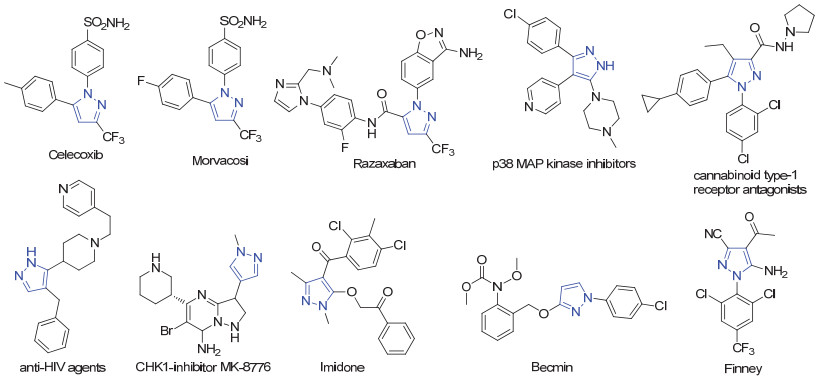
图 1.
一些重要的吡唑化合物
Figure 1.
Some important pyrazoles

含氮杂环类结构单元作为基础的杂环, 广泛存在于天然产物、药物及功能材料中[1~5], 对于现代药物化学和有机化学有着非常重要的作用.正是由于上述原因, 发展含氮杂环类化合物的高效合成方法一直是有机化学家和药物化学家们研究的热点[6~10].在众多的含氮杂环中, 吡唑是重要的成员之一, 作为关键的结构单元, 广泛存在于许多药物和生物活性分子中(图 1), 如具有止痛[11]、退热[12]、抗炎[13]、抗菌[14]和抗高血糖[15]等药用活性分子.很多已成为市售药品, 具有代表性的有塞来昔布(celecoxib)[16]、吗伐烤昔(mavacoxib)[17]、雷扎沙班(razaxaban)[18, 19]等(图 1).随着对吡唑类化合物合成及其临床应用研究的深入, 近年来科研工作者也发展出很多含吡唑结构的生物活性分子, 如p38激酶抑制剂[20]、大麻素1型受体拮抗剂[21]、抗HIV病毒试剂[22]和细胞周期检测点激酶抑制剂(CHK1)[23]等.上述研究结果将推动基于吡唑结构的创新药物的发展.此外, 吡唑类化合物除了临床用药外, 也因其具有高效、低毒等特点, 被广泛应用于农药领域, 如除草剂、杀菌剂和杀虫杀螨剂.市场上已经商品化的代表性农药有吡草酮(benzofenap)、唑菌胺酯(pyraclostrobin)、氟虫腈(fipronil)等.
鉴于吡唑类化合物极其重要的作用, 发展高效地合成吡唑的方法一直以来是有机化学以及药学工作者所关注的热点.常见的合成策略有:由1, 3-二羰基化合物及类似物出发, 通过环化反应获得吡唑, 或者由腙出发制得吡唑等.本文介绍了近些年来有关吡唑类化合物的合成方法, 希望能对本领域的研究起到一定的辅助与推动作用.
1, 3-二羰基化合物和肼类化合物环化是一种沿用至今的合成吡唑类化合物的重要合成方法. 1, 3-亲电性化合物包括1, 3-二羰基化合物、α, β-不饱和羰基化合物以及β-位上带有离去基团的α, β-不饱和羰基化合物.
1883年, Knorr[24]报道了取代的1, 3-二羰基化合物和肼反应生成1, 3, 5-三取代的吡唑.该反应由于区域选择性不好, 会生成两种取代基位置不同的异构体1和2 (Eq. 1).
|
|
(1) |
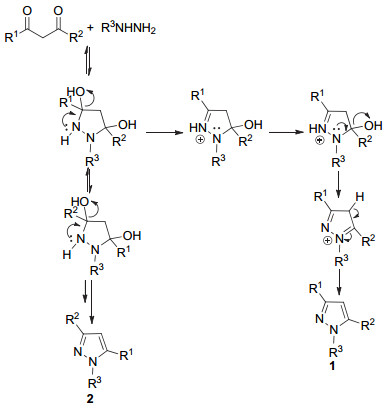
2000年, Elguero等[25]提出了不同取代基的1, 3-二羰基化合物与肼类化合物反应合成吡唑的通用反应机理.取代的肼上两个氮原子都可能与1, 3-二羰基化合物的1, 3-位发生反应, 该过程类似羟醛缩合反应, 可逆地生成两种取代基位置不同的异构体, 然后再不可逆地脱除一分子水, 最终得到取代基位置不同的吡唑类化合物1和2 (Scheme 1).
2015年, Alves课题组[26]报道了在路易斯酸作用下“一锅法”反应合成含硒多取代吡唑的合成方法.以1, 3-二羰基类化合物、苯肼、取代的二硒醚为原料, CuBr为催化剂, 2, 2'-联吡啶(bpy)为配体, 空气条件下在二甲基亚砜(DMSO)中加热至100 ℃, 最终以98%的收率成功地构建了C-4位碳硒键的吡唑化合物3 (Eq. 2).该反应底物适用范围广, 产率良好.
|
|
(2) |
2016年, Kivrak课题组[27]对上述方法进行了改进.首先使用α, β-不饱和醛4和苯肼反应生成α, β-不饱和腙, 然后在碱的作用下和苯硒氯进行环化反应, 以较高的收率合成4-苯硒吡唑5 (Eq. 3).
|
|
(3) |
2017年, 王华课题组[28]报道了“一锅法”反应合成含硫多取代吡唑的方法, 该反应以乙酰乙酸乙酯、苯肼、对甲基苯硫醇为原料, 在I2的催化下, DMSO作氧化剂, 加热反应至70 ℃, 最终以56%~84%的收率得到C-4含硫的多取代吡唑6 (Eq. 4).该方法无需金属催化剂和溶剂, 且具有良好的选择性.
|
|
(4) |
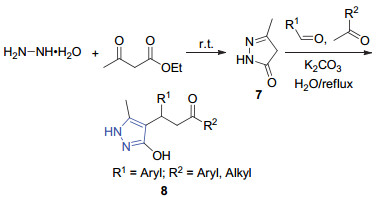
2017年, Deka等[29]报道了“一锅多组分反应”合成吡唑的方法.该方法以乙酰乙酸乙酯和水合肼为底物, 先在室温下反应2 min生成中间体7, 然后再和醛(或者酮)在K2CO3为催化剂、水为溶剂的条件下回流2 h, 以85%的收率得到3, 4, 5-取代的吡唑8 (Scheme 2).该方法以廉价易得的K2CO3为催化剂和水作为反应溶剂, 底物范围广、产量高、操作简单、产物易于分离.
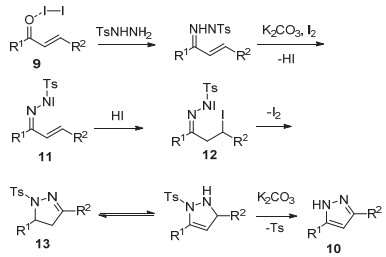
2016年, 王保民课题组[30]发展了碘催化下, 乙醇作溶剂, 以α, β-不饱和羰基化合物9和对甲苯磺酰肼为底物, 75 ℃反应2 h生成3, 5-二取代的吡唑10的方法(Eq. 5).该方法简单, 条件温和, 反应时间短, 底物范围广, 产率高.
|
|
(5) |
上述反应的可能机理见Scheme 3所示.首先, 在碘的催化下, α, β-不饱和酮和对甲苯磺酰肼反应生成α, β-不饱和腙, 再在K2CO3和碘的作用下, 失去一分子HI, 得到中间体11.之后HI和C=C进行加成, 生成中间体12, 随后进行分子内的环化得到4, 5-二氢-1H-吡唑13.碱性条件下, 脱去对甲苯磺酰基, 从而得到3, 5-二取代吡唑10.
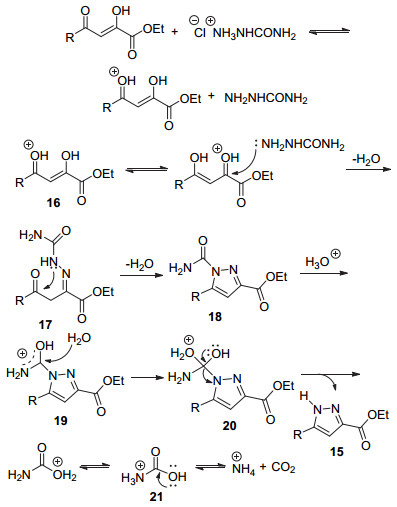
2015年, Markovic和Joksovic[31]报道了以水作为溶剂, 用氨基脲盐酸盐代替肼, 与底物14发生反应, 经过回流2 h制得5-取代-3-羧酸酯吡唑15 (Eq. 6).该方法绿色, 条件温和, 产率高, 并且产物易于分离.
|
|
(6) |
该反应的可能机理为:第一步, 氨基脲和质子化的酯16进行亲核加成反应, 得到亚胺中间体17, 再经分子内环化脱水, 得到环状缩合的吡唑-1-酰胺衍生物18, 然后经质子化得到中间体19.在酸性条件下, 水亲核进攻19生成中间体20.由于中间体20不稳定, 分解得到5-取代-3-羧酸酯吡唑15和质子化的氨基甲酸21.未取代的氨基甲酸不稳定, 在质子作用下, 氮原子发生质子化生成阳离子, 随后分解生成氨基阳离子和CO2 (Scheme 4).
二氧化硫脲(TUD)是便宜易得的有机催化剂, 反应完成后, 通过过滤分离产物, 剩余的TUD水溶液可以重复使用数次, 其催化活性不会减弱. 2016年, Vekariya课题组[32]报道了使用TUD作催化剂, 以乙酰乙酸乙酯、水合肼、苯甲醛和丙二腈为底物, “一锅法”四组分反应制得1, 4-二氢吡喃并[2, 3-c]吡唑-5-甲腈衍生物22 (Scheme 5).该方法产率高, 后处理过程简单, 避免使用有机溶剂, 反应时间短, 催化剂可重复使用.
同年, Banerjee小组[33]报道了使用ZrO2纳米颗粒催化的“一锅法”四组分反应合成吡喃并[2, 3-c]吡唑衍生物23 (Eq. 7).该反应以乙酰乙酸乙酯、肼、芳香甲醛、丙二腈为底物, ZrO2纳米颗粒作为催化剂, EtOH-H2O为溶剂, 在室温下反应5 min, 以95%的收率得到单一产物23.该反应实验操作简单、反应快、产率高、产物可直接从乙醇中重结晶获得, 避免了使用柱色谱进行分离纯化. ZrO2NPs催化剂既无毒又可重复使用, 是一种绿色高效合成吡唑环的方法.
|
|
(7) |
2014年, 王晓龙小组[34]在干燥的二氯甲烷中加入卤代的腙、β-酮磷酸酯24和2 equiv.的氢氧化锂, 室温条件下反应10 h得到1, 3, 5-三取代吡唑25 (Eq. 8).该反应必须在无水条件下才能实现.对底物的范围和官能团兼容性研究发现, 当腙苯环上R1为缺电子取代基时, 反应收率不受影响; 当β-酮磷酸酯24上的R3取代基为吸电子基团, 如酯基、氨基、酮、三氟甲基等时, 都能以较好的收率(55%~95%)合成相应的3-位取代的吡唑.该方法原料易得, 在室温条件下就能反应, 区域选择性高, 底物范围广, 产率高.
|
|
(8) |
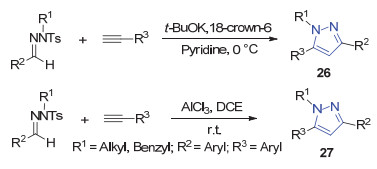
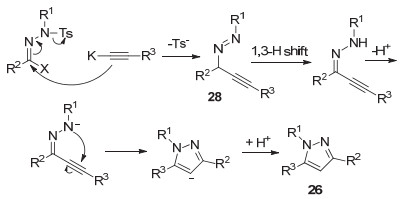
2014年, 唐萌课题组[35]在碱的作用下, N-烷基对甲苯磺酰腙和末端炔烃反应得到了1, 3, 5-三取代的吡唑26, 随后该课题组[36]对之前方法进行改进, 使用AlCl3催化N-烷基对甲苯磺酰腙和末端炔烃合成1, 3, 5-三取代的吡唑27 (Scheme 6).该方法反应条件温和, 在室温条件下就能进行反应.
可能的机理见Scheme 7所示.炔基钾和N-烷基对甲苯磺酰腙进行亲核加成, 失去一分子对甲苯磺酰基阴离子生成中间体28, 然后经1, 3-氢转移失去一个质子, 最后经分子内环化再获得一个质子, 得到1, 3, 5-三取代的吡唑26.该方法可以进行克级规模制备1, 3, 5-取代的吡唑类化合物, 并且底物适用范围广, 区域选择性好, 产率高.
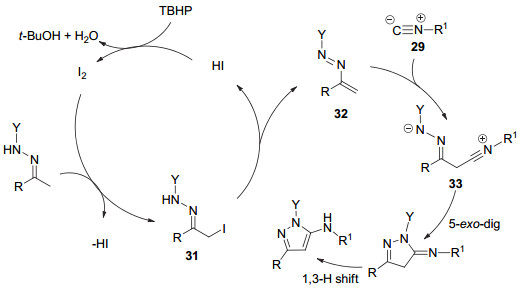
2015年, Wang课题组[37]报道了I2-叔丁基过氧化氢(TBHP)催化的N-磺酰腙和取代异氰29合成5-氨基吡唑类化合物30 (Eq. 9).该方法不需要金属催化, 底物适用性范围广, 原子利用率高, 反应时间短, 产率高.
|
|
(9) |
作者推测反应机理为:在I2催化下, N-磺酰腙先转化为α-碘取代的腙31, 随后失去HI得到偶氮烯烃32, 异氰和偶氮烯烃进行共轭加成生成中间体33, 最后进行分子内环化和1, 3-氢转移合成得到5-氨基吡唑类化合物(Scheme 8).
2016年, Alizadeh课题组[38]使用乙醇作溶剂, 先将氯代的腙和Et3N的混合物在室温下搅拌10 min, 然后将3-甲酰基色酮34加入到反应体系中, 并将反应物搅拌4 h, 高产率地得到化合物35 (Eq. 10).该课题组研究了当X为H、Cl、Me; Ar为Ph、4-ClC6H4等5种取代基的反应情况, 都能以82%~86%的良好收率得到产物.该反应优点:区域选择性高, 实验操作简单, 产物收率高, 易于纯化和反应条件温和.
|
|
(10) |
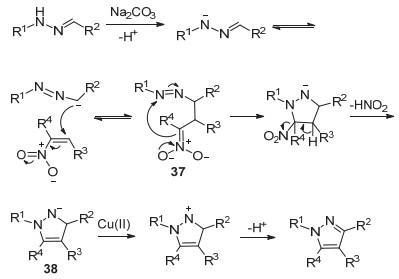
2016年, 黄国生课题组[39]报道了铜催化的腙和硝基烯的[3+2]反应, 合成了1, 3, 4-三取代和1, 3, 4, 5-四取代吡唑36 (Eq. 11).
|
|
(11) |
该反应机理如Scheme 9所示.首先腙在碱的作用下失去质子, 再和硝基烯进行Michael加成得到中间体37, 随后进行分子内环化, 脱去一分子亚硝酸得到中间体38, 然后经二价铜氧化, 获得一个质子生成1, 3, 4, 5-四取代吡唑(Scheme 9).该反应原料易得, 底物适用范围广, 产率较好.
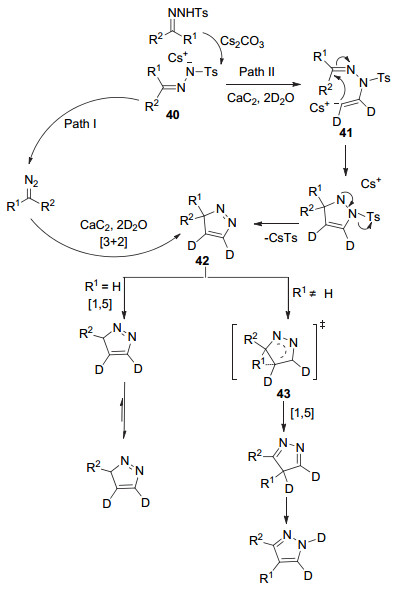
2016年, 江焕峰课题组[40]报道了一种更加绿色的合成吡唑的方法, 即使用对甲苯磺酰腙和碳化钙为底物, 在碱和水为溶剂条件下合成3, 5-二取代吡唑39 (Eq. 12).该反应不需要金属催化剂, 产率良好, 并且可以用于工业生产, 但区域选择性不好.
|
|
(12) |
作者推测在Cs2CO3作用下, 对甲苯磺酰腙去质子化得到铯盐40.该反应机理有两种可能路径(Scheme 10).一条可能路径是, 在加热条件下, 铯盐40先生成重氮化合物, 然后和[D2]-乙炔气体[3+2]环化生成3H-吡唑化合物42; 另一条可能路径是:铯盐40的氮负阴离子和[D2]-乙炔气体进行亲核加成反应, 生成碳负离子中间体41, 分子内的碳负离子进行亲和进攻, 随后对甲苯磺酰基阴离子和铯离子结合离去, 得到3H-吡唑化合物42.当R1=H时, 分子内氢经过[1, 5]氢转移和异构化得到产物.当R1≠H是富电子基团时, 先形成转移状态43, R1进行C4位迁移, 随后进行1, 5-σ键转移生成产物.
同年, Pramanik课题组[41]也报道了一种绿色合成吡唑类化合物的方法, 用磁性可分离的Fe3O4@SiO2-SO3H纳米粒子, 高效地合成了吡唑稠合的异香豆素类化合物(Eq. 13).该反应无需溶剂, 以44和芳基腙为底物, 使用磁性可分离的Fe3O4@SiO2SO3H纳米颗粒固体酸作为催化剂, 以良好的收率合成吡唑稠合的异香豆素类化合物45.此反应无需使用溶剂, 底物适用范围, 催化剂可以分离和回收.
|
|
(13) |
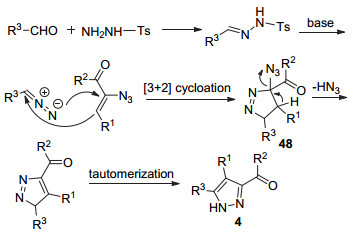
2013年, 俞永平课题组[42]报道了使用乙烯基叠氮46, 醛和对甲苯磺酰肼, “一锅法”多组分合成了3, 4, 5-三取代吡唑47 (Eq. 14).该方法底物适用范围广、产率高.
|
|
(14) |
上述反应的可能机理为:醛和对甲苯磺酰肼缩合生成对甲苯磺酰腙, 随后在碱性和加热条件下生成重氮化合物, 重氮化合物和乙烯基叠氮46发生[3+2]环化反应得到中间体48, 48失去叠氮酸后再进行异构化得到产物(Scheme 11).
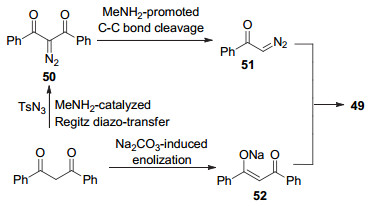
2017年, 胡跃飞课题组[43]报道了一种四步串联反应合成吡唑类化合物的方法. 1, 3-二芳基-1, 3-二酮、TsN3、甲胺水溶液、Na2CO3在N, N-二甲基甲酰胺(DMF)溶液加热至85 ℃, 反应3 h后得到成3, 5-二芳酰基-4-芳基吡唑49 (Eq. 15).
|
|
(15) |
该反应首先在甲胺的作用下, 1, 3-二芳基-1, 3-二酮和对甲苯磺酰基叠氮反应生成重氮化合物50, 随后在甲胺的作用下进行C—C键断裂, 得到重氮化合物51. 51与1, 3-二芳基-1, 3-二酮在碱的作用下得到的烯醇化合物52进行1, 3-偶极环加成反应, 最终得到吡唑产物3, 5-二芳酰基-4-芳基吡唑49 (Scheme 12).
2014年, Namboothiri课题组[44]报道, 在搅拌下, α-重氮基-β-酮砜(54)首先生成重氮甲基砜阴离子, 再和乙烯砜55进行1, 3-偶极环加成, 然后在碱性条件下选择性地消除苯磺酰基, 最后得到产物55 (Eq. 16).该方法反应条件温和, 底物适用范围广, 产率高.
|
|
(16) |
2016年, 该课题组[45]也报道了由查尔酮和α-重氮基-β-酮酯区域选择性地合成吡唑酮酯56的方法(Eq. 17).该方法与之前报道的方法类似, 尽管产率中等, 但是区域选择性高.
|
|
(17) |
2015年, Mohanan课题组[46]在碱性条件下, 用Bestmann-Ohira试剂(含氧的丙基膦酸酯57)和α, β-不饱和醛进行反应, 合成了乙烯基吡唑58 (Eq. 18).
|
|
(18) |
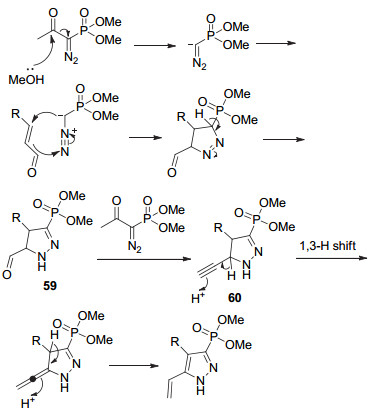
该反应可能机理如Scheme 13所示. BOR在甲醇溶液中形成重氮甲基阴离子, 再和肉桂醛进行共轭加成、环化生成中间体59, 然后59和另一分子的BOR反应生成二氢化吡唑炔60, 随后进行1, 3-氢转移、芳构化生成乙烯基吡唑.该方法也可以用于合成磷酰基类的吡唑生物碱.
2016年, 万小兵课题组[47]报道, 在四丁基碘化铵和TBHP催化作用下, 烯烃和重氮乙酸乙酯经过[3+2]环化、氧化脱氢过程, 最终得到吡唑产物61 (Eq. 19).该反应原料易得, 原子利用率高, 底物范围广, 反应条件环保.
|
|
(19) |
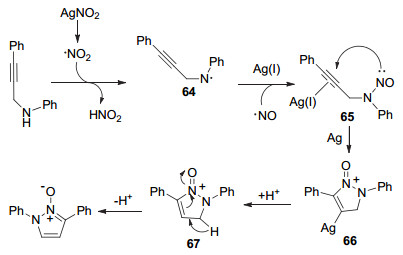
2016年, 严汝龙课题组[48a]报道了一种简单的合成吡唑N-氧化物的方法.在四氢呋喃(THF)溶剂中, 以N-(3-苯基丙-2-炔-1-基)苯胺(62)和AgNO2为底物, 在100 ℃下反应6 h, 以85%产率得到1, 3-二苯基-1H-吡唑N-氧化物63 (Eq. 20).该反应首次将AgNO2用作合成吡唑N-氧化物的NO源, 含N-炔丙基胺上的各种取代基在反应中都不受影响, 以较高的收率得到吡唑N-氧化物.
|
|
(20) |
作者推测该反应可能机理为:在高温条件下, AgNO2分解产生硝基自由基.硝基自由基获得N-(3-苯基丙-2-炔)-1-苯胺上的氢自由基生成HNO2和含氮自由基64, 随后64与HNO2分解产生的硝基自由基结合, 再与一价的银配位得到中间体65.在银催化下, 65分子内环化生成66, 再在质子的作用下进行结构互变, 最终生成吡唑N-氧化物(Scheme 14).
同年, Saikia小组[48b]使用AgOTf为催化剂, 在乙酸和氯仿的混合溶剂中加入炔丙基胺68和3 equiv.的亚硝酸钠, 在0 ℃缓慢恢复至室温的条件下反应3 h, 以85%的产率得到吡唑N-氧化物69 (Eq. 21).该方法产率高, 反应条件温和.
|
|
(21) |
2015年, Yadav等[49]在eosinY(一种有机光催化剂)和空气(O2)作为氧化剂条件下, 以乙醇为溶剂, 以苯甲醛、丙二腈和苯肼为底物, 于室温下, 使用简单的家用紧凑型荧光灯(CFL, 22 W)照射该反应体系, 最终以95%收率得到吡唑类化合物70 (Eq. 22).该方法无需金属条件, 原料简单易得, 反应时间短, 后处理简单, 产物易于分离, 无需柱色谱, 产物产率高.
|
|
(22) |
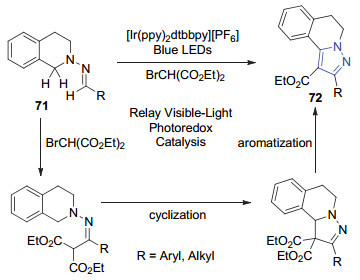
2017年, 朱成建课题组[50]使用腙71和2-溴-1, 3-二羰基化合物为反应底物, 在可见光催化下, 经过[4+1]环化、芳构化合成吡唑衍生物72 (Scheme 15).该反应过程经历了一个连续的偶联/环化/芳构化过程, 反应条件温和, 以高效且经济的方式快速获得吡唑骨架.
2014年, Dhage课题组[51]报道了一种合成吡唑-4-羧酸酯的新方法, 即用过渡金属Pd(II)作为催化剂, α, β-炔腙为底物, 由配体控制来合成吡唑类化合物73或74 (Eq. 23).优化反应条件发现, 当催化剂为Pd(TFA)2, 且使用配体L1时, 该反应以91%的收率得到产物73.不加配体, 使用DMSO/MeOH (V:V=2.5:3)混合溶剂时, 在室温下反应72 h, 没有73生成, 仅以优异的产率(86%)得到化合物74.几种不同底物的α, β-炔烃腙都能以良好的收率得到相应的产物(76%~98%).当以烷基代替炔末端的芳基时, 产物收率相对较低(76%), 但是当R1=Me, R2=Ph, Ar=4-CF3C6H4时, 产物的收率达到98%.
|
|
(23) |
2014年, 刘培念课题组[52]用吖啶铑催化的肼和炔烃进行加成-环化串联反应合成了四取代吡唑76 (Scheme 16).该串联反应通过酰肼的C—N键和炔烃进行[3+2]环化, 随后酰胺的C—N键断裂, 再环化脱水.通过对反应条件的优化筛选, 发现在[Cp*RhCl2]2 (2.5 mol%)和NaOAc (25 mol%)存在下, MeCN作溶剂在60 ℃下反应5 h, 以最高88%产率得到单一产物76.该课题组在最优的反应条件下, 探究了这种串联转化的底物适用范围.首先当苯环对位取代基R为给电子基团时, 其产率优于苯环对位是吸电子基团的产率.当邻位有Me取代时, 产率有所降低, 可能是环化作用时空间位阻的影响所致.接下来, 将R2变换为不同的芳基取代基, 均可得到预期的产物, 产率在91%~96%之间.
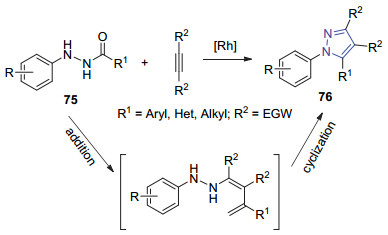
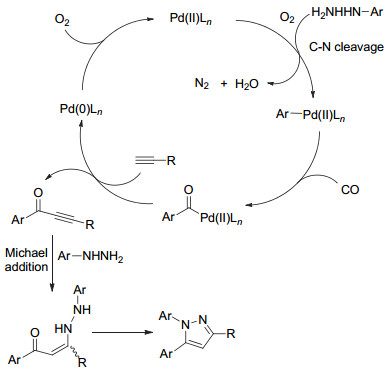
2017年, 赵军锋课题组[53]在钯催化下, 芳基肼、末端取代炔烃和CO/氧气进行“一锅法”多组分串联反应, 合成了三取代吡唑77 (Eq. 24).
|
|
(24) |
上述反应经过五步的串联反应, 包括: C—N断裂、CO插入、Sonogashira偶联、Michael加成环化(Scheme 17).该方法区域选择性好、产率高.
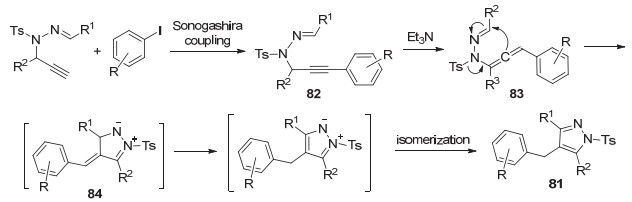
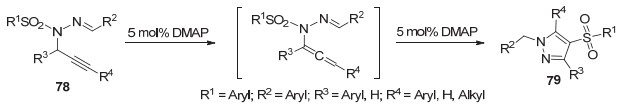
2013年, 詹庄平课题组[54]报道了路易斯碱作用下炔丙基腙合成吡唑的方法.该反应经历分子内环化、1, 3-磺酰基转移, 合成了多取代4-磺酰基-1H-吡唑79 (Scheme 18).
2017年, 该课题组[55]对此方法进行改进, 使用N-炔丙基磺酰腙通过Pd(II)/Cu(I)-催化的Sonogashira/环化反应, 高效地合成了多取代吡唑(Eq. 25).上述反应的机理为:底物80和取代的碘苯通过Sonogashira偶联反应生成化合物82.在三乙胺作用下, 82异构化得到联烯83. 83经分子内环化得到中间体84, 再经两次异构化, 最终生成产物81 (Scheme 19).该反应底物适用范围广、产率高.
2015年, Schmitt课题组[56]使用过渡金属钌催化的2-烷基-1, 3-二醇和肼反应, 合成了1, 4二取代吡唑85 (Eq. 26).该方法具有良好的立体选择性, 也适用于β-羟基酮立体选择性的合成不对称的吡唑.
|
|
(25) |
|
|
(26) |
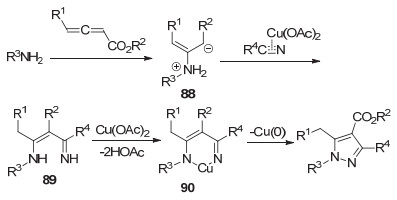
2014年, 麻生明课题组[57]使用铜催化剂, 以联烯86、胺和取代腈类为反应底物, “一锅法”合成吡唑类化合物87 (Eq. 27).
|
|
(27) |
反应的可能机理如Scheme 20所示.联烯86和胺进行Michael加成得到中间体88, 然后经过醋酸铜活化的腈类反应, 生成中间体89.中间体89在醋酸铜作用下, 失去两分子醋酸, 得到金属环化的中间体90, 随后进行还原消除得到吡唑类化合物.该反应原料易得、产率高.
2014年, 江焕峰课题组[58]在铜催化下, 以肟乙酸盐91、苯胺和多聚甲醛为底物, DMSO为溶剂, 加热至120 ℃反应12 h, 得到1, 3, 4-取代的吡唑92 (Eq. 28).该反应操作简单, 底物适用范围广.
|
|
(28) |
使用离子液体作为反应介质, 可以为溶剂的排放和催化剂的回收提供解决方案.离子液体已经成为有机溶剂和催化剂的优选替代物, 被用作有机反应的反应介质和催化剂.为了扩大离子液体[DMBSI]HSO4在杂环化合物合成中的应用. 2014年, Manouchehr课题组[59]使用[DMBSI]HSO4作催化剂, 以1, 3-二羰基化合物、水合肼、丙二睛、苯甲醛为底物, 60 ℃条件下, 反应10 min, 以95%的收率得到吡喃并[2, 3-c]吡唑93 (Eq. 29).该反应催化剂经过三次连续循环使用, 催化剂的活性并没有发生明显损失.
|
|
(29) |
该课题组继续扩大底物醛和腈类化合物的研究的范围, 使用1, 2-二酮化合物为底物, 在上述相同的条件下, 以85%~96%的产率得到螺环结构的吡喃并[2, 3-c]吡唑94 (Eq. 30).
|
|
(30) |
2015年, Nageswar课题组[60]报道了一种新颖、温和、简单高效和绿色合成吡唑衍生物的方法, 即用聚乙二醇(PEG-400)为反应介质, 一锅四组分合成螺[二氢吲哚-3, 4-吡喃并[2, 3-c]吡唑]-3'-羧酸酯衍生物95 (Eq. 31).该方法底物范围广谱, 产率高, 易于操作, PEG-400具有回收性.
|
|
(31) |
通过对吡唑及其衍生物合成方法的介绍, 发现其合成方法众多, 反应的种类也各种各样.对其总结发现:第一, 自从Knorr合成吡唑后, 由于其区域选择性不好, 化学家对其不断改进, 随后一些区域选择性高、底物适用范围广的方法被开发.第二, 腙及其类似物, 叠氮或者重氮化合物大都经过环化或者环加成反应构建吡唑, 吡唑产物区域选择性高.腙及其类似物底物容易制备, 但是叠氮或者重氮化合物由于其毒性大、易爆炸, 使用率较低.第三, 近些年光催化、过渡金属催化C—N、N—N键构建方法的发展, 也为吡唑的合成提供了一些新的、简便的方法.随着人类对于环境保护的重视以及高效合成的不断追求, 绿色合成将是未来发展方向.在化学工作者的不断探索下, 必将进一步简化吡唑类药物的合成与生产工艺, 助力新的吡唑类功能分子的发现.
(a) Dedeian, K.; Shi, J.; Shepherd, N.; Forsythe, E.; Morton, D. C. Inorg. Chem. 2005, 44, 4445.
(b) Chang, S. Y.; Chen, J. L.; Chi, Y.; Cheng, Y. M.; Lee, G. H.; Jiang, C. M.; Chou, P. T. Inorg. Chem. 2007, 46, 11202.
(c) Cavero, E.; Uriel, S.; Romero, P.; Serrano, J. L.; Giménez, R. J. Am. Chem. Soc. 2007, 129, 11608.
(d) Ye, C.; Gard, G. L.; Winter, R. W.; Syvret, R. G.; Twamley, B.; Shreeve, J. M. Org Lett. 2007, 9, 3841.
(e) Yang, L.; Okuda, F.; Kobayashi, K.; Nozaki, K.; Tanabe, Y.; Ishii, Y.; Haga, M. Inorg Chem. 2008, 47, 7154.
(f) Seltzman, H. H. Drug Dev. Res. 2009, 70. 601.
(g) Dai, H. X.; Stepan, A. F.; Plummer, M. S.; Zhang, Y. H.; Yu, J. Q. J. Am. Chem. Soc. 2011, 133, 7222.
(h) Jones, L. H.; Allan, G.; Corbau, R.; Middleton, D. S.; Mowbray, C. E.; Newman, S. D.; Phillips, C.; Webster, R.; Westby, M. Chem. Biol. Drug Des. 2011, 77, 393.
(i) Kumar, V.; Kaur, K.; Gupta, G. K.; Sharma, A. K. Eur. J. Med. Chem. 2013, 69, 735.
(a) Fache, F.; Schulz, E.; Tommasino, M. L.; Lemaire, M. Chem. Rev. 2000, 100, 2159.
(b) Rosiak, A.; Hoenke, C.; Christoffers, J. Eur. J. Org. Chem. 2007, 26, 4376.
(c) Fustero, S.; Sanchez-Rosello, M.; Barrio, P.; Simon-Fuentes, A. Chem. Rev. 2011, 111, 6984.
(d) Hu, J.; Cheng, Y.; Yang, Y.; Rao, Y. Chem. Commun. 2011, 47, 10133.
(e) Lominac, W. J.; D'Angelo, M. L.; Smith, M. D.; Ollison, D. A.; Hanna, J. J. M. Tetrahedron Lett. 2012, 53, 906.
(f) Peng, J.; Xie, Z.; Chen, M.; Wang, J.; Zhu. Q. Org Lett. 2014, 16, 4702.
(g) Tang, M.; Kong, Y.; Chu, B.; Feng, D. Adv. Synth. Catal. 2016, 358, 926.
耿瑞, 赵宇, 李益豪, 刘鑫磊, 王明安, 有机化学, 2019, 39, 3574. doi: 10.6023/cjoc201906017Geng, R.; Zhao, Y.; Li, Y.; Liu, X.; Wang, M. Chin. J. Org. Chem. 2019, 39, 3574(in Chinese). doi: 10.6023/cjoc201906017
钟良坤, 江涛, 张帆, 付庆, 刘幸海, 许天明, 丁成荣, 陈杰, 袁静, 谭成侠, 有机化学, 2019, 39, 2655. doi: 10.6023/cjoc201903056Zhong, L.; Jiang, T.; Zhang, F.; Fu, Q.; Liu, X.; Xu, T.; Ding, C.; Chen, J.; Yuan, J.; Tan, C. Chin. J. Org. Chem. 2019, 39, 2655(in Chinese). doi: 10.6023/cjoc201903056
龚超超, 谈寒一, 张倩, 有机化学, 2018, 38, 3086. doi: 10.6023/cjoc201805020Gong, C. C.; Tan, H. Y.; Zhang, Q. Chin. J. Org. Chem. 2018, 38, 3086(in Chinese). doi: 10.6023/cjoc201805020
何波, 王大伟, 杨文超, 陈琼, 杨光富, 有机化学, 2017, 37, 2895. doi: 10.6023/cjoc201705031He, B.; Wang, D. W.; Yang, W. C.; Chen, Q.; Yang, G. F. Chin. J. Org. Chem. 2017, 37, 2895(in Chinese). doi: 10.6023/cjoc201705031
Zhong, Y. Y.; Yu, L. J.; He, Q. Y.; Zhu, Q. Y.; Zhang, C. G.; Cui, X. P.; Zheng, J. X.; Zhao, S. Q. ACS Appl. Mater. Inter. 2019, 11, 32769. doi: 10.1021/acsami.9b11754
孙娜波, 沈钟华, 翟志文, 韩亮, 翁建全, 谭成侠, 刘幸海, 有机化学, 2017, 37, 2705. doi: 10.6023/cjoc201704032Sun, N. B.; Shen, Z. H.; Zhai, Z. W.; Han, L.; Weng, J. Q.; Tan, C. X.; Liu, X. H. Chin. J. Org. Chem. 2017, 37, 2705(in Chinese). doi: 10.6023/cjoc201704032
石玉军, 周钱, 王杨, 钱宏炜, 叶林玉, 冯霞, 陈辉, 李雅婷, 戴红, 魏中昊, 吴锦明, 有机化学, 2018, 38, 2450. doi: 10.6023/cjoc201806030Shi, Y. J.; Zhou, Q.; Wang, Y.; Qian, H. W.; Ye, L. Y.; Feng, X.; Chen, H.; Li, Y. T.; Dai, H.; Wei, Z. H.; Wu, J. M. Chin. J. Org. Chem. 2018, 38, 2450(in Chinese). doi: 10.6023/cjoc201806030
Zhu, Q.; Yang, Y.; Lao, Z.; Zhong, Y.; Zhang, B.; Cui, X.; O'Neill, P.; Hong, D.; Zhang, K.; Zhao, S. Pest. Manage. Sci. 2019, DOI: 10.1002/ps.5559.
Abd El Razik, H. A.; Badr, M. H.; Atta, A. H.; Mouneir, S. M.; Abu-Serie, M. M. Arch. Pharm. 2017, 350, e1700026. doi: 10.1002/ardp.201700026
Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 1423. doi: 10.2174/138527211795378263
(a) Bekhit, A. A.; Abdel-Aziem, T. Bioorg. Med. Chem. 2004, 12, 1935.
(b) Selvam, C.; Jachak, S. M.; Thilagavathi, R.; Chakraborti, A. K. Bioorg. Med. Chem. Lett. 2005, 15, 1793.
(c) El-Sayed, M. A. A.; Abdel-Aziz, N. I.; Abdel-Aziz, A. A. M.; El-Azab, A. S.; ElTahir, K. E. H. Bioorg. Med. Chem. 2012, 20, 3306.
Tanitame, A.; Oyamada, Y.; Ofuji, K.; Fujimoto, M.; Iwai, N.; Hiyama, Y.; Suzuki, K.; Ito, H.; Terauchi, H.; Kawasaki, M.; Nagai, K.; Wachi, M.; Yamagishi, J. J. Med. Chem. 2004, 47, 3693. doi: 10.1021/jm030394f
Bhosle, M. R.; Mali, J. R.; Pal, S.; Srivastava, A. K.; Mane, R. A. Bioorg. Med. Chem. Lett. 2014, 24, 2651. doi: 10.1016/j.bmcl.2014.04.064
Penning, T. D.; Talley, J. J.; Bertenshaw, S. R.; Carter, J. S.; Collins, P. W.; Docter, S.; Graneto, M. J.; Lee, L. F.; Malecha, J. W.; Miyashiro, J. M.; Rogers, R. S.; Rogier, D. J.; Yu, S. S.; Anderson, G. D.; Burton, E. G.; Cogburn, J. N.; Gregory, S. A.; Koboldt, C. M.; Perkins, W. E.; Seibert, K.; Veenhuizen, A. W.; Zhang, Y. Y.; Isakson, P. C. J. Med. Chem. 1997, 40, 1347. doi: 10.1021/jm960803q
Cox, S. R.; Lesman, S. P.; Boucher, J. F.; Krautmann, M. J.; Hummel, B. D.; Savides, M.; Marsh, S.; Fielde, A.; Stegemann, M. R. J. Vet. Pharmacol. Ther. 2010, 33, 461. doi: 10.1111/j.1365-2885.2010.01165.x
Zhang, D.; Raghavan, N.; Chen, S. Y.; Zhang, H.; Quan, M.; Lecureux, L.; Patrone, L. M.; Lam, P. Y. S.; Bonacorsi, S. J.; Knabb, R. M.; Skiles, G. S.; He, K. Drug Metab. Dispos. 2008, 36, 303. doi: 10.1124/dmd.107.018416
Lange, J. H. M.; van Stuivenberg, H. H.; Coolen, H. K. A. C.; Adolfs, T. J. P.; McCreary, A. C.; Keizer, H. G.; Wals, H. C.; Veerman, W.; Borst, A. J. M.; de Looff, W.; Verveer, P. C.; Kruse, C. G. J. Med. Chem. 2005, 48, 1823. doi: 10.1021/jm040843r
Graneto, M. J.; Kurumbail, R. G.; Vazquez, M. L.; Shieh, H. S.; Pawlitz, J. L.; Williams, J. M.; Stallings, W. C.; Geng, L.; Naraian, A. S.; Koszyk, F. J.; Stealey, M. A.; Xu, X. D.; Weier, R. M.; Hanson, G. J.; Mourey, R. J.; Compton, R. P.; Mnich, S. J.; Anderson, G. D.; Monahan, J. B.; Devraj, R. J. Med. Chem. 2007, 50, 5712. doi: 10.1021/jm0611915
Barth, F.; Rinaldi-Carmona, M. Curr. Med. Chem. 1999, 6, 745.
Cox, B. D.; Prosser, A. R.; Sun, Y.; Li, Z.; Lee, S.; Huang, M. B.; Bond, V. C.; Snyder, J. P.; Krystal, M.; Wilson, L. J.; Liotta, D. C. ACS Med. Chem. Lett. 2015, 6, 753. doi: 10.1021/acsmedchemlett.5b00036
Labroli, M. A.; Dwyer, M. P.; Poker, C.; Keertikar, K. M.; Rossman, R.; Guzi, T. J. Tetrahedron Lett. 2016, 57, 2601. doi: 10.1016/j.tetlet.2016.04.102
Knorr, L. Ber. Dtsch. Chem. Ges. 1883, 16, 2593. doi: 10.1002/cber.188301602193
Singh, S. P.; Kumar, D.; Batra, H.; Naithani, R.; Rozas, I.; Elguero, J. Can. J. Chem. 2000, 78, 1109. doi: 10.1139/v00-104
Oliveira, D. H.; Aquino, T. B.; Nascimento, J. E. R.; Perin, G.; Jacob, R. G.; Alves, D. Adv. Synth. Catal. 2015, 357, 4041. doi: 10.1002/adsc.201500625
Zora, M.; Demirci, D.; Kivrak, A.; Kelgokmen, Y. Tetrahedron Lett. 2016, 57, 993. doi: 10.1016/j.tetlet.2016.01.071
Sun, P.; Yang, D.; Wei, W.; Sun, X.; Zhang, W.; Zhang, H.; Wang, Y.; Wang, H. Tetrahedron 2017, 73, 2022. doi: 10.1016/j.tet.2017.02.046
Kalita, S. J.; Bayan, R.; Devi, J.; Brahma, S.; Mecadon, H.; Deka, D. C. Tetrahedron Lett. 2017, 58, 566. doi: 10.1016/j.tetlet.2016.12.084
Zhang, H.; Wei, Q.; Zhu, G.; Qu, J.; Wang, B. Tetrahedron Lett. 2016, 57, 2633. doi: 10.1016/j.tetlet.2016.05.020
Markovic, V.; Joksovic, M. D. Green Chem. 2015, 17, 842. doi: 10.1039/C4GC02028F
Vekariya, R. H.; Patel, K. D.; Patel, H. D. Res. Chem. Intermed. 2016, 42, 4683. doi: 10.1007/s11164-015-2308-7
Saha, A.; Payra, S.; Banerjee, S. Green Chem. 2015, 17, 2859. doi: 10.1039/C4GC02420F
Sun, A.; Ye, J. H.; Yu, H.; Zhang, W.; Wang, X. Tetrahedron Lett. 2014, 55, 889. doi: 10.1016/j.tetlet.2013.12.045
Kong, Y.; Tang, M.; Wang, Y. Org. Lett. 2014, 16, 576. doi: 10.1021/ol403447g
Tang, M.; Wang, Y.; Wang, H.; Kong, Y. Synthesis 2016, 48, 3065. doi: 10.1055/s-0035-1561646
Senadi, G. C.; Hu, W. P.; Lu, T. Y.; Garkhedkar, A. M.; Vandavasi, J. K.; Wang, J. J. Org Lett. 2015, 17, 1521. doi: 10.1021/acs.orglett.5b00398
Alizadeh, A.; Moafi, L.; Zhu, L. G. Synlett 2016, 27, 595.
Shi, C.; Ma, C.; Ma, H.; Zhou, X.; Cao, J.; Fan, Y.; Huang, G. Tetrahedron 2016, 72, 4055. doi: 10.1016/j.tet.2016.05.034
Yu, Y.; Huang, W.; Chen, Y.; Gao, B.; Wu, W.; Jiang, H. Green Chem. 2016, 18, 6445. doi: 10.1039/C6GC02776H
Mukherjee, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2016, 57, 2103. doi: 10.1016/j.tetlet.2016.04.002
Zhang, G.; Ni, H.; Chen, W.; Shao, J.; Liu, H.; Chen, B.; Yu, Y. Org. Lett. 2013, 15, 5967. doi: 10.1021/ol402810f
Zhang, J.; Chen, W.; Huang, D.; Zeng, X.; Wang, X.; Hu, Y. Tetrahedron Lett. 2017, 58, 4133. doi: 10.1016/j.tetlet.2017.09.050
Kumar, R.; Nair, D.; Namboothiri, I. N. N. Tetrahedron 2014, 70, 179.
Nair, D.; Pavashe, P.; Katiyar, S.; Namboothiri, I. N. N. Tetrahedron Lett. 2016, 57, 3146. doi: 10.1016/j.tetlet.2016.06.020
Ahamad, S.; Gupta, A. K.; Kant, R.; Mohanan, K. Org. Biomol. Chem. 2015, 13, 1492. doi: 10.1039/C4OB02365J
Shao, Y.; Tong, J.; Zhao, Y.; Zheng, H.; Ma, L.; Ma, M.; Wan, X. Org. Biomol. Chem. 2016, 14, 8486. doi: 10.1039/C6OB01522K
(a) Yuan, B.; Zhang, F.; Li, Z.; Yang, S.; Yan, R. Org. Lett. 2016, 18, 5928.
(b) Unnava, R.; Saikia A. K. ChemistrySlect 2016, 1, 1816.
Yadav, S.; Rai, P.; Srivastava, M.; Singh, J.; Tiwari, K. P.; Singh, J. Tetrahedron Lett. 2015, 56, 5831. doi: 10.1016/j.tetlet.2015.07.039
Cheng, J.; Li, W.; Duan, Y.; Cheng, Y.; Yu, S.; Zhu, C. Org. Lett. 2017, 19, 214. doi: 10.1021/acs.orglett.6b03497
Dhage, Y. D.; Daimon, H.; Peng, C.; Kusakabe, T.; Takahashi, K.; Kanno, Y.; Inouye, Y.; Kato, K. Org. Biomol. Chem. 2014, 12, 8619. doi: 10.1039/C4OB01576B
Li, D. Y.; Mao, X. F.; Chen, H. J.; Chen, G. R.; Liu, P. N. Org. Lett. 2014, 16, 3476. doi: 10.1021/ol501402p
Tu, Y.; Zhang, Z.; Wang, T.; Ke, J.; Zhao, J. Org. Lett. 2017, 19, 3466. doi: 10.1021/acs.orglett.7b01447
Zhu, Y.; Lu, W. T.; Sun, H. C.; Zhan, Z. P. Org. Lett. 2013, 15, 4146. doi: 10.1021/ol401818m
Yang, Y.; Hu, Z. L.; Li, R. H.; Chen, Y. H.; Zhan, Z. P. Org. Biomol. Chem. 2018, 16, 197. doi: 10.1039/C7OB02576A
Schmitt, D. C.; Taylor, A. P.; Flick, A. C.; Kyne, R. E. Org. Lett. 2015, 17, 1405. doi: 10.1021/acs.orglett.5b00266
Chen, B.; Zhu, C.; Tang, Y.; Ma, S. Chem. Commun. 2014, 50, 7677. doi: 10.1039/c4cc02856b
Tang, X.; Huang, L.; Yang, J.; Xu, Y.; Wu, W.; Jiang, H. Chem. Commun. 2014, 50, 14793. doi: 10.1039/C4CC06747A
Mamaghani, M.; Hossein, R.; Shirini, F.; Tabatabaeian, K.; Rassa, M. Med. Chem. Res. 2015, 24, 1916. doi: 10.1007/s00044-014-1271-y
Karnakar, K.; Ramesh, K.; Reddy, K. H. V.; Anil Kumar, B. S. P.; Nanubonula, J. B.; Nageswar, Y. V. D. New J. Chem. 2015, 39, 8978. doi: 10.1039/C5NJ01448D

图式 1 1, 3-二羰基化合物与肼合成吡唑的机理
Scheme 1 Plausible mechanism for synthesis of pyrazole with 1.3-diketone and hydrazine

图式 3 α, β-不饱和酮和对甲苯磺酰肼合成吡唑的机理
Scheme 3 Plausible mechanism for the synthesis of pyrazole with unsaturated ketones and TsNHNH2

图式 4 氨基脲盐酸盐合成3, 5-二取代吡唑的机理
Scheme 4 Plausible mechanism for synthesis of 3, 5-disubstituted pyrazoles with semicarbazide hydrochloride

图式 5 乙酰乙酸乙酯、水合肼、苯甲醛和丙二腈多组分反应合成吡唑
Scheme 5 Synthesis of pyrazoles from ethyl acetoacetate, hydrazine hydrate, benzaldehyde and malononitrile

图式 6 N-烷基对甲苯磺酰腙和末端炔烃合成吡唑
Scheme 6 Synthesis of pyrazoles from N-alkyl-p-toluene-sulfonylhydrazone and terminal alkyne

图式 7 N-烷基对甲苯磺酰腙和末端炔烃合成1, 3, 5-三取代吡唑的机理
Scheme 7 Plausible mechanism for the synthesis of 1, 3, 5-trisubstituted pyrazoles with N-alkylated tosylhyrazones and terminal alkynes

图式 8 N-磺酰腙和取代异氰合成5-氨基吡唑的机理
Scheme 8 Plausible mechanism for the synthesis of 5-aminopyrazoles with N-sulfonyl hydrazones and isocyanides

图式 9 腙和硝基烯合成1, 3, 4-三取代和1, 3, 4, 5-四取代吡唑的机理
Scheme 9 Plausible mechanism for the synthesis of 1, 3, 4-trisubstituted and 1, 3, 4, 5-tetrasubstituted pyrazoles with hydrazones and nitroolefins

图式 10 对甲苯磺酰腙和碳化钙合成3, 5-二取代吡唑的机理
Scheme 10 Plausible mechanism for synthesis of 3, 5-disubstituted pyrazoles with tosylhyrazones and calcium carbide

图式 11 乙烯基叠氮、醛和对甲苯磺酰肼合成3, 4, 5-三取代吡唑的机理
Scheme 11 Plausible mechanism of 3, 4, 5-trisubstituted 1H-pyrazoles with vinyl azide, aldehyde, and tosylhydrazine

图式 12 串联合成3, 5-二芳酰基-4-芳基吡唑的反应途径
Scheme 12 Plausible pathway for the tandem synthesis of 3, 5-diaroyl-4-arylpyrazoles

图式 13 Bestmann-Ohira试剂和α, β-不饱和醛合成乙烯基吡唑的反应机理
Scheme 13 Plausible mechanism for the synthesis of vinylpyrazoles with BOR and α, β-unsaturated aldeydes

图式 14 N-(3-苯基丙-2-炔-1-基)苯胺合成吡唑N-氧化物的反应机理
Scheme 14 Plausible mechanism for the synthesis of substituted pyrazole N-oxides from N-propargylamines

图式 15 2-溴-1, 3-二羰基化合物合成吡唑的机理
Scheme 15 Plausible mechanism for the synthesis of pyrazole by 2-bromo-1, 3-dicarbonyl compound

图式 16 肼和炔烃合成四取代吡唑
Scheme 16 Cascade process for synthesis of tetra-substituted pyrazole from hydrazine and alkyne

图式 17 串联反应合成三取代吡唑
Scheme 17 Plausible cascade process for synthesis of trisubstituted pyrazole

图式 19 N-炔丙基磺酰腙合成吡唑反应机理
Scheme 19 Plausible mechanism for the synthesis of pyrazole with N-propargyl sulfonylhydrazones

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
































 下载:
下载:



 下载:
下载: