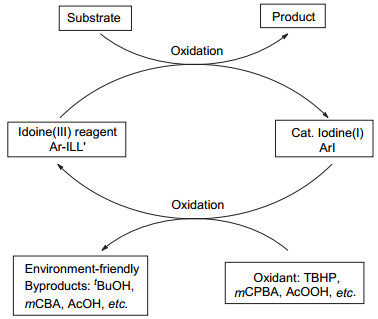
图 1.
芳基碘作为前催化剂原位生成三价碘化合物示意图
Figure 1.
Aryl iodide as a procatalyst in-situ generation of hypervalent iodine(Ⅲ) reagent

高价碘试剂被称为超价碘试剂, 这是一类特殊的含有碘元素的有机分子.与一般的含碘化合物相比, 此类分子中碘元素在形成化合物时, 自身的价电子总数超过了八隅体规则所允许的数目, 此时碘显现为高价态[1].一般高价碘可分为三价碘试剂或五价碘试剂.高价碘试剂本身是一种氧化剂, 其氧化能力与二价汞(Hg, Ⅱ)、四价铅(Pb, Ⅳ)、六价铬(Cr, Ⅵ)和八价锇(Os, Ⅷ)等金属氧化剂相近, 但高选择性、性能温和等优点则是上述金属氧化剂所不具备的[2]. 19世纪以来, 高价碘试剂在氧化、加成、重排、取代以及不对称合成反应中都已有应用[3], 作者曾利用三价碘试剂完成了四氢异喹啉中苄位亚甲基的分子内官能团化[4].随着社会对环保的要求越来越高, 广泛使用的传统高价碘试剂显露了它的缺点, 如: (1)五价碘试剂具有潜在的爆炸性, 不易保存, 且价格较昂贵; (2)很多氧化反应需要使用过量的高价碘试剂参与反应, 造成资源浪费; (3)传统高价碘试剂在反应中主要被分解为芳基碘, 不易回收再利用, 且需要较复杂的无害化后处理过程[5]; (4)高价碘试剂在有机溶剂中溶解性不好, 限制了它们的使用范围.这些问题成为设计清洁、低成本的化学反应的瓶颈, 限制了高价碘试剂在工业领域中的应用[6].
因此, 开发出既具有较高活性又能实现“原子经济性”[7, 8], 符合绿色化学理念的高价碘试剂成为合成领域研究热点之一.随着研究的深入, 人们发现原位生成的高价碘不仅保留了高价碘试剂的所有优点, 而且催化剂还可以被循环使用[9].原位生成高价碘策略是在反应中使用催化量的低价碘(碘为负一价)与等物质的量的氧化剂反应, 原位生成高价碘替代直接使用高价碘的一种合成策略.反应中常用催化剂有碘苯、取代碘苯、碘盐和单质碘等碘化物, 氧化剂则包括间氯过氧苯甲酸(mCP-BA)、过氧化氢(H2O2)等过氧化物以及N-溴代琥珀酰亚胺(NBS)、过硫酸氢钾复合盐(Oxone)等.详细介绍了原位生成高价碘的机理, 反应类型, 以及近年来原位生成的三价碘、五价碘以及手性高价碘试剂在有机合成反应中的应用, 并对原位生成高价碘试剂的发展做了展望.
首先利用氧化剂将催化量的低价碘氧化为高价态的碘, 生成的高价碘直接参与反应, 然后又释放出低价碘, 如此反复循环.在低价碘被氧化成高价碘的过程中, 不同的前催化剂产生的高价碘不同, 以下分别对芳基碘和碘盐这两大类前催化剂原位生成高价碘的机理进行阐述.
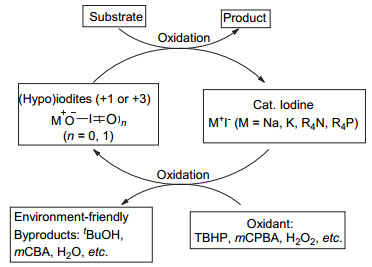
芳基碘作为前催化剂原位生成高价碘的机理见图 1.价廉环保的过氧化物氧化剂(mCPBA、H2O2、过氧叔丁醇TBHP、过氧乙酸AcOOH等)先将催化量的碘苯氧化成三价碘试剂, 后者参与反应将原料氧化成相应的产物, 而后释放出的碘苯又进一步被氧化, 进而实现碘苯的循环再利用.另外反应生成的副产物安全可控, 如间氯苯甲酸(mCBA)、水、叔丁醇、乙酸等.在催化量的低价碘介导的循环体系中, 选择不同的氧化剂和催化剂可分别得到三价碘或者五价碘中间体, 低价态的碘氧化到高价态的过程是该反应的关键步骤.碘盐作为前催化剂原位生成高价碘的机理如图 2.过氧化物(mCPBA, H2O2, TBHP等)将催化量的碘盐(NaI, KI, Bu4NI, Bu4PI等)氧化为正一价或正三价的高价碘(IO-或IO2-), 与图 1过程类似, 后者参与氧化反应并释放出安全可控的副产物[10].
近年来原位生成三价碘试剂的催化循环反应已被成功地应用到了杂环和螺环等化合物的构建中, 其中芳基碘试剂作为催化剂在催化循环反应中尤为常见.总结了芳基碘试剂作为催化剂, 不同类型的氧化剂(如mCPBA, AcOOH, H2O2, TBHP等)原位生成三价碘在有机合成中的应用.
间氯过氧苯甲酸(mCPBA)是一种过氧羧酸, 可被广泛地应用于有机合成的氧化反应中.主要应用领域有酮转化为酯(Baeyer-Villiger氧化)、烯烃环氧化(Prilezhaev反应)和甲硅烷基烯醇醚转化为甲硅烷基α-羟基酮(Rubottom氧化)等.因mCPBA氧化性过强, 易破坏原料, 降低收率, 所以在原位生成反应过程中不直接用于氧化原料, 而是使用氧化剂mCPBA将催化量的碘试剂氧化为高价碘, 后者作为氧化剂参与原料的氧化反应.这类反应中可使用的氧化剂种类很多如Oxone、H2O2等, 因mCPBA氧化性强, 相对易处理, 所以应用较多.但是mCPBA在使用过程中要避免碰撞, 防止发生爆炸.以下对以mCPBA为氧化剂原位生成三价碘介导的去芳构化反应、偶联反应、重排反应等作简要介绍.
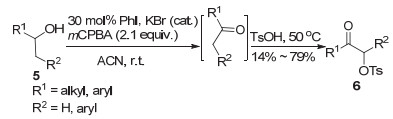
2005年, Ochiai课题组[11]首次运用原位生成三价碘策略, 即以0.1 equiv.的碘苯作为催化剂, 2.0 equiv. mCPBA作为氧化剂, 醋酸为原料, 在羰基化合物1的α位引入乙酰氧基生成目标化合物2 (Eq. 1).在反应过程中mCPBA将催化量的碘苯氧化成三价碘二醋酸碘苯(PIDA), PIDA参与酮1的α位氧化, 生成的碘苯进一步被氧化循环为三价碘.该反应在室温条件下进行, 产率中等(43%~63%), 反应时间较长(超过20 h).在2007年, Togo团队[12]研究发现原位生成高价碘条件下, 用甲磺酸替代醋酸同样可以实现酮类化合物3的α-对甲苯磺酰氧基化, 产率可达85%且时间缩短为5 h (Eq. 2).同时该课题组团队在原有基础上对醇类化合物5的α-甲苯磺酰氧基化进行了研究, 研究表明, 通过“一锅法”, 原位生成的三价碘将醇氧化为酮, 随后实现酮类的甲磺酰氧化(Scheme 1).产物α-甲苯磺酸酮是重要的合成前体, 可以用于构建多种芳杂环化合物, 如噻唑、咪唑和噁唑等.
|
|
(1) |
|
|
(2) |
原位生成三价碘介导下的酚类去芳构化反应是合成螺杂环化合物的重要方法之一, 可应用于呋喃衍生物的骨架构建. 2005年, Kita等[13]报道了原位生成三价碘试剂介导下由酚类化合物7构建二氢吡喃酮型螺杂环化合物8的反应.该方法成功实现了原位生成高价碘介导的酚类去芳构化, 具有反应时间短、产率高等优点, 最高收率可达到91% (Eq. 3).原位生成三价碘介导的去芳构化还可以用于吡咯烷酮的构建. 2007年, Kita课题组[14]以0.1 equiv.的4-甲基碘苯为催化剂, 1.5 equiv.的mCPBA在室温条件下将原料酰胺类化合物9氧化完成, C—N键的构建, 合成了N-稠合螺内酰胺10 (Eq. 4).
|
|
(3) |
|
|
(4) |
原位生成三价碘介导的氧化反应可以实现C(sp3)—H和C(sp2)—H的氨基化, 且产率较高. 2015年, Shi课题组[15]研究发现, 使用碘代芳烃和mCPBA组合在温和条件下短时间可以完成分子内的叔C—H(sp3)键的胺化.通过密度泛函理论(M06-2X)计算表明, 反应过程经历了原位生成的三价碘介导的碘鎓阳离子中间体和C—H活化/C—N键生成的历程.该方法可用于γ-内酰胺13合成方法研究(Eq. 5). 2017年, Xue小组[16]研究发现在催化量的碘苯和氧化剂mCPBA原位生成三价碘介导下, 芳基酰胺类化合物14实现了分子内C(sp2)—N键的偶联, 构建菲啶酮15骨架.该方法底物应用范围较广, 条件温和, 室温空气中即可进行, 最高产率可达98%, 且菲啶酮类化合物是一种重要的三元环化合物, 常作为医药原料或中间体应用于制备一系列药物或活性功能分子(Eq. 6).
|
|
(5) |
|
|
(6) |
2013年, Kita课题组[17]第一次报道了原位生成三价碘介导的分子间芳烃氧化交叉偶联反应.该研究发现磺酰苯胺类化合物16在催化剂2, 2-二碘联苯和氧化剂mCPBA作用下, 芳烃类化合物经过分子间选择性的C—C键偶联得到联苯类化合物17, 产率在54%~99%之间(Eq. 7).
|
|
(7) |
mCPBA作为氧化剂原位生成三价碘, 不仅可以用于C—C, C—O和C—N键的构建, 还可以应于部分重排反应.早在2010年, Miyamoto团队[18]就报道了使用催化量的PhI和氧化剂mCPBA原位生成高价碘介导的霍夫曼重排反应.反应中需要添加HBF4试剂, 在反应中起诱导作用.碘苯、水先被氧化成四配位的三价碘PhI+(H2O)2OHBF4-, 然后作为氧化剂使原料酰胺完成1, 2迁移, 生成RN=C=O, 再脱去CO2生成伯胺, 产率较高(Eq. 8).
|
|
(8) |
过氧乙酸(AcOOH)是一种过氧化物, 生活中常用于杀菌消毒, 在化学反应中常用作烯烃的环氧化剂[19].其氧化能力低于mCPBA, 但在原位生成高价碘反应中仍是常用的氧化剂, 可用于C—C, C—O和C—N键的构建.由于该化合物不稳定, 浓度大于45%或者遇高热易爆炸, 所以AcOOH不能长时间储存, 且参与的反应加热温度不能过高.
2011年, Antonchick小组[20]以碘化物21为催化剂, 过氧乙酸为氧化剂, 使2-氨基联苯20发生分子内的氧化环合反应, 构建了C—N键.该反应在室温下进行, 最高产率可达98%, 是合成咔唑的一种新方法(Eq. 9). 2013年, 朱强课题组[21]则报道了以碘苯、过氧乙酸构成的催化氧化体系, 通过对N-苯基-2-氨基吡啶类化合物23进行氧化, 完成C(sp2)—N键的构建.该反应最高产率可达99%, 可适用于多种官能团, 为苯并咪唑的基本骨架的建立开辟了一种新的途径(Eq. 10).
|
|
(9) |
|
|
(10) |
在2014年, Martin课题组[22]报道了在非金属条件下, 取代碘苯作为催化剂, 过氧乙酸为氧化剂原位生成三价碘介导的sp2或sp3C—H键和羧酸之间的偶联反应.该方法成功构建了C—O键, 实现了分子内酯化反应, 且条件温和, 底物使用范围广泛(Eqs. 11, 12).
|
|
(11) |
|
|
(12) |
原位生成三价碘介导的C—O键偶联反应同样可以用于化合物的磺化反应. 2017年, 沈超课题组[23]报道了在碘苯-过氧乙酸组合作用下喹啉酰胺类化合物29的磺化反应.该反应在室温下溶剂HFIP中进行.经过氧化之后, 以72%~95%的收率得到一系列芳基磺酸酯类化合物31.合成得到的部分磺化喹诺酮类化合物可用于生物和医药领域(Eq. 13).
|
|
(13) |
N-溴代琥珀酰亚胺(NBS)一般用于有机化学中的自由基取代、亲电加成、溴代反应以及氧化反应.在原位生成高价碘反应中, NBS不仅可以作为氧化剂还可以作为溴的引入剂, 而且与其它过氧化物相比, 无爆炸危险, 更加安全.目前文献报道NBS参与的原位生成高价碘反应多为氧化反应、重排反应和溴代反应.在2006年, Braddock课题组[24]报道了以邻位取代的碘苯作为有机催化剂, NBS作为氧化剂, 实现了烯烃基羧酸的溴代内酯化反应.随后Gulder课题组[25~28]报道了一系列NBS作为氧化剂原位生成高价碘介导的一系列反应.
2012年, Gulder课题组[25]研究发现, 甲基丙烯胺化合物32在NBS和催化剂邻碘苯酰胺衍生物33的共同作用下, 完成了溴代环合反应, 得到目标产物3, 3-二取代的吲哚类化合物34, 产率在59%~94%之间.该环合方法已经用于乙酰胆碱酯酶抑制剂毒扁豆碱的制备中(Eq. 14).
|
|
(14) |
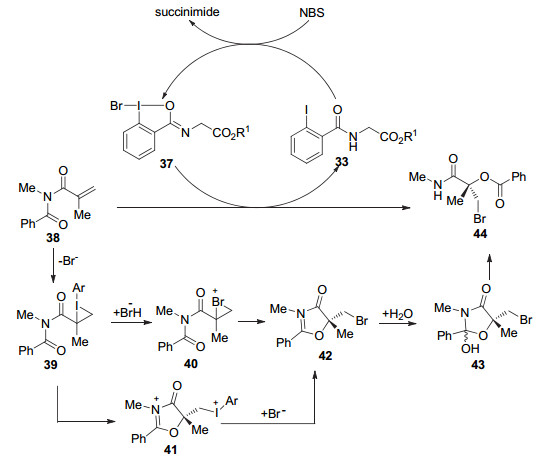
NBS氧化剂不仅可以用于C—C键氧化偶联反应, 而且还可以用于重排反应中. 2015年, Gulder课题组[26]报道了原位生成三价碘介导的酰亚胺类化合物35发生重排反应, 得到了一系列的α, α-二取代-α-羟基羧酰胺类化合物36 (Eq. 15).反应在室温下进行, 产率可高达98%, 并且在未用手性添加剂情况下, 诱导产生单一的手性化合物.值得一提的是, 该报道用1H NMR, 13C NMR和ESI-MS捕捉到了三价碘37, 证实了原位生成的三价碘化合物参与了催化过程, 其反应机理如Scheme 2所示.这为将来根据不同反应的特点设计和选择有效的催化剂提供了理论支撑.
|
|
(15) |
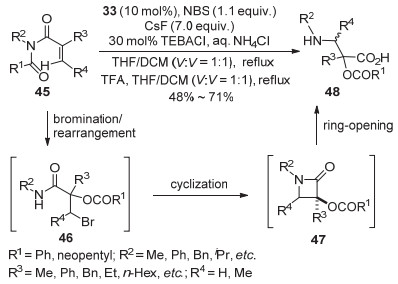
基于前面的报道, 2016年, 该课题组[27]用Eq. 14中相同的原料、氧化剂和催化剂, 对一锅法制备异丝氨酸类衍生物方法进行了深入的研究.反应中原料酰胺类化合物45在氧化剂NBS和催化剂取代碘苯33作用下发生溴化重排反应, 得到β-卤代羧基酰胺中间体46(即Scheme 2中产物), 而后加入碱CsF, 溶剂加热回流得到β-内酰胺类环合中间体47, 最后在酸性TFA作用下开环得到一系列异丝氨酸衍生物48.该方法利用原位生成高价碘策略, 成功地实现了一锅四步的反应, 为β-内酰胺类化合物及异丝氨酸类化合物的合成提供了新途径(Scheme 3).
Gulder课题组[28]对原位生成三价碘介导的区域选择性的双卤代反应也进行了研究.反应选用N-丁基-邻苯酰亚胺50作为催化剂, NBS或者KBr-Oxone组合作为氧化剂来代替溴单质, 将一系列烯烃或炔烃49转化为反式的双溴代化合物51, 发现产物中未发生芳香溴化.进一步研究表明, 在相同的反应条件下, 当反应中的氧化剂为KCl-Oxone时, 可以实现炔烃或者烯烃的双氯代, 产率中等偏上(Eq. 16).
|
|
(16) |
可用于原位生成高价碘的氧化剂有很多种, 除常用的mCPBA, AcOOH和NBS外, 另有文献报道了NaBO3• H2O、Selectfluor (1-氯甲基-4-氟-1, 4-重氮化二环[2.2.2]辛烷双(四氟硼酸)盐)以及Oxone(过硫酸氢钾复合盐)等都可以作为氧化剂参与反应, 分别完成C—C和C—O键的氧化偶联.而且Oxone参与原位生成高价碘可以使酰胺类化合物完成霍夫曼重排反应, 且产率较高.
一些无机碘盐或者碘单质同样可以代替芳基碘作为催化剂在反应中生成高价碘[10], 相关文献中的机理推测以生成三价碘中间体最为常见.该类反应中常用的碘盐有四丁基碘化铵(TBAI)、KI和NaI, 这些碘盐在反应中通常被氧化为一价的IO-或者三价的IO2-, 后者随后参与氧化偶联反应.而碘单质则是在反应中先形成自由基, 再被氧化为高价碘参与反应(表 1).
 下载:
导出CSV
下载:
导出CSV

|
2011年, Ishihara课题组[33]报道了分子内和分子间的羰基α-位氧酰化反应.使用TBAI作为催化剂, 过氧化氢作为氧化剂完成分子间和分子内酯化反应.酮类、醛类、二羰基化合物都可以发生反应, 且反应条件温和, 产率较高, 最高可达99% (Eqs. 17, 18).
|
|
(17) |
|
|
(18) |
2011年, 于炜和韩丙课题组[34]报道了一种原位生成高价碘介导的C—N偶联反应.原料氨基吡啶56与β-酮酯或1, 3-二酮57在催化量的TBAI和过氧叔丁醇(TBHP)作用下反应, 生成了一系列的咪唑并[1, 2-a]吡啶58 (Eq. 19).
|
|
(19) |
同年, 魏运洋课题组[35]将四丁基碘化铵和过氧化氢在反应体系中原位生成高价碘, 以邻苯二胺59和苯甲醛60为原料构建苯并咪唑61.值得一提的是, 反应过程首次用ESI-MS检测到了中间体三价碘[Bu4N]+-[IO2]-, 这为反应机理提供了可信的实验证据.该反应不使用酸和过渡金属, 且唯一的副产物是水(Eq. 20).
|
|
(20) |
Togo课题组[12]已经报道了过碘苯作为催化剂介导的羰基α位苯甲磺酰化.在此基础上, 2014年, Shafir课题组[36]提出KI或者I2可以代替碘苯完成此反应, 且产率中等(Eq. 21).随后, 2015年张敏和苏伟平课题组[37]报道了NaI-mCPBA组合原位生成高价碘介导的酮类化合物62的α-烷氧基化反应.该反应最关键的步骤是由NaI和mCPBA催化氧化酮生成中间体α-碘酮, 随后该中间体上的碘被甲醇亲核取代得到最终产物65 (Eq. 22).
|
|
(21) |
|
|
(22) |
2018年, Reiher和Muniz等[38]的研究表明, 酰胺类脂肪烃化合物66可在原位生成高价碘介导下发生分子内的碳-氮键氧化偶联, 形成四氢吡咯类化合物67.该反应以单质碘作为催化剂, 完成了Hofmann-Löffler反应(Eq. 23).
|
|
(23) |
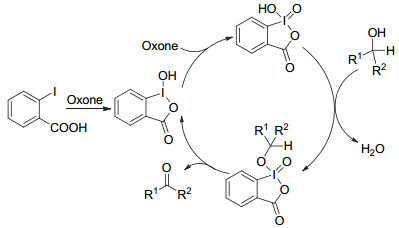
原位生成五价碘试剂与原位生成三价碘过程类似, 即催化量的碘试剂在氧化剂作用下, 被氧化成五价碘, 后者参与氧化反应.两者的区别主要在于使用的催化剂不同.原位生成五价碘大部分使用邻碘苯甲酸及其衍生物为催化剂, 被氧化后生成五价碘中间体IBX (2-碘酰基苯甲酸)或者PhIO2, 后者参与氧化反应(机理如Scheme 4, 以醇氧化为羰基化合物为例).因为五价碘在大部分溶剂中溶解度不高, 目前原位生成五价碘试剂应用范围较窄, 大部分用于醇类氧化反应.
2005年, Vinod课题组[39]首次实现了原位生成五价碘介导的醇类的氧化.该反应中催化量的2-碘苯甲酸和氧化剂Oxone加热到70 ℃生成中间体五价碘IBX, 随后在IBX的氧化作用下, 伯醇68转化为对应的酸69, 最高产率可达97% (Eq. 24). 2006年, Giannis课题组[40]报道了类似的反应.反应中使用混合溶剂MeCN/H2O (V:V=2:1), 因此添加了催化量的Bu4NHSO4作为相转移催化剂, 使一系列苄醇70以72%~93%的产率转化为苯甲醛71.同时, 该反应仲醇只能转化为酮类, 不可进一步转化为酸(Eq. 25).
|
|
(24) |
|
|
(25) |
2009年, Ishihara课题组[41]研究发现, 醇类可以在2-碘苯磺酸(IBS)与Oxone原位生成五价碘的介导下氧化成酮及酸.此外, 环己醇72在相似条件下, 使用过量Oxone可氧化成相应的环烯酮73, 产率中等(Eq. 26).同年, 该课题组[42]又报道了IBS和Oxone催化介导下, 叔丁基醇74氧化重排成烯酮75 (Eq. 27).
|
|
(26) |
|
|
(27) |
2009年, Yusubov课题组[43]报道了以催化量的PhI, RuCl3以及氧化剂Oxone原位生成五价碘PhIO2的方法.反应体系中, PhI和Oxone首先生成PhIO, 随后PhIO在RuCl3作用下发生歧化反应产生PhIO2.反应中各类醇可以被氧化为相应的酮或羧酸, 产率较高(80%以上).值得注意的是, 该反应中丙基苯以及部分芳环相邻α位的C—H键同样可以被氧化为羰基(Eq. 28).
|
|
(28) |
众所周知, 手性化合物在医药、农药、激素和食品添加剂等精细化工以及具有生理活性物质的生产上具有广泛的应用, 但是手性化合物在合成过程中会面临手性拆分成本高, 工业化难的问题.因此研究有机高价碘试剂介导的不对称合成反应具有重要的实用价值.该类方法即通过外加氧化剂将带有手性的芳基碘(碘为负一价)氧化成手性高价碘化物, 进而参与催化不对称氧化反应.该方法可以合成单一构型的手性化合物, 且无需外加的手性配体协同参与手性控制, 转化途径绿色环保, 但是氧化过程的效率和立体选择性还有待提高.
C(sp3)—H键官能团化是一种非常重要的有机反应类型, 原位生成的手性高价碘可以介导苄基、羰基的α位等活性亚甲基的氧化官能团化反应, 并且诱导产生单一手性化合物.
2007年, Wirth课题组[44]报道了原位生成手性高价碘介导的对映选择性的合成反应.该反应以0.1 equiv.的取代手性碘苯79作为催化剂, 3 equiv. m-CPBA作为氧化剂, 在室温条件下实现了酮类化合物78的α位不对称甲磺酰基化反应.该反应产率适中, 缺点是立体选择性不高(24%~28%), 但是该方法为C(sp3)—H官能团化的不对称催化反应提供了研究基础(Eq. 29).随后熊燕课题组[45]报道了在原料α-取代-β-酮酸81的C-2位置引入羟基或者苯甲磺酸的反应, 实现了C2-位立体中心的转化, 最高ee值达到69%, 但是该类反应仍需提高对应异构体的产率(Eq. 30).
|
|
(29) |
|
|
(30) |
2018年, Rueping课题组[46]也报道了类似的反应.茚满酮β-酮酯类化合物84可以在原位生成的高价碘体系中实现羰基α位的氟化, 得到手性产物86 (Eq. 31), ee值高达91.5%.
|
|
(31) |
去芳构化为苯酚或苯胺的芳环失去芳香性的过程, 去芳构化的过程中旧键断去, 新键生成, 易于构建螺环、桥环或者稠环等复杂环系.实现芳构化的方法有多种, 如氧化、加成、取代、还原等反应, 其中原位生成高价碘试剂介导的氧化芳构化是比较经典的, 该过程主要经历了原位生成高价碘、高价碘试剂与酚羟基的络合、亲核试剂进攻芳环去芳构化反应等过程.
据Harned课题组[47]研究表明, 手性碘代芳烃是可以用作分子间氧化偶联反应的催化剂, 该反应可以使酚类化合物87选择性氧化脱芳构化并引入羟基, 得到相应的环己二烯酮类衍生物89, 产率和对映选择性中等.该反应中的催化剂也可用于酚类化合物分子内的螺环化反应(Eq. 32).
|
|
(32) |
原位生成手性高价碘的使用能够实现酚类化合物的芳构化, 诱导产生螺环中心对映选择性, 这类反应已经被很多课题组报道[47~52]. 2007年, Kita课题组[48]选择具有螺二茚骨架的手性三价碘作为氧化剂, 实现了原料萘酚类化合物的脱芳构化.随后, 该组[49]改进了手性催化方法.使用邻位取代的螺二茚芳基碘苯92作为催化剂, 在mCPBA的氧化作用下原位生成中间体三价碘91, 后者将原料萘酚90氧化为螺环化合物93, 该反应在0 ℃下进行, 对映选择性良好, ee值可达92% (Eq. 33).在Kita课题组研究基础上, 2010年Ishihara课题组[50]研究表明, 使用一种C2轴对称的手性芳基碘化合物95作为催化剂, 同样可以实现原料的去芳构化, 并且ee值很高, 在83%~90%之间(Eq. 34).最近, Kita课题组[51]研究发现2, 2-二甲基8, 8-二碘萘衍生物97可以作为催化剂, 原位生成手性高价碘, 并完成了萘酚96的螺环化反应, 且对映选择性良好(Eq. 35).
|
|
(33) |
|
|
(34) |
|
|
(35) |
2015年, 龚流柱课题组[52]报道了另外一种合成手性螺环化合物的方法.以Ishihara课题组使用的手性芳基碘化物95作为催化剂, 在反应体系中原位生成手性高价碘, 将原料-羟基-N-芳基-萘甲酰胺衍生物98进行去芳构化, 构建C—C键, 得到螺杂环产物99, 该方法产率高, 对映性好, 但是要在低温下-30 ℃下进行反应(Eq. 36).
|
|
(36) |
2014年, Wirth课题组[53]以手性碘苯101作为催化剂, 将原位生成手性高价碘策略成功地应用到烯烃类衍生物100的不对称双胺化反应中, 得到一系列手性双环产物102, 产率为45%~72%, 最高ee值可达86%.该反应的部分产物在还原性条件下转化为游离的二胺(Eq. 37).
|
|
(37) |
龚流柱课题组[54]还报道了原位生成手性高价碘试剂介导的一系列串联螺环化反应. N, N-二苯基丙二酰胺类原料103在C2轴对称的手性碘代芳烃104催化氧化作用下, 活化四个C—H键, 发生分子内的C—C键的氧化偶联反应, 以中等及以上收率和较高的ee值得到具有对映选择性的吲哚酮类化合物105, 最高ee值可达90% (Eq. 38). 2016年, 杜云飞课题组[55]使用Gong课题组相同的手性碘代芳烃104, 以mCPBA为氧化剂将一系列5-(N-甲基苯胺)-3, 5-二羰基戊酸乙酯类底物106不对称合成了螺吲哚呋喃类化合物107, 该反应在室温下进行, 收率中等偏上, 对应选择性好(Eq. 39).对照试验结果表明, 该不对称螺环化反应先进行的是碳氧键的偶联再进行碳碳键的偶联, 碳碳键偶联反应是决定手性的关键步骤.
|
|
(38) |
2018年, Jacobsen等[56]发现烯丙基胺类化合物108在手性高价碘109和氧化剂mCPBA作用下, 与吡啶氢氟酸可发生氧化环合生成β-氟唑啶110.该反应需要在低温下进行, 产率中等, 但立体选择性较高(Eq. 40).
|
|
(39) |
|
|
(40) |
原位生成高价碘作为高价碘领域的一个新兴分支, 引发了许多化学家浓厚兴趣, 近十几年来围绕原位生成高价碘介导的催化循环反应的研究成果层出不穷, 主要包括原位生成三价碘、五价碘、手性高价碘在去芳构化、环合反应和不对称催化等各种氧化反应中的应用.
原位生成策略采用催化量的芳基碘代物或者无机碘化物代替等物质的量高价碘, 这不仅避免了高价碘试剂自身的不稳定性, 也实现了原子经济性.常用的氧化剂反应生成的副产物多为常见的物质, 如叔丁醇、甲酸、水等, 安全可控且绿色环保.该方法既保留了高价碘的优点, 又符合绿色化学的理念, 已被广泛应用于C—C, C—N, C—O键以及手性化合物的构建中.但是原位生成高价碘仍然面临很多挑战: (1)大部分原位生成反应需要使用过氧化物作为氧化剂, 过氧化物易爆炸, 如何寻找更多种类的氧化剂需要科研工作者进一步研究. (2)反应过程不够高效, 部分反应需要时间过长, 尤其生成手性化合物的过程.还需深入研究反应条件, 开发高效氧化剂和添加剂. (3)与传统高价碘相比, 原位生成高价碘介导的反应类型偏简单, 在四元环、七元环、多元杂环和复杂多环化合物的构建报道较少.因此需要研究新的合成方法制备不同类型的化合物, 更大发挥原位生成高价碘在不同领域的应用.
随着高价碘化学、计算化学和应用化学的不断发展, 各种由原位生成的高价碘试剂介导的非金属条件下的氧化反应有望不断涌现, 为具有药理活性的药物分子提供重要的化合物来源.
Musher, J. I. Angew. Chem. Int. Ed. 1969, 8, 54.
(a) Sandin, R. B. Chem. Rev. 1943, 32, 249.
(b) Banks, D. F. Chem. Rev. 1966, 66, 243.
(c) Varvoglis, A. Tetrahedron 1997, 53, 1179.
(d) Tohma, H.; Kita, Y. Adv. Synth. Catal. 2004, 346, 111.
(e) Zhou, W.; Zhang, L.; Jiao, N. Angew. Chem., Int. Ed. 2009, 48, 7094.
(f) Mu, X.; Wu, T.; Wang, H.; Guo, Y.; Liu, G. J. Am. Chem. Soc. 2012, 134, 878.
(g) Duan, X.; Yang, K.; Wang, Z.; Zhang, L.; Tong, Y.; Liu, H.; Du, K.; Tang, R. Chin. J. Org. Chem. 2015, 35, 2552(in Chinese).
(段希焱, 杨坤, 王志成, 张璐, 佟悦, 刘浩哲, 杜珂, 唐榕泽, 有机化学, 2015, 35, 2552.)
(a) Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123.
(b) Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523.
(c) Moriarty, R. M. J. Org. Chem. 2005, 70, 2893.
(d) Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299.
(e) Zhdankin, V. V. ARKIVOC 2009, i, 1.
(f) Kuepper, F. C.; Feiters, M. C.; Olofsson, B.; Kaiho, T.; Yanagida, S.; Zimmermann, M. B.; Carpenter, L. J.; Luther, G. W.; Lu, Z.; Jonsson, M.; Kloo, L. Angew. Chem., Int. Ed. 2011, 50, 11598.
(g) Charpentier, J.; Fruh, N.; Togni, A. Chem. Rev. 2015, 115, 650.
(h) Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328.
(i) Duan, Y.; Jiang, S.; Han, Y.; Sun, B.; Zhang, C. Chin. J. Org. Chem. 2016, 36, 1973(in Chinese).
(段亚南, 姜山, 韩永超, 孙博, 张弛, 有机化学, 2016, 36, 1973.)
(j) Zhang, X.; Cong, Y.; Lin, G.; Guo, X.; Cao, Y.; Lei, K.; Du, Y. Chin. J. Org. Chem. 2016, 36, 2513(in Chinese).
(张翔, 丛颖, 林光宇, 郭旭亮, 曹阳, 雷坤华, 杜云飞, 有机化学, 2016, 36, 2513.)
(k) Han, Y.; Zhang, C. Tetrahedron Lett. 2018, 59, 3052.
(l) Cai, Q.; Ma, H. Acta Chim. Sinica 2019, 77, 213(in Chinese).
(蔡倩, 马浩文, 化学学报, 2019, 77, 213.)
(m) Liu, Dan.; He J.; Zhang, C. Univ. Chem. 2019, 34, 1(in Chinese).
(刘丹, 贺家豪, 张弛, 大学化学, 2019, 34, 1.)
Yang, L.; Zhang-Negrerie, D.; Zhao, K.; Du, Y. J. Org. Chem. 2016, 81, 3372. doi: 10.1021/acs.joc.5b02443
Yusubov, M. S.; Zhdankin, V. V. Curr. Org. Synth. 2012, 9, 247. doi: 10.2174/157017912799829021
Uyanik M, Ishihara K. ChemCatChem 2012, 4, 177. doi: 10.1002/cctc.201100352
Trost, B. M. Science 1991, 254, 1471.
Trost, B. M. Angew. Chem., Int. Ed. 1995, 34, 259.
(a) Zheng, Z.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Sci. China, Chem. 2014, 57, 189.
(b) Zheng, Z. Ph.D. Dissertation, Tianjing University, Tianji, 2014(in Chinese).
(郑子圣, 博士论文, 天津大学, 天津, 2014.)
(c) Singh, F. V.; Wirth, T. Chem.-Asian J. 2014, 9, 950.
(d) Parra, A.; Reboredo, S. Chem.-Eur. J. 2013, 19, 17244.
Uyanik, M.; Ishihara, K. Chem. Cat. Chem. 2012, 4, 177.
Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K. J. Am. Chem. Soc. 2005, 127, 12244. doi: 10.1021/ja0542800
Yamamoto, Y.; Kawano, K.; Toy, P. H.; Togo, H. Tetrahedron 2007, 63, 4680. doi: 10.1016/j.tet.2007.03.091
Dohi, T.; Maruyama, A.; Yoshimura, M.; Morimoto, K.; Tohma, H.; Kita, Y. Angew. Chem., Int. Ed. 2005, 44, 6193. doi: 10.1002/anie.200501688
Dohi, T.; Maruyama, A.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Chem. Commun. 2007, 1224.
Zhu, C.; Liang, Y.; Hong, X.; Sun, H.; Sun, W.; Houk. K.; Shi, Z. J. Am. Chem. Soc. 2015, 137, 7564 doi: 10.1021/jacs.5b03488
Liang, D.; Yu, W.; Nguyen, N.; Deschamps, J. R.; Imler, G. H.; Li, Y.; MacKerell, A. D.; Jiang, C.; Xue, F. J. Org. Chem. 2017, 82, 3589.
Ito, M.; Kubo, H.; Itani, I.; Morimoto, K.; Dohi, T.; Kita, Y. J. Am. Chem. Soc. 2013, 135, 14078. doi: 10.1021/ja407944p
Miyamoto, K.; Sakai, Y.; Goda, S.; Ochiai, M. Chem. Commun. 2012, 48, 982. doi: 10.1039/C2CC16360H
Philips, B.; Frostick, F. C.; Starcher, P. S. J. Am. Chem. Soc. 1957, 79, 5982. doi: 10.1021/ja01579a037
Antonchick, A. P.; Samanta, R.; Kulikov, K.; Lategahn, J. Angew. Chem., Int. Ed. 2011, 50, 8605. doi: 10.1002/anie.201102984
He, Y.; Huang, J.; Liang, D.; Liu, L.; Zhu, Q. Chem. Commun. 2013, 49, 7352.
Wang, X.; Gallardo-Donaire, J.; Martin, R. Angew. Chem., Int. Ed. 2014, 53, 11084. doi: 10.1002/anie.201407011
Shen, C.; Yang, M.; Xu, J.; Chen, C.; Zheng, K.; Shen, J.; Zhang, P. RSC Adv. 2017, 7, 49436.
Braddock, D. C.; Cansell, G.; Hermitage, S. A. Chem. Commun. 2006, 2483.
Fabry, D. C.; Stodulski, M.; Hoerner, S.; Gulder, T. Chem.-Eur. J. 2012, 18, 10834.
Ulmer, A.; Stodulski, M.; Kohlhepp, S. V.; Patzelt, C.; Poethig, A.; Bettray, W.; Gulder, T. Chem.-Eur. J. 2015, 21, 1444. doi: 10.1002/chem.201405888
Patzelt, C.; Alexander, P.; Gulder, T. Org. Lett. 2016, 18, 3466. doi: 10.1021/acs.orglett.6b01658
Stodulski, M.; Goetzinger, A.; Kohlhepp, S. V.; Gulder, T. Chem. Commun. 2014, 50, 3435. doi: 10.1039/C3CC49850F
Liu, H.; Tan, C.-H. Tetrahedron Lett. 2007, 48, 8220. doi: 10.1016/j.tetlet.2007.09.078
Alhalib, A.; Kamouka, S.; Moran, W. J. Org. Lett. 2015, 17, 1453. doi: 10.1021/acs.orglett.5b00333
Morimoto, K.; Sakamoto, K.; Ohshika, T.; Dohi, T.; Kita. Y. Angew. Chem., Int. Ed. 2016, 55, 3652. doi: 10.1002/anie.201511007
Yoshimura, A.; Middleton, K. R.; Luedtke, M. W.; Zhu, C.; Zhdankin, V. V. J. Org. Chem. 2012, 77, 11399. doi: 10.1021/jo302375m
Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2011, 50, 5331. doi: 10.1002/anie.201101522
Ma, L.; Wang, X.; Yu, W.; Han, B. Chem. Commun. 2011, 47, 11333. doi: 10.1039/c1cc13568f
Zhu, C.; Wei, Y. ChemSusChem 2011, 4, 1082. doi: 10.1002/cssc.201100228
Guo, W.; Vallcorba, O.; Vallribera, A.; Shafir, A.; Pleixats, R.; Rius, J. ChemCatChem 2014, 6, 468. doi: 10.1002/cctc.201300774
Zhu, C.; Zhang, Y.; Zhao, H.; Huang, S.; Zhang, M.; Su, W. Adv. Synth. Catal. 2015, 357, 331.
Duhamel, T.; Stein, C. J.; Martínez, C.; Reiher, M.; Muñiz. K. ACS Catal. 2018, 8, 3918.
Thottumkara, A. P.; Bowsher, M. S.; Vinod, T. K. Org. Lett. 2005, 7, 2933.
Schulze, A.; Giannis, A. Synthesis 2006, 257.
Uyanik, M.; Akakura, M.; Ishihara, K. J. Am. Chem. Soc. 2009, 131, 251. doi: 10.1021/ja807110n
Uyanik, M.; Fukatsu, R.; Ishihara, K. Org. Lett. 2009, 11, 3470. doi: 10.1021/ol9013188
Yusubov, M. S.; Zagulyaeva, A. A.; Zhdankin, V. V. Chem.-Eur. J. 2009, 15, 11091. doi: 10.1002/chem.200901953
Richardson, R. D.; Page, T. K.; Altermann, S.; Paradine, S. M.; French, A. N.; Wirth, T. Synlett 2007, 538.
Fenga, Y.; Huanga, R.; Hua, L.; Xiong, Y.; Coeffard, V. Synthesis 2016, 48, 2637. doi: 10.1055/s-0035-1561442
Pluta, R.; Krach, P. E.; Cavallo, L.; Falivene, L.; Rueping, M. ACS Catal. 2018, 8, 2582. doi: 10.1021/acscatal.7b03118
Volp, K. A.; Harned, A. M. Chem. Commun. 2013, 49, 3001. doi: 10.1039/c3cc00013c
Dohi, T.; Maruyama, A.; Takenaga, N.; Senami, K.; Minamitsuji, Y.; Fujioka, H.; Caemmerer, S. B.; Kita, Y. Angew. Chem., Int. Ed. 2008, 47, 3787. doi: 10.1002/anie.200800464
Dohi, T.; Takenaga, N.; Nakae, T.; Toyoda, Y.; Yamasaki, M.; Shiro, M.; Fujioka, H.; Maruyama, A.; Kita, Y. J. Am. Chem. Soc. 2013, 135, 4558. doi: 10.1021/ja401074u
Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2010, 49, 2175. doi: 10.1002/anie.200907352
Dohi, T.; Sasa, H.; Miyazaki, K.; Fujitake, M.; Takenaga, N.; Kita, Y. J. Org. Chem. 2017, 82, 11954. doi: 10.1021/acs.joc.7b02037
Zhang, D.-Y.; Xu, L.; Wu, H.; Gong, L.-Z. Chem.-Eur. J. 2015, 21, 10314. doi: 10.1002/chem.201501583
Mizar, P.; Laverny, A.; Mohammad. E. S.; Farid, U.; Brown, M.; Malmedy, F.; Wirth, T. Chem. Eur. J. 2014, 20, 9910. doi: 10.1002/chem.201403891
Wu, H.; He, Y.-P.; Xu, L.; Zhang, D.-Y.; Gong, L.-Z. Angew. Chem., Int. Ed. 2014, 53, 3466. doi: 10.1002/anie.201309967
Cao, Y.; Zhang, X.; Lin, G.; Zhang-Negrerie, D.; Du, Y. Org. Lett. 2016, 18, 5580. doi: 10.1021/acs.orglett.6b02816
Mennie, K. M.; Banik, S. M.; Reichert, E. C.; Jacobsen, E. N. J. Am. Chem. Soc. 2018, 140, 4797. doi: 10.1021/jacs.8b02143

图 1 芳基碘作为前催化剂原位生成三价碘化合物示意图
Figure 1 Aryl iodide as a procatalyst in-situ generation of hypervalent iodine(Ⅲ) reagent

图 2 碘盐作为前催化剂原位生成高价碘化合物示意图
Figure 2 Iodide salts as precatalysts in-situ generation of hypervalent iodine reagent

图式 1 mCPBA作为氧化剂原位生成三价碘介导的羰基α位官能团化
Scheme 1 mCPBA as an oxidant in situ to generate iodine(Ⅲ) reagents-mediated oxidative functionalization at α positions of carbonyls

图式 2 NBS作为氧化剂原位生成三价碘介导的重排反应
Scheme 2 NBS as an oxidant in situ to generate iodine(Ⅲ) reagents-mediated rearrangement reaction

图式 3 NBS作为氧化剂原位生成三价碘介导的异丝氨酸类衍生物的合成
Scheme 3 NBS as an oxidant in situ to generate iodine(Ⅲ) reagents-mediated synthesis of isoflavin derivatives
表 1 NaBO3•H2O, selectfluor, oxone作为氧化剂原位生成三价碘在有机合成中的应用
Table 1. Application of NaBO3•H2O, selectfluor, oxone as oxidant in situ to generate trivalent iodine in organic synthesis
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们









































 下载:
下载: