 1991年, Lindquist等[22]从菲律宾海鞘(Dizona Chinensis)中分离得到一种次生代谢产物, 命名为Diazonamide A I-12, 这是一种结构复杂的Pimprinine类天然产物.体外实验表明Diazonamide A具有优异的HCT-116(人结肠癌细胞核)和B-16(鼠黑色素瘤细胞)抑制活性, 其中对HCT-116的IC50值小于15 ng/mL, 具有很好的抗癌药物研发潜力.它的合成研究也成为近年来的热点领域[23].
由于3-(噁唑-5-基)吲哚类天然产物显示出广泛的生物活性, 在医药和农药开发中有着重要的研究价值, 如何快速高效地构建其骨架是合成这类天然产物的关键.由于在3-(噁唑-5-基)吲哚类天然产物中, 吲哚为主体结构, 且吲哚衍生物在自然界中广泛存在, 吲哚作为原料便宜易得, 所以文献报道的方法基本上都是取代吲哚为原料进行关环或者偶联等方法来得到该类化合物.经过大量文献调研, 总结出3-(噁唑-5-基)吲哚构建的方法主要有酰胺酮关环、TosMIC关环、金属催化偶联、α-氨基酸关环等.
1963年, Joshi等[5]首次完成Pimprinine结构表征之后, 又首次完成了Pimprinine的全合成(Scheme 2).作者以1-(3'-吲哚基)-2-氨基-乙酮氢溴酸盐(II-1)为原料, 在醋酸酐和吡啶条件下, 在吲哚NH和氨基上同时进行乙酰化得到化合物II-2, 然后在三氯氧磷加热回流条件下关噁唑环得到化合物II-3, 再在酸性条件下水解得到目标化合物Pimprinine I-1.虽然该合成路线较简短, 但是产率太低, 实际应用价值不大.
1985年, Somei等[24]首次报道了甲氧基取代的Pimprinine衍生物, 即5-(1'-甲氧基吲哚-3'-基)噁唑的合成方法(Scheme 3).作者以2-硝基甲苯为原料关环得到1-甲氧基吲哚, 然后在吲哚3号位引入氯乙酰基, 氨水氨化之后得到α-氨基酮, 不经分离直接与乙酰氯一锅法反应.最后在三氯氧磷回流条件下得到目标化合物5-(1'-甲氧基吲哚-3'-基)噁唑, 催化水解即可得到天然产物Pimprinine.
1998年, Wipf课题组[25]报道Pimprinine类化合物的合成(Scheme 4).以色胺为原料, 用氯甲酸苄酯(CbzCl)将氨基保护后用二氯二氰基苯醌(DDQ)苄位氧化, 三氟乙酸钛和碘化亚铜催化下在吲哚4位碘取代, 然后与叔丁基二苯基硅醚(BDPSO)保护的乙醇酸反应得到α-酰胺酮结构, 最后在三苯基膦作用下关环得到3-(噁唑-5-基)吲哚骨架.作者将此方法运用到了复杂天然产物Diazonamide A中, 有效构建了吲哚多噁唑的结构[26].
2000年, Moody课题组[27]报道了3-(噁唑-5-基)吲哚类天然产物Diazonamide A合成过程中一个重要中间体的合成方法(Scheme 5), 其关键步骤是在醋酸铑的催化下重氮与酰胺的偶联反应, 得到的α-酰胺酮再在碘和三苯基膦条件下关噁唑环, 得到3-(噁唑-5-基)吲哚骨架.
2001年, Magnus课题组[28]报道了一种使用伯吉斯试剂氧化关噁唑环构建3-(噁唑-5-基)吲哚的方法(Scheme 6).取代色胺酮与羧酸取代的噁唑在1-乙基-(3-二甲氨基丙基)碳酰二亚胺盐酸盐(EDCI)的作用下脱水形成α-酰胺酮结构, 然后在伯吉斯试剂作用下关噁唑环, 从而合成得到Pimprinine类衍生物.

2004年, 闻韧等[12]完成了含取代苯基的3-(噁唑-5-基)吲哚衍生物的合成(Scheme 7), 并测定了其抗氧化活性.作者以色氨酸为原料, 与取代的苯甲酸在二环己基碳二亚胺(DCC)催化下脱水缩合得到酰胺, 再通过DDQ进行Yonemitsu苄位氧化和分子内环化, 生成3-(噁唑-5-基)吲哚, 然后用1, 1-二苯基-2-三硝基苯肼(DPPH)法体外抗氧化模型测定化合物的抗氧化活性, 并得到若干个抗氧化活性较好的目标化合物.

2005年, Moody等[19]在总结3-(噁唑-5-基)吲哚的合成方法基础上[27]深入研究了金属铑(Rhodium)催化的NH插入反应, 形成α-酰胺酮结构, 进而关环形成噁唑的反应(Scheme 8).通过该方法可以有效合成天然产物Pimprinine及其噁唑2-位取代的衍生物.
2006年, Gribble课题组[8]完成了双吲哚基噁唑的合成(Scheme 9).以色氨盐酸盐为原料, 用Boc2O将吲哚3位支链的氨基保护起来, 然后以NaH为碱的条件下用溴代烷基腈吲哚NH上引入烷基腈, 然后在DDQ的条件下进行Yonemitsu氧化, 把吲哚3位上取代的亚甲基氧化为羰基.值得一提的是, 吲哚NH取代基的电性对该氧化反应影响较大, 由于腈的吸电子效应, 烷基链越短, 氧化越困难, 所获得的II-41中间体再在三氟乙酸的条件下去保护得到氨盐II-42, 再与吲哚乙酸反应得到酰胺中间体II-43, 最后在三苯基膦、碘和三乙胺的作用下生成Pimprinine类目标产物II-44[29, 30].
同时作者也尝试了吲哚NH不保护条件下的反应, 也顺利得到目标化合物(Scheme 10).首先吲哚傅克酰基化引入氯乙酰基, 然后与叠氮化钠反应引入叠氮基, 再与吲哚乙酸反应得到重要中间体酰胺, 最后在三氯氧磷和吡啶作用下氧化关噁唑环得到目标化合物 II-49[31~36].

2008年, Kumar等[37]报道了一种简单高效地合成3-(噁唑-5-基)吲哚类天然产物的方法(Scheme 11).该方法中的关键步骤就是吲哚-3-乙酰基引入OTs取代基, 1-苯磺酰基-3-氨基乙酰基吲哚盐酸盐的生成, 以及α-乙酰胺基酮的关环形成噁唑结构[38~45].

2010年, Horne课题组[46]报道了一种简便高效地合成3-(噁唑-5-基)吲哚的方法(Scheme 12).作者以吲哚-3-甲酰腈为原料, Pd/C为催化剂, 用H2将其还原为色氨酮, 接着与酰氯反应后, 室温下与三氯氧磷回流得到目标化合物.该方法的特点是反应条件较温和, 并且保护吲哚的游离NH, 省去了保护基取代的步骤, 一定程度上提高了总产率.
2015年, Lee课题组[17]报道了3-(噁唑-5-基)吲哚衍生物HIS (5-hydroxy-2'-isobutylstreptochlorin)的合成(Scheme 13), 并测定了其抑制炎症的活性.作者以5-甲氧基吲哚-3-丙胺为原料, 与3-甲基丁酸催化下脱水缩合得到酰胺, 再通过DDQ进行Yonemitsu苄位氧化和分子内环化生成3-(噁唑-5-基)吲哚, 随后用叔丁氧羰基(Boc)将吲哚NH保护并在噁唑环4号位上引入Cl原子, 最后分别脱去Boc和甲基, 得到目标化合物.通过一系列测试表明, 该化合物表现出了很好的抗炎作用.
2017年, Lade等[47]报道了3-(噁唑-5-基)吲哚衍生物Almazole D (II-71)及其对映异构体Almazole R (II-74)的全合成(Scheme 14).作者以N-Boc-L-苯丙氨酸与L-色氨酸甲酯脱水缩合, 再通过DDQ进行Yonemitsu苄位氧化和分子内环化生成3-(噁唑-5-基)吲哚.再通过脱保护基、甲基化、水解反应得到目标化合物.值得一提的是, 该路线对映选择性好、产率高, 反应物Boc保护的苯丙氨酸的构型能直接决定最终产物的构型.

2002年, Dhar等[48]报道了基于NH保护的吲哚-3-甲醛和对甲苯磺酰甲基异腈(TosMIC)的[3+2]环加成方法(Scheme 15), 并用于合成新型的次黄嘌呤单核苷酸脱氢酶(IMPDH)抑制剂3-(噁唑-5-基)吲哚类化合物[49], 测试了该系列化合物的离体活性, 总结了构效关系[50].此类方法的关键步骤是Van Leusen噁唑合成反应, 具体反应机理见Scheme 16.值得注意的是, 该方法用甲基或者酰基保护了吲哚NH, 但是后期进行NH衍生反应要脱去这些基团较为困难.另外, 该反应还会产生一定量的关环后对甲苯磺酰基未脱去的副产物, 原子经济性不高.

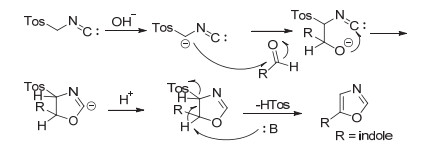
2005年, Chakrabarty等[51]报道了一种可以直接获得3-(噁唑-5-基)吲哚结构的方法(Scheme 17).作者用取代吲哚-3-甲醛与对甲苯磺酰甲基异腈(TosMIC)在碳酸钾作碱的条件下冷凝回流, 高效实现了由醛基到噁唑环一步构建, 当吲哚NH上为烷基取代基时, 反应仅生成未脱保护的关环产物; 而当吲哚NH上为Tos、Boc等易离去基团时, 反应同时生成脱保护和未脱保护的关环产物.某种程度上为后续的吲哚NH上的结构改造造成了一定的不便, 且降低了纯度和产率.

2012年, 杨光富等[52]报道了3-(噁唑-5-基)吲哚衍生物Streptochlorin的合成方法(Scheme 18).作者以廉价易得的吲哚为原料, 通过Vilsmeier-Haack反应得到吲哚-3-甲醛, 随后用苯磺酰氯将游离NH保护起来, 在碱性条件下与TosMIC发生[3+2]关环反应, 再在噁唑环的4号位引入Cl或者Br, 完成了Streptochlorin基本骨架的构建, 最后在吲哚NH位置引入不同的取代基对其进行结构优化改造, 得到了一系列杀菌活性较好的化合物.值得一提的是, 作者使用强碱性阴离子交换树脂, 不仅作为碱吸附反应的副产物对甲苯磺酸, 同时增大了反应的接触面积, 促进反应顺利进行.与前人使用碳酸钾作为碱相比, 提高了反应的选择性和产品产率.

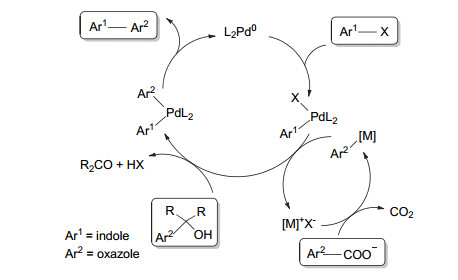
1998年, Pattenden课题组[53]尝试合成复杂的3-(噁唑-5-基)吲哚类天然产物Diazonamide A, 并总结了合成该类天然产物的方法(Scheme 19).其中介绍的一种就是基于烷基锡试剂和金属钯催化偶联的方法[54~56], 该合成方法一步即可实现天然产物骨架的合成, 简单高效, 但是所需的两个中间体都较为昂贵, 使该方法应用受限.

2009年, Primas等[57]报道了一种基于Suzuki交叉偶联反应的在卤代芳杂环上高效引入噁唑结构的方法, 其中包括在NH保护的吲哚5位引入噁唑(Scheme 20).偶联反应用的噁唑硼酸酯是通过噁唑的官能化实现的, 首先用三异丙基硅基(TIPS)将噁唑较为活泼的2位保护之后, 再在噁唑5位上引入硼酸频呐酯, 最后与卤代的芳杂环进行Suzuki交叉偶联反应, 得到目标化合物[15, 57, 68], 该方法也具有一定的借鉴意义.

2010年, Zhang等[69]发展了一种基于金属催化的偶联脱羧反应构建Pimprinine骨架的方法(Scheme 21).作者利用在钯和银的催化下, 1-苯磺酰基-3-溴吲哚与2-甲基-5-甲酸噁唑偶联脱羧形成3-(噁唑-5-基)吲哚骨架, 氢氧化钠条件下水解脱去苯磺酰基保护, 得到天然产物Pimprinine.脱羧偶联反应机理如Scheme 22所示.该方法简单高效, 一步脱羧偶联构建3-(噁唑-5-基)吲哚结构, 但是其原料1-苯磺酰基-3-溴吲哚和2-甲基-5-甲酸噁唑价格较贵, 还使用了钯和银两种贵金属, 反应温度较高, 反应时间较长, 不适合应用于实际生产中.

2012年, 吴运东等[70]报道了一种一价金配合物催化的简便高效且环境友好的3-(噁唑-5-基)吲哚类天然产物的合成方法(Scheme 23).该方法以吲哚为原料, 经过碘化、N-Boc保护、Sonogashira偶联、金催化成环反应、脱保护5步反应合成目标物.关键反应是在金催化下8-甲基喹啉氮氧化物与炔产生α-羰基金卡宾中间体.

2015年, 吴晓明课题组[71]开发了一种钯催化的C—H氧化交叉偶联反应, 实现了3-(噁唑-5-基)吲哚骨架的构建(Scheme 24).作者用苯环取代的吲哚和2, 4-二取代噁唑在钯和铜的协同催化下于135 ℃下反应24 h, 以较好的产率得到了包括Pimprinine在内的一系列衍生物.该反应的特点是具有良好的官能团耐受性和良好的区域选择性.但也存在金属催化偶联普遍存在的中间体价格昂贵、反应时间长、反应温度高等问题.

2013年, 吴安心课题组[72a]开发了一种I2/DMSO组合体系作为工具化试剂, 主要研究思路是以分子集群的自分类原理探寻小分子自组装合成复杂结构体的内在规律, 从而产生新颖的合成设计策略和探索新型合成方法学, 并由此探求药物分子高效简洁的合成新方法, 实现活性天然产物的直接全合成. Xiang课题组[72b]于2014年发表了一篇关于一锅法合成Pimprinine及其天然同系物及其衍生物的文章.以3-乙酰吲哚、氨基酸和I2为原料, DMSO为溶剂兼氧化剂, 在110 ℃的条件下搅拌反应1 h, 完成了Pimprinine及其天然同系物简易结构的合成.该反应的核心是通过碘化和Kornblum氧化方法在吲哚3位引入β-羰基醛的结构, 然后再与α-氨基酸进行关环(Scheme 25).作者也在文中提出了可能的机理(Scheme 26).该方法简单高效地构建取代3-(噁唑-5-基)吲哚结构, 且原料3-乙酰吲哚和氨基酸价格便宜易得, 反应时间短.


2019年, 汪清民课题组[73]报道了有关吲哚3位杂环取代化合物相关的合成, 包括噁唑、1, 2, 3-噁二唑等杂环的合成, 并对其进行结构改造, 最后测定了杀菌活性, 得到了一系列杀菌活性高的分子.其中在构建3-(噁唑-5-基)吲哚时, 作者以廉价易得的吲哚为原料, 与乙二酰二氯在吲哚3号位发生傅克酰基化得到吲哚的β-羰基酰氯, 然后用烷基锡试剂将其还原成醛, 最后再与α-氨基酸关噁唑环得到目标化合物(Scheme 27).值得一提的是, 作者用傅克酰基化和烷基锡试剂得到吲哚的二羰基取代物, 并且得到的β-羰基醛还可与TosMIC反应得到羰基噁唑的结构.

1994年, Moody课题组[19, 74]报道了一种基于重氮化反应和铑催化构建3-(噁唑-5-基)吲哚方法(Scheme 28).该方法以Boc保护的3-乙酰基吲哚为起始原料, 酰基α位的碳氢三氟乙酰化后使用甲磺酰基重氮(MsN3)进行α位叠氮化, 得到重要中间体重氮酮结构, 然后再金属铑的催化下关噁唑环, 脱去Boc保护之后即可得到目标化合物[70, 75].

2000年, Vedejs等[76]报道了基于Schölkopf反应的3-(噁唑-5-基)吲哚合成方法(Scheme 29).作者通过制备得到1-三异丙基硅基-3-甲酸甲酯-4-苄氧基吲哚, 然后与甲基异腈锂(LiCH2NC)发生Schölkopf反应, 但是得到大部分产物还是未关环的异腈, 如果继续加入吡啶对甲苯磺酸盐, 则全部转化为噁唑, 得到Pimprinine类化合物的3-(噁唑-5-基)吲哚骨架结构[77~79].
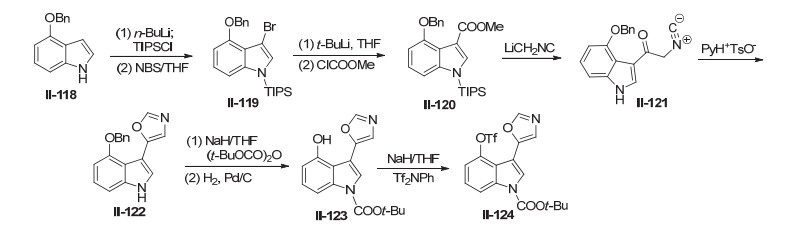
2001年, Liebscher课题组[80]报道了氯代噁唑相关的天然产物的合成方法, 其中包含有天然产物Diazona- mide A的亚结构氯代吲哚噁唑(Scheme 30).作者以吲哚啉-2-酮为起始原料, 首先利用Vilsmeier-Haack反应在吲哚3位引入醛基, 用氯甲酸-2, 2, 2-三氯乙酯(Troc-Cl)将吲哚NH保护起来, 再用亚氯酸钠将醛基氧化为羧酸, 在草酰氯和三甲基氰硅烷作用下生成重要中间体酰腈, 最后在三氟化硼乙醚和通氯化氢气体条件下和各种醛反应关环生成氯代噁唑.值得一提的是, 含卤素取代的醛在反应关环后, 继续反应转化为醛, 醛又可以转化为双噁唑的结构[35, 81, 82].
2016年, 贺峥杰课题组[83]报道了一种基于Huisgen内盐构建3-(噁唑-5-基)吲哚的方法(Scheme 31). Huisgen内盐是指偶氮二甲酸酯与叔膦经亲核加成反应形成的加合物, 它被认为是著名的Mitsunobu反应的关键中间体.研究表明, Huisgen内盐可与缺电子烯、亚胺、醛酮等多种亲电试剂发生关环反应, 是一种高效构建五元氮杂环的方法.利用Huisgen内盐这种独特性质, 作者用3-酰基甲叉基吲哚酮与Huisgen内盐的环化反应, 通过向反应体系中加入碱的策略, 实现了两步一锅法合成3-(噁唑-5-基)吲哚衍生物.该合成方法具有反应条件温和、高效以及底物适用范围广等特点.

2018年, 刘培均课题组[84]发展了一种构建3-(噁唑-5-基)吲哚结构的新方法(Scheme 32).与吴安心课题组相似的是, 作者也用到了I2作为反应催化剂, 开发了取代吲哚-3-甲酰腈与芳基甲胺的氧化环化反应, 获得了各种Pimprinine型的化合物.该反应具有反应条件温和、环境友好、底物范围宽、官能团相容性好等特点.

由于3-(噁唑-5-基)吲哚类天然产物表现出广泛的生物活性, 开展该类化合物骨架结构的合成方法探索, 具有非常重要的研究意义和潜在的应用价值.获取3-(噁唑-5-基)吲哚类天然产物的衍生物分子库进行生物活性筛选, 需要选用步骤简短、条件温和、原料便宜和产率高的合成方法, 以及更容易产生多样性取代的合成路线.文中总结了五种常见合成方法类别, 包括酰胺酮关环、TosMIC关环、金属催化偶联、α-氨基酸关环以及其它合成方法.其中酰胺酮关噁唑环的合成方法是最早被研究出来的, 但是该方法反应路线相对较长, 且反应过程中有DDQ氧化和POCl3关环等不友好不绿色的条件, 并且产率普遍较低, 不利于后期的结构改造和取代衍生.金属催化的交叉偶联反应虽然产率高, 合成步骤简便, 但是包括卤代噁唑、金属钯、金等反应试剂价格昂贵, 在克级规模的制备成本高, 偶联反应也存在耗时长、条件苛刻等不足. TosMIC关环反应是利用异腈与醛基进行[3+2]关环的方法, 原料吲哚-3-甲醛廉价易制备, 且反应路线短, 产率中等到较好. α-氨基酸关环的合成方法简单高效地构建2-取代3-(噁唑-5-基)吲哚结构, 且原料3-乙酰吲哚和氨基酸易制备或价格低廉.由此, 利用TosMIC关环反应或α-氨基酸关环可以简单高效地构建2-取代3-(噁唑-5-基)吲哚结构, 且原料吲哚、氨基酸等价格便宜, 易得, 符合高效构建多样性导向的衍生物分子库的要求.
随着有机合成和药物化学的发展以及新方法、新技术的出现, 3-(噁唑-5-基)吲哚类天然产物衍生物的化学合成将更高效更简便, 通过设计和筛选获取高活性的3-(噁唑-5-基)吲哚类化合物, 进而研究其作用靶标和作用机理.
Zhang, M. Z.; Chen, Q.; Yang, G. F. Eur. J. Med. Chem. 2015, 89, 421. doi: 10.1016/j.ejmech.2014.10.065
李庆欣, 史雪凤, 黄智, 田新朋, 王发左, 热带海洋学报, 2013, (1), 35. doi: 10.3969/j.issn.1009-5470.2013.01.005Li, Q. X.; Shi, X. F.; Huang, Z.; Tian, X. P.; Wang, F. Z. J. Trop. Oceanogr. 2013, (1), 35 (in Chinese). doi: 10.3969/j.issn.1009-5470.2013.01.005
Bhate, D. S.; Hulyalkar, R. K.; Menon, S. K. Experientiu 1960, 16, 504. doi: 10.1007/BF02158365
Bhate, D. S.; Ambekar, G. R.; Bhatnagar, K. K.; K., H. R. Hind. Antibiot. Bull. 1961, 4, 139.
Joshi, B. S.; Taylor, W. I.; Bhate, D. S.; Karmarkar, S. S. Tetrahedron 1963, 19, 1437. doi: 10.1016/S0040-4020(01)98569-2
Koyama, Y.; Yokose, K.; Dolby, L. J. Agric. Biol. Chem. 1981, 45, 1285.
Naik, S. R.; Harindran, J.; Varde, A. B. J. Biotechnol. 2001, 88, 1. doi: 10.1016/S0168-1656(01)00244-9
Roy, S.; Haque, S.; Gribble, G. W. Synthesis 2006, 3948.
Khan, S.; Ahmed, M. S.; Arakawa, O.; Onoue, Y. J. J. Aquacult. Bamidgeh. 1995, 47, 137.
Khan, S.; Arakawa, O.; Onoue, Y. Phycologia 1996, 35, 239. doi: 10.2216/i0031-8884-35-3-239.1
Pettit, G. R.; Knight, J. C.; Herald, D. L.; Davenport, R.; Pettit, R. K.; Tucker, B. E.; Schmidt, J. M. J. Nat. Prod 2002, 65, 1793. doi: 10.1021/np020173x
Miao, Y. P.; Wen, R.; Hitoshi, A.; Zhou, P. G. Acta Pharm. Sinica 2004, 39, 37.
Watanabe, H.; Amano, S.; Yoshida, J.; Takase, Y.; Miyadoh, S.; Sasaki, T.; Hatsu, M.; Takeuchi, Y.; Komada, Y. Meiji Seika Kenkyu Nenpo 1988, 27, 55.
Shin, H. J.; Jeong, H. S.; Lee, H. S.; Park, S. K.; Kim, H. M.; Kwon, H. J. J. Microbiol. Biotechnol. 2007, 17, 1403.
Romero, F. A.; Du, W.; Hwang, I.; Rayl, T. J.; Kimball, F. S.; Leung, D.; Hoover, H. S.; Apodaca, R. L.; Breitenbucher, J. G.; Cravatt, B. F.; Boger, D. L. J. Med. Chem. 2007, 50, 1058. doi: 10.1021/jm0611509
Shin, D. Y.; Shin, H. J.; Kim, G. Y.; Cheong, J.; Choi, I. W.; Kim, S. K.; Moon, S. K.; Kang, H. S.; Choi, Y. H. J. Microbiol. Biotechnol. 2008, 18, 1862.
Shim, D. W.; Shin, H. J.; Han, J. W.; Ji, Y. E.; Jang, C. H.; Koppula, S.; Kang, T. B.; Lee, K. H. Toxicol. Appl. Pharm. 2015, 284, 227. doi: 10.1016/j.taap.2015.02.006
Takahashi, S.; Matsunaga, T.; Hasegawa, C.; Saito, H.; Fujita, D.; Kiuchi, F.; Tsuda, Y. Chem. Pharm. Bull. 1998, 46, 1527. doi: 10.1248/cpb.46.1527
Davies, J. R.; Kane, P. D.; Moody, C. J.; Slawin, A. M. Z. J. Org. Chem. 2005, 70, 5840. doi: 10.1021/jo050303h
缪宇平, 博士论文, 复旦大学, 上海, 2003.Miao, Y. P. Ph.D. Dissertation, Fudan University, Shanghai, 2003 (in Chinese).
Diaye, N.; Guella, G.; Mancini, I.; Pietra, F. Tetrahedron Lett. 1996, 37, 3049. doi: 10.1016/0040-4039(96)00466-2
Lindquist, N.; Fenical, W.; Vanduyne, G. D.; Clardy, J. J. Am. Chem. Soc. 1991, 113, 2303. doi: 10.1021/ja00006a060
Nicolaou, K. C.; Chen, D. Y.; Huang, X.; Ling, T.; Bella, M.; Snyder, S. A. J. Am. Chem. Soc. 2004, 126, 12888. doi: 10.1021/ja040092i
Somei, M.; Sato, H.; Komura, N.; Kaneko, C. Heterocycles 1985, 23, 1101. doi: 10.3987/R-1985-05-1101
Wipf, P.; Yokokawa, F. Tetrahedron Lett. 1998, 39, 2223. doi: 10.1016/S0040-4039(98)00231-7
Wipf, P.; Methot, J. L. Org. Lett. 2001, 3, 1261. doi: 10.1021/ol0157196
Bagley, M. C.; Hind, S. L.; Moody, C. J. Tetrahedron Lett. 2000, 41, 6897. doi: 10.1016/S0040-4039(00)01120-5
Kreisberg, J. D.; Magnus, P.; McIver, E. G. Tetrahedron Lett. 2001, 42, 627. doi: 10.1016/S0040-4039(00)02023-2
Roy, S.; Eastman, A.; Gribble, G. W. Org. Biomol. Chem. 2006, 4, 3228. doi: 10.1039/b607504e
Wipf, P.; Miller, C. P. J. Org. Chem. 1993, 58, 3604. doi: 10.1021/jo00066a004
Bergman, J.; Backvall, J. E.; Lindstrom, J. O. Tetrahedron 1973, 29, 971. doi: 10.1016/0040-4020(73)80047-X
Deng, W. P.; Nam, G.; Fan, J. F.; Kirk, K. L. J. Org. Chem. 2003, 68, 2798. doi: 10.1021/jo020731c
Jiang, B.; Gu, X. H. Bioorgan. Med. Chem. 2000, 8, 363. doi: 10.1016/S0968-0896(99)00290-4
Fresneda, P. M.; Molina, P.; Sanz, M. A. Synlett 2001, 218.
Radspieler, A. Ph.D. Dissertation, Humboldt University, Berlin, 2000.
Oikawa, Y.; Yonemitsu, O. J. Org. Chem. 1977, 42, 1213. doi: 10.1021/jo00427a024
Kumar, D.; Sundaree, S.; Patel, G.; Rao, V. S. Tetrahedron Lett 2008, 49, 867. doi: 10.1016/j.tetlet.2007.11.173
Koser, G. F.; Relenyi, A. G.; Kalos, A. N.; Rebrovic, L.; Wettach, R. H. J. Org. Chem. 1982, 47, 2487. doi: 10.1021/jo00133a053
oser, G. F. Adv. Heterocycl. Chem. 2004, 86, 225. doi: 10.1016/S0065-2725(03)86004-X
Kelly, T. R.; Fu, Y.; Xie, R. L. Tetrahedron Lett. 1999, 40, 1857. doi: 10.1016/S0040-4039(99)00074-X
Doyle, K. J.; Moody, C. J. Synthesis 1994, 1021.
Brain, C. T.; Paul, J. M. Synlett 1999, 1642.
Nishida, A.; Fuwa, M.; Naruto, S.; Sugano, Y.; Saito, H.; Nakagawa, M. Tetrahedron Lett. 2000, 41, 4791. doi: 10.1016/S0040-4039(00)00677-8
Liu, S. F.; Wu, Q. G.; Schmider, H. L.; Aziz, H.; Hu, N. X.; Popovic, Z.; Wang, S. N. J. Am. Chem. Soc. 2000, 122, 3671. doi: 10.1021/ja9944249
Ketcha, D. M.; Gribble, G. W. J. Org. Chem. 1985, 50, 5451. doi: 10.1021/jo00350a001
Miyake, F.; Hashimoto, M.; Tonsiengsom, S.; Yakushijin, K.; Horne, D. A. Tetrahedron 2010, 66, 4888. doi: 10.1016/j.tet.2010.03.109
Lade, D. M.; Krishna, V. S.; Sriram, D.; Rode, H. B. Chemistryselect 2017, 2, 1250. doi: 10.1002/slct.201601821
Dhar, T. G. M.; Shen, Z. Q.; Fleener, C. A.; Rouleau, K. A.; Barrish, J. C.; Hollenbaugh, D. L.; Iwanowicz, E. J. Bioorg. Med. Chem. Lett. 2002, 12, 3305. doi: 10.1016/S0960-894X(02)00748-5
Van Leusen, D.; Hessen, B. Organometallics 2001, 20, 224. doi: 10.1021/om000678n
Houwing, H. A.; Wildeman, J.; Van Leusen, A. M. J. Heterocycl. Chem. 1981, 18, 1133. doi: 10.1002/jhet.5570180615
Chakrabarty, M.; Basak, R.; Harigaya, Y.; Takayanagi, H. Tetrahedron 2005, 61, 1793. doi: 10.1016/j.tet.2004.12.022
Zhang, M. Z.; Chen, Q.; Mulholland, N.; Beattie, D.; Irwin, D.; Gu, Y. C.; Yang, G. F.; Clough, J. Eur. J. Med. Chem. 2012, 53, 283. doi: 10.1016/j.ejmech.2012.04.012
Boto, A.; Ling, M.; Meek, G.; Pattenden, G. Tetrahedron Lett. 1998, 39, 8167. doi: 10.1016/S0040-4039(98)01819-X
Barrett, G. M.; Kohrt, J. T. Synlett 1995, 415.
Dondoni, A.; Fantin, G.; Fogagnolo, M.; Medici, A.; Pedrini, P. Synthesis 1987, 693.
Kelly, T. R.; Lang, F. R. J. Org. Chem. 1996, 61, 4623. doi: 10.1021/jo960433d
Primas, N.; Bouillon, A.; Lancelot, J. C.; Rault, S. Tetrahedron 2009, 65, 6348. doi: 10.1016/j.tet.2009.06.023
Besselievre, F.; Mahuteau-Betzer, F.; Grierson, D. S.; Piguel, S. J. Org. Chem. 2008, 73, 3278. doi: 10.1021/jo7027135
Ohnmacht, S. A.; Mamone, P.; Culshaw, A. J.; Greaney, M. F. Chem Commun 2008, 1241.
Do, H. Q.; Daugulis, O. J. Am. Chem. Soc. 2007, 129, 12404. doi: 10.1021/ja075802+
Bellina, F.; Calandri, C.; Cauteruccio, S.; Rossi, R. Tetrahedron 2007, 63, 1970. doi: 10.1016/j.tet.2006.12.068
Bellina, F.; Cauteruccio, S.; Rossi, R. Eur. J. Org. Chem. 2006, 1379.
Del Zotto, A.; Amoroso, F.; Baratta, W.; Rigo, P. Eur. J. Org. Chem. 2009, 110.
Liu, W. J.; Xie, Y. X.; Yun, L. A.; Li, J. H. Synthesis 2006, 860.
Alimardanov, A.; de Vondervoort, L. S. V.; de Vries, A. H. M.; de Vries, J. G. Adv. Synth. Catal. 2004, 346, 1812. doi: 10.1002/adsc.200404210
Leadbeater, N. E.; Marco, M. Org. Lett. 2002, 4, 2973. doi: 10.1021/ol0263907
Tanaka, A.; Terasawa, T.; Hagihara, H.; Sakuma, Y.; Ishibe, N.; Sawada, M.; Takasugi, H.; Tanaka, H. J. Med. Chem. 1998, 41, 2390. doi: 10.1021/jm9800853
Lancelot, J. C.; Prunier, H.; Robba, M.; Delagrange, P.; Renard, P.; Adam, G. EP623620 A1, 1994.
Zhang, F. Z.; Greaney, M. F. Org. Lett. 2010, 12, 4745. doi: 10.1021/ol1019597
吴运东, 彭莎, 欧阳跃军, 钱鹏程, 何卫民, 向建南, 化学学报, 2012, 70, 47.Wu, Y. D.; Peng S.; Ou-yang, Y. J.; Qian, P. C.; He, W. M.; Xiang, J. N. Acta Chim. Sinica 2012, 70, 47 (in Chinese).
Zhou, H. P.; Gai, K.; Lin, A. J.; Xu, J. Y.; Wu, X. M.; Yao, H. Q. Org. Biomol. Chem. 2015, 13, 1. doi: 10.1039/C5OB90001H
(a) Zhu, Y. P.; Fei, Z.; Liu, M. C.; Jia, F. C.; Wu, A. X. Org. Lett. 2013, 15, 378.
(b) Xiang, J. C.; Wang, J. G.; Wang, M.; Meng, X. G.; Wu, A. X. Tetrahedron 2014, 70, 7470.
Liu, B.; Li, R.; Li, Y. A.; Li, S. Y.; Yu, J.; Zhao, B. F.; Liao, A. C.; Wang, Y.; Wang, Z. W.; Lu, A. D.; Liu, Y. X.; Wang, Q. M. J. Agric. Food Chem. 2019, 67, 1795. doi: 10.1021/acs.jafc.8b06175
Danheiser, R. L.; Miller, R. F.; Brisbois, R. G.; Park, S. Z. J. Org. Chem. 1990, 55, 1959. doi: 10.1021/jo00293a053
Konopelski, J. P.; Hottenroth, J. M.; Oltra, H. M.; Veliz, E. A.; Yang, Z. C. Synlett 1996, 609.
Vedejs, E.; Barda, D. A. Org. Lett. 2000, 2, 1033. doi: 10.1021/ol005548p
Schöllkopf, U.; Schröder, R. Angew. Chem., Int. Ed. 1971, 10, 333. doi: 10.1002/anie.197103331
Batcho, A. D.; Leimgruber, W. Org. Synth. 1985, 63, 214. doi: 10.15227/orgsyn.063.0214
Amat, M.; Hadida, S.; Pshenichnyi, G.; Bosch, J. J. Org. Chem. 1997, 62, 3158. doi: 10.1021/jo962169u
Radspieler, A.; Liebscher, J. Synthesis 2001, 745.
Adreani, A.; Bonazzi, D.; Rambaldi, M.; Guarnieri, A. J. Med. Chem. 1977, 20, 1344. doi: 10.1021/jm00220a023
Olah, G. A.; Arranaghi, M.; Prakash, G. K. Synthesis 1983, 8, 636.
Yang, C. J.; Chen, X. Y.; Tang, T.; He, Z. J. Org. Lett. 2016, 18, 1486 doi: 10.1021/acs.orglett.6b00456
Liu, X. Z.; Zhou, Y. X.; Chen, G. J.; Yang, Z. Q.; Li, Q.; Liu, P. J. Org. Biomol. Chem. 2018, 16, 3572. doi: 10.1039/C8OB00833G

图式 6 伯吉斯试剂氧化关噁唑环构建3-(噁唑-5-基)吲哚结构
Scheme 6 Construction of 3-(5-oxazolyl)indole by Burgess reagent reaction

图式 7 3-(2-取代苯基噁唑-5-基)吲哚衍生物的合成
Scheme 7 Synthesis of 3-(2-substituted phenyl-(5-oxazolyl)in- dole derivatives

图式 10 吲哚NH不保护的双吲哚基噁唑的合成
Scheme 10 Synthesis of indole substituted 3-(5-oxazolyl)indole with unprotected NH of indole

图式 11 天然物质3-(噁唑-5-基)吲哚的合成
Scheme 11 Synthesis of natural substances 3-(5-oxazolyl)- indole

图式 14 Almazole D和Almazole R的全合成
Scheme 14 Total synthesis of almazole D and almazole R

图式 15 TosMIC环加成法合成3-(噁唑-5-基)吲哚类化合物
Scheme 15 Synthesis of 3-(5-oxazolyl)indole compounds by TosMIC cycloaddition reaction

图式 17 TosMIC和碳酸钾构建3-(噁唑-5-基)吲哚结构
Scheme 17 Construction of 3-(5-oxazolyl)indole structure with TosMIC and potassium carbonate

图式 18 Streptochlorin及其衍生物的合成路线
Scheme 18 Synthetic route of streptochlorin and its derivatives

图式 19 基于烷基锡试剂合成3-(噁唑-5-基)吲哚结构
Scheme 19 Synthesis of 3-(5-oxazolyl)indole based on organotin reagent

图式 20 Suzuki交叉偶联反应构建5-(噁唑-5-基)吲哚
Scheme 20 Construction of 5-(5-oxazolyl)indole by Suzuki Cross coupling reaction

图式 21 Pd和Ag催化的偶联反应构建3-(噁唑-5-基)吲哚骨架
Scheme 21 Construction of 3-(5-oxazolyl)indole by coupling reaction catalyzed with Pd and Ag

图式 23 金催化的偶联反应构建3-(噁唑-5-基)吲哚骨架
Scheme 23 Construction of 3-(5-oxazolyl)indole skeleton by Gold-catalyzed coupling reaction

图式 24 Pd催化的C—H氧化交叉偶联反应构建3-(噁唑-5-基)吲哚骨架
Scheme 24 Construction of 3-(5-oxazolyl)indole by Pd-catalyz- ed C—H oxidation cross coupling reaction

图式 25 Kornblum氧化和α-氨基酸构建3-(噁唑-5-基)吲哚骨架
Scheme 25 Construction of 3-(5-oxazolyl)indole by Kornblum oxidation and α-amino acids

图式 27 烷基锡试剂和α-氨基酸构建3-(噁唑-5-基)吲哚骨架
Scheme 27 Construction of 3-(5-oxazolyl)indole by organotin reagents and α-amino acids

图式 28 重氮化反应和铑催化构建3-(噁唑-5-基)吲哚
Scheme 28 Construction of 3-(5-oxazolyl)indole by diazotization reaction and Rhodium catalyst

图式 29 基于Schölkopf反应构建3-(噁唑-5-基)吲哚骨架
Scheme 29 Construction of 3-(5-oxazolyl)indole through Schölkopf reaction

图式 31 Huisgen内盐构建3-(噁唑-5-基)吲哚
Scheme 31 Construction of 3-(5-oxazolyl)indole with Husigen internal salt

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:










 下载:
下载:













 下载:
下载: