C—H广泛存在于各种有机化合物中, 通过直接活化分子中的C—H来构建新的有机分子是极具优势的合成策略.近20年来, 过渡金属参与的C—H直接插入反应成为活化分子中C—H的重要手段, 长期研究表明如何高效打开惰性的C—H键和有效控制反应的区域选择性成为该领域重点关注的两大难题, 而导向基团辅助的特定位置C—H活化成为解决这些问题的重要策略[1 . 20年来, 大量的优秀导向基团被成功引入到有机体系中, 并在辅助活化特定位置C—H键和构建新的有机分子中发挥着重要的作用[2 .
在众多辅助导向基团之中, 羧酸作为一种高效导向基团被广泛应用于各类C—H活化反应[3 .与其它导向基团相比, 羧基具有以下优势: (1)羧酸广泛存在于自然界, 具有来源广泛、价廉和低毒的特点; (2)羧酸作为一种活性的“合成子”容易制备, 同时催化反应后也容易进一步官能团化; (3)羧基作为一种“无痕”导向基团, 容易以CO2 的形式脱去.按照羧基导向C—C偶联、C—N偶联、C—O与C—S偶联、其它类型偶联及无痕导向C—H活化几个方面对近年来以羧基为导向的C—H活化官能团化反应研究进展进行介绍.
1.
羧基导向的C—H偶联
1.1
C—C偶联
1.1.1
烯基化偶联
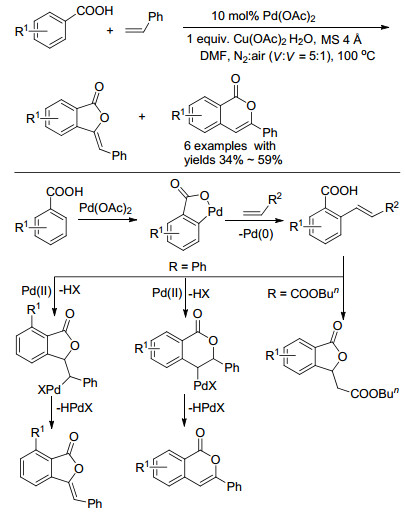
早在1998年, Miura小组[4 就首次报道了Pd(OAc)2 催化苯甲酸邻位导向与烯烃的烯基化反应(Scheme 1 2 为催化剂, Cu(OAc)2 ·H2 O为氧化剂.研究发现, 当苯甲酸邻位有取代基时, 由于位阻的影响烯烃迁移插入易形成五元杂环, 得到3-苯乙烯基苯并呋喃酮.反之, 则容易形成六元杂环, 得到异香豆素.值得注意的是, 作者报道了一例以丙烯酸正丁酯为反应物的例子, 反应通过羧基对烯烃的迈克尔加成得到烷基化产物3-乙酸正丁酯基苯并呋喃酮.
图式 1
2012年, Swamy小组[5 报道了Pd(OAc)2 催化2-吲哚甲酸导向与苯基丙二烯偶联关环生成吲哚并[2, 3-c ]吡喃酮类衍生物的反应(Scheme 2
图式 2
2013年, Lee小组[6 也报道了Pd(OAc)2 催化苯甲酸邻位C—H活化与苯乙烯的C—C和C—O偶联生成杂环的反应(Scheme 3
图式 3
Ackermann小组[7 在2012年报道了Ru(Ⅲ)催化苯甲酸与二苯乙炔关环偶联生成苯并吡喃酮的反应(Eq. 1).反应在以[RuCl2 (p- cymene)]2 与KPF6 为催化剂, 以Cu(OAc)2 ·H2 O为氧化剂的条件下, 实现了苯甲酸与二苯乙炔的C—C与C—O偶联.体系对芳基炔烃与烷基炔烃均有较好兼容性, 同时作者报道了三例以杂环羧酸为底物的反应.不足的是反应仅对富电子的甲酸与对称性炔烃效果较好.
几乎同时, Jeganmohan小组[8 也对该反应进行了研究(Eq. 2).与前面报道不同的是, 反应在[RuCl2 (p -cymene)]2 与AgSbF6 为催化剂的条件下, 仅需催化量的Cu(OAc)2 ·H2 O作为氧化剂.反应对芳基炔烃、烷基炔烃和不对称炔烃均有较好兼容性.当底物为不对称炔烃时, 炔烃在迁移插入到C—Rh(Ⅱ)键的过程中, Rh能选择性地插入到位阻较大的一端, 而苯环则插入位阻较小的一端.同时, 还报道了两例丙烯酸为反应底物的反应.他们认为, 催化量的Cu(OAc)2 ·H2 O能作氧化剂可能是反应生成的Cu(Ⅰ)被空气中的氧气氧化得到了Cu(Ⅱ), 使得催化循环得以继续.
2015年, Ackermann小组[9 在O2 作为氧化剂的温和条件下再次对该体系进行了研究(Eq. 3).反应以101 kPa O2 为氧化剂, NaOAc为碱的温和条件下即可高效进行.体系使用O2 作为唯一的氧化剂, 副产物为H2 O, 因而具有较好的原子经济性.
2016年, Gooßen小组[10 也报道了Ru(Ⅱ)催化苯甲酸与炔烃的C—C偶联烯基化反应(Eq. 4).反应以[Ru(p -cymene)I2 ]2 为催化剂, 在碳酸胍为碱的条件下, 对非对称的炔烃具有较好的选择性, Ru(Ⅱ)倾向于插入位阻较大的一端, 而苯环倾向插入位阻较小的一端.
同年, 游劲松小组[11 报道了Rh(Ⅲ)催化苯甲酸与端炔烃C—C与C—O关环偶联生成3-苯乙烯基苯并呋喃酮的反应(Eq. 5).反应在Rh(Ⅲ)与AgSbF6 为催化剂, Ag2 O为氧化剂, 特戊酸为添加剂的条件下, 对各种端炔均具有较好的普适性.机理研究表明, 端炔在Ag(Ⅰ)的作用下产生炔基自由基, 对C—Rh(Ⅲ)中间体进行氧化加成, 然后还原消除实现C—C的偶联, 因此该反应是Rh(Ⅲ)-Rh(Ⅳ)-Rh(Ⅱ)的催化循环.
2017年, 钟国富小组[12 报道了Rh(Ⅲ)催化的α , β 不饱和羧酸与丙烯酸丁酯的C—C及C—O偶联关环反应(Eq. 6).反应以Cu(OAc)2 ·H2 O为氧化剂, 无需其它任何添加剂的介入, 产物Z 式与E 式构型的比例为3:1.
同年, 石先莹小组[13 也报道了Rh(Ⅲ)催化的苯甲酸与丙烯酸酯C—C与C—O偶联关环的反应(Eq. 7).体系中添加了Cu2 O为助催化剂, Ag2 CO3 与空气为氧化剂, 反应高选择性地得到E 式关环的产物, 有较好的底物普适性, 且能有效抑制迈克尔加成副产物的产生.
2018年, Gooßen小组[14 报道了Ru(Ⅱ)催化的苯甲酸邻位C—H活化与乙酸丙烯醇酯C—C偶联烯基化的反应(Scheme 4 β -O的消除, 脱去乙酸基进而得到丙烯基化的目标产物.
图式 4
几乎同时, Jeganmohan小组[15 报道了Ru(Ⅱ)催化的苯甲酸与乙酸丙烯醇酯C—C和C—O偶联关环为内酯的环烯基化反应(Eq. 8).与Gooßen小组报道不同的是, 体系使用K2 CO3 为碱和N , N -二甲基酰胺为溶剂, 得到苯甲酸邻位丙烯基化产物后, 在Ru(Ⅱ)的作用下羧基会进一步与烯基发生分子内关环得到苯并环内酯.值得注意的是, 当苯甲酸邻位没有取代基时, 关环产物会进一步C—H活化得到苯并环内酯邻位丙烯基化的产物.
同样在2018年, Gooßen小组[16 又分别将烯基源扩展到了丙烯胺和丙烯醇类化合物(Eqs. 9, 10).以丙烯胺为烯基源时, 添加剂为三氟乙酸; 以丙烯醇为烯基源时, 添加剂为磷酸三钾.两反应具有类似的反应机理, 丙烯胺(丙烯醇)迁移插入Ru—C键后, 经β 胺(β 氧)的消除后得到目标产物.
Baidya小组[17 在2018年直接采用苯乙烯为烯基来源, 实现了Ru(Ⅱ)催化的苯甲酸邻位烯基化反应(Eq. 11).反应体系中加入CuO为氧化剂, K2 HPO4 为碱, 反应结束后直接在体系中添加K2 CO3 和MeI可“一锅法”得到羧酸的甲酯化产物.
1.1.2
芳基化偶联
羧基导向C—H直接芳基化是羧基作为导向基团研究得最多的一类工作.早在2007年, 余金权课题组[18 报道了以芳基硼酸酯和芳基碘为芳基源对苯甲酸和脂肪羧酸的芳基化反应(Eqs. 12~14).以芳基硼酸酯为芳基源时, 反应在以苯醌和Ag2 CO3 为氧化剂, K2 HPO4 为碱的条件下对苯甲酸邻位C—H和脂肪酸的β 位C—H均能有效实现芳基化(Eqs. 12, 13).值得注意的是, 当脂肪酸的β 位有多个甲基时, 体系能高选择性地得到单芳基化产物, 而双芳基化产物均少于2% (Eq. 13).以芳基碘为芳基来源时, 在类似的条件下反应得到的是脂肪酸的β 位C—H单芳基化和双芳基化的混合产物(Eq. 14).作者发现K2 HPO4 或NaOAc为碱对促进羧酸导向的C—H活化具有关键性的作用.
与此同时, Daugulis小组[19 也报道了Pd(OAc)2 催化苯甲酸邻位导向芳基化反应(Scheme 5 2 CO3 , 则得到单芳基化和双芳基化的产物, 反应在催化量的BuAd2 P作用下经历的是Pd(0)和Pd(Ⅱ)的催化循环, 反应对富电子和缺电子的苯甲酸均有较好的效果.
图式 5
2013年, 周向葛小组[20 以二芳基碘盐为芳基的来源, 以Pd(OAc)2 为催化剂, 在水相体系中实现了该类反应(Eq. 15).反应无需其它添加剂的介入即可在水溶液中高效进行, 但体系仅对富电子的底物有较好的效果.
2015年, 苏伟平小组[21 使用氨基酸为配体和六氟异丙醇(HFIP)为溶剂, 在室温条件下实现了苯甲酸邻位的芳基化反应(Eq. 16).反应体系对富电子和缺电子的底物均有较好的催化效果, 底物适应性较广.同时作者证实反应体系中催化量的N -乙酰-L -异亮氨酸配体和六氟异丙醇溶剂能有效地促进苯甲酸邻位C—H活化的速率, 对实现温和条件下的苯甲酸邻位C—H芳基化具有决定性的作用.
同年, Gooßen小组[22 以[{IrCp*Cl2 }2 ]为催化剂, 以芳基重氮盐为芳基来源, 在温和条件下实现了苯甲酸邻位C—H直接芳基化反应(Scheme 6 2 CO3 和Li2 CO3 为碱, 在丙酮溶剂中60 ℃的条件下即可高效进行苯甲酸邻位芳基化反应, 对富电子和缺电子的苯甲酸均有中等以上的收率.与之前报道不同的是, 他们认为反应中Ir(Ⅲ)的金属有机配合物依次经过芳基自由基和Ag(Ⅱ)的氧化, 分别形成Ir(Ⅳ)和Ir(Ⅴ)的配合物中间体, 最后经过还原消除得到目标产物和Ir(Ⅲ)的配合物, 并完成整个催化循环.故体系是Ir(Ⅲ)-Ir(Ⅳ)-Ir(Ⅴ)的催化循环.
图式 6
2017年, Larrosa小组[23 采用Ru催化体系对该类反应进行了研究(Eq. 17).反应在5 mol%的[Ru(t- BuCN)6 ]-(BF4 )2 为催化剂, K2 CO3 和KOC(CF)3 为碱的条件下即可高效进行, 避免了AgOAc的使用.同时反应的普适性较广, 对邻位有取代基的苯甲酸和碘苯以及杂环底物均有较好的效果, 这进一步推动了该反应的实用性.
2015年, 李朝军小组[24 创造性地提出了Rh催化的苯甲酸邻位导向C—H/C—H自偶联反应(Scheme 7 2 ]为催化剂, 在MnO2 和空气为共同氧化剂的条件下, 可高效进行羧基导向的自偶联反应.他们认为Rh(Ⅲ)促使苯甲酸邻位双重C—H活化形成双五元环的C-Rh(Ⅲ)配合物, 然后经历还原消除得到偶联的产物并释放出Rh(Ⅰ), 最后MnO2 将Rh(Ⅰ)氧化为Rh(Ⅲ)使催化循环得以进行.值得注意的是, 作者还报道了一例不同苯甲酸交叉偶联的反应, 这将为合成非对称联苯甲酸类化合物提供重要的参考.
图式 7
同年, 游劲松小组[25 报道了苯甲酸邻位导向与芳香杂环的C—H/C—H芳基化反应(Eq. 18).反应在Rh(Ⅲ)催化体系作用下, 以Ag2 O为氧化剂, K2 HPO4 为碱, 实现了苯甲酸邻位C—H与噻吩杂环或呋喃杂环的2号位C—H的交叉偶联.同位素动力学研究表明, 苯甲酸邻位C—H活化为反应的决速步, 反应经历了Rh(Ⅲ)-Rh(Ⅰ)的催化循环.
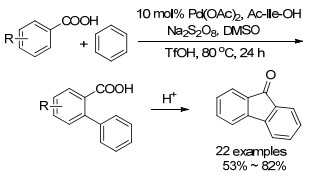
2016年, 该小组[26 又报道了以苯为芳基源的苯甲酸邻位芳基化反应(Scheme 8 2 和N -乙酰-L -异亮氨酸组合的催化体系, 在Na2 S2 O8 和二甲基亚砜(DMSO)作为氧化剂的条件下高效地实现了苯甲酸邻位C—H/C—H的直接芳基化.值得注意的是, 作者通过同位素动力学实验证明了整个反应的决速步为苯环上的C—H活化, 而不是苯甲酸邻位的C—H活化, 说明了N -乙酰-L -异亮氨酸作为配体能有效促进苯甲酸邻位C—H活化过程.
图式 8
2018年, 游劲松小组[27 又以Ir(Ⅲ)为催化剂研究了苯甲酸与噻吩杂环的芳基化反应(Scheme 9 2 ]2 }为催化剂, 以Ag2 O为氧化剂, 无需任何酸与碱的作用即可高效的实现苯甲酸与噻吩杂环的C—H/C—H芳基化以及羧基与噻吩的分子内关环.值得注意的是, 当底物为α , β 不饱和羧酸与2取代噻吩时, 在该条件下能直接得到含噻吩环的螺环化合物.同时证实了噻吩环上C—H活化和关环过程都经历了类似heck偶联的过程, 而并非经典的亲电取代(SE
图式 9
1.1.3
炔基化偶联
2017年, Ackermann小组[28 报道了Ru(Ⅱ)催化的苯甲酸与溴代三异丙基硅基炔的炔基化反应(Eq. 19).体系条件简单且具有良好底物适应性, 反应以5 mol%的[Ru-(p -cymene)Cl2 ]2 为催化剂, 仅需要加入2 equiv.的K2 CO3 作碱.当苯甲酸邻位没有取代基时可以得到双炔基化的产物.
1.1.4
烷基化偶联
前面已经介绍, 早在1998年, Miura小组[4 在苯甲酸邻位烯基化偶联反应的研究中就报道了一例Pd(OAc)2 催化苯甲酸与丙烯酸正丁酯关环偶联生成3-乙酸正丁酯基苯并呋喃酮的反应.虽然只有一例, 却是苯甲酸邻位导向烷基化偶联的最早报道.
2007年, 余金权小组[18 在Pd(OAc)2 催化苯甲酸邻位C—H芳基化偶联反应的研究中也报道了两例苯甲酸邻位烷基化的反应(Eq. 20).反应以甲基硼酸为甲基源, 分别以邻甲基苯甲酸和间甲氧基苯甲酸为底物, 对应的甲基化产物产率分别为75%与71%.
2009年, 余金权小组[29 又报道了Pd(OAc)2 催化苯甲酸邻位导向与卤代烷烃的烷基化反应(Eqs. 21, 22).他们发展了二氯乙烷和二溴甲烷两种烷基源, 分别与苯甲酸C—C和C—O偶联关环生成对应的六元环内酯和五元环内酯.研究表明, 加入的无机碱对反应有关键性的促进作用, 能高效地促进反应进行的碱分别为K2 HPO4 、KHCO3 和Na2 CO3 .反应对含推电子基和吸电子基的底物均有较好的适应性.值得注意的是, 以1-氯正戊烷为烷基源时, 反应也能得到26%的邻正戊烷基苯甲酸正戊酯, 这为合成含有长脂肪链的苯甲酸类物质提供了一种新方法.
2011年, Ackermann小组[30 报道了Ru(Ⅱ)催化苯甲酸与丙烯酸酯的C—C偶联与迈克尔加成的反应(Eq. 23).反应在水相中以Cu(OAc)2 ·H2 O为氧化剂的条件下高选择性地得到了3-乙酸乙酯基苯并呋喃酮类化合物.
2012年, 李朝军小组[31 报道了首例Rh(Ⅲ)催化苯甲酸邻位C—H活化与醛加成生成苯并呋喃酮类化合物的反应(Eq. 24).反应使用价廉易得的醛为反应物, 芳香醛和脂肪醛均能得到对应的苯并呋喃酮类化合物.同时反应使用Ag2 CO3 为添加剂, 仅对富电子的苯甲酸与缺电子的醛类有较好的效果.
2013年, 余金权小组[32 以Pd(OAc)2 为催化剂, 以烷基硼酸盐为烷基来源实现了苯甲酸邻位烷基化反应(Eq. 25).研究表明, N -叔丁氧羰基-O -叔丁基苏氨酸作为配体对反应有较大的促进作用, 反应对苯甲酸和苯乙酸均具有较好的催化效果.同时反应具有较好的官能团容忍性, 但对二级碳和三级碳的硼酸盐底物效果稍差.
2015年, 该小组[33 又将烷基源拓展到了环氧丙烷类化合物(Scheme 10 2 的作用下即可高效进行, 且对富电子和缺电子的苯甲酸以及各种类型的环氧丙烷均具有较好的催化反应效果.其中1 equiv. KOAc为碱对该反应至关重要, 同时乙酰基保护的叔丁基异亮氨酸对反应也有明显的促进作用.机理研究表明, 反应经历了Pd(Ⅱ)配合物促进的环氧丙烷SN 2亲核取代的开环过程.
图式 10
2018年, 石先莹小组[34 采用Ru(Ⅱ)催化体系, 以α , β 不饱和酮为烷基来源实现了苯甲酸邻位烷基化反应(Eq. 26).反应将[RuCl2 (p -cymene)]2 的用量降到了1.5 mol%, 无需其它添加剂介入, 在水溶剂和空气中即可高效进行烷基化反应, 反应有效避免了β 氢消除和迈克尔加成的五元杂环副产物的生成.
几乎同时, Kapur小组[35 报道了以丙烯醇为烷基源的苯甲酸邻位烷基化反应(Scheme 11 p -cymene)Cl2 ]2 的催化作用下发生邻位C—H活化, 生成五元环的C-Ru(Ⅱ)配合物, 进一步与α , β 不饱和酮生成烯基钌配合物, 当体系以AgSbF6 为添加剂, Cu(OAc)2 · H2 O为氧化剂的时候, 烯基迁移插入C-Ru(Ⅱ)后生成酮式七元环C-Ru(Ⅱ)配合物C ; 当体系以Cu(OAc)2 为添加剂, KOAc为碱的时候, 在碱的作用下, 烯基迁移插入C-Ru(Ⅱ)生成醇式七元环C-Ru(Ⅱ)配合物C' .醇式七元环C-Ru(Ⅱ)配合物C' 能有效避免体系发生β 氢的消除, 故醇式七元环C-Ru(Ⅱ)配合物C' 发生Ru(Ⅱ)的解离得到烷基化产物, 而酮式七元环的C-Ru(Ⅱ)配合物C 则发生β 氢的消除后进一步发生迈克尔加成得到苯并呋喃酮的衍生物.
图式 11
1.1.5
羰基化偶联
2008年, 余金权小组[36 报道了首例以CO为羧基源的苯甲酸邻位羧基化的反应(Eq. 27).反应体系中加入Ag2 CO3 为氧化剂, NaOAc为碱, 在1, 4-二氧六环溶剂中高效地得到了邻苯二甲酸产物.体系对苯甲酸与β 位为季碳的苯乙酸均有较好效果.
2013年, 李朝军小组[37 使用异氰酸酯为羰基来源, Rh(Ⅲ)为催化剂, NaOAc为碱, 对苯甲酸进行邻位C—H活化羰基化, 一步得到邻苯二甲酰亚胺类化合物(Eq. 28).反应对富电子的苯甲酸具有较好效果, 但对缺电子的苯甲酸效果稍差.机理研究表明, 反应首先经历Rh(Ⅲ)诱导的C—H活化, 接着C-Rh(Ⅲ)中间体对异氰酸酯迁移插入得到苯甲酸邻位酰胺化产物, 最后分子内关环得到邻苯二甲酰亚胺.
同年, Gooßen小组[38 以羧酸酐为羰基源, 以Cs2 CO3 为碱, 在均三甲苯中实现了苯甲酸邻位的羰基化反应(Scheme 12
图式 12
与此同时, 葛海波小组[39 采用苯乙酮酸为羰基源对苯甲酸进行了邻位C—H羰基化反应(Eq. 29).反应以Pd(TFA)2 为催化剂, Ag2 CO3 同时作为碱和氧化剂, 在乙二醇二甲醚中高选择性地得到了苯甲酸单边苯甲酰基化产物.机理研究表明, 反应经历了Pd(Ⅱ)-Pd(0)的催化循环.
1.2
C—N偶联
与羧基导向的C—C偶联工作相比, C—N偶联的工作报道相对要少得多.在1981年, Rauch等[40 报道了CuO催化苯甲酸邻位氨基化生成苯胺的工作(Scheme 13
图式 13
直到2012年, Yu小组[41 报道了Pd(OAc)2 催化苯甲酸邻位C—H的氨基甲酸酯基化反应(Scheme 14 2 的作用下经过金属协同脱氢(CMD)过程得到Pd(Ⅱ)-C的双核二聚体配合物, 然后甲磺酸氨基甲酸乙酯脱去甲磺酸根后与Pd形成氮烯, 最后氮烯插入Pd-C得到目标产物.
图式 14
2014年, 该小组[42 将氨基甲酸酯的来源扩大到了N -氯氨基甲酸甲酯.反应采用[RhCl2 (Cp*)]2 为催化剂, 以AgOAc为碱, 以叔丁醇为溶剂, 在60 ℃的温和条件下实现了苯甲酸邻位的氨基甲酸酯基化反应(Eq. 30).同时, 作者推测反应过程中可能形成了氮烯中间体, 然后氮烯插入Rh-C进而得到氨基甲酸酯基化产物.
2015年, Chang小组[43 在Ir(Ⅲ)催化体系中以苯磺酰叠氮化物为磺酰胺来源实现了苯甲酸邻位C—H磺酰胺基化反应(Eq. 31).体系以[IrCl2 (Cp*)]2 为催化剂, 以AgNTf2 为催化助剂, 以LiOAc为碱, 在温和条件下即可高效地实现苯甲酸邻位的苯磺酰胺基化.
1.3
C—O和C—S偶联
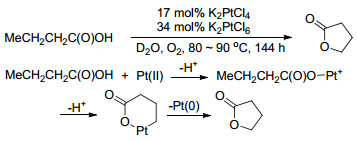
最早在1991年, Sen小组[44 就报道了Pt(Ⅱ)催化链状脂肪羧酸C—H活化分子内关环生成环内酯的反应(Scheme 15 2 PtCl4 为催化剂, 以34 mol%的K2 PtCl6 和O2 为氧化剂, 作者认为K2 PtCl6 的作用是将Pt(0)重新氧化为Pt(Ⅱ).虽然体系需要消耗近50 mol%的铂盐, 且反应时间为144 h, 但却是最早羧基导向C—O偶联的工作.
图式 15
2001年, Sames小组[45 也报道了Pt(Ⅱ)催化氨基酸分子中羧基导向C—H活化分子内关环生成氨基酸内酯的反应(Eq. 32).与Sen的报道不同, 该体系以5 mol%的K2 PtCl4 为催化剂, 以7倍CuCl2 为氧化剂, 减少了贵金属的使用.但体系需要在160 ℃的高温下才能有效反应, 且产物的分离产率均小于30%.
2006年, Chang小组[46 报道了Pt(Ⅱ)催化邻甲基苯甲酸C(sp3 )—H活化分子内关环生成苯甲酸内脂的反应(Scheme 16 2 的用量减小到3倍, 苯甲酸与苯乙酸均能有效反应.不足的是产率均只有中等左右, 且只报道了5例.
图式 16
2011年, Martin小组[47 在Pd(OAc)2 催化体系中对该反应进行了进一步的研究(Eq. 33).与Pt(Ⅱ)催化体系不同, Pd(OAc)2 催化体系以Ag2 CO3 为氧化剂, 同时加入K2 HPO4 为碱, 并且乙酰亮氨酸作为配体对反应的进行也具有重要的促进作用.与之前的工作相比, 该体系具有更高的产率和底物兼容性.
2006年, White小组[48 报道了Pd(Ⅱ)催化长链不饱和羧酸分子内C—O偶联关环生成大环内脂的反应(Scheme 17 2 的作用下得到中间体配合物complex 1, 而中间体配合物complex 1经历还原消除得到大环的内酯化合物, 进一步在苯醌的氧化作用下完成整个催化循环.该反应为合成大环内酯类化合物提供了一种简洁的新方法.
图式 17
2011年, Sasai小组[49 也研究了Pd(Ⅱ)催化含烯丙基羧酸化合物分子内C—O偶联关环生成环内酯的反应(Eq. 34).与White小组的报道不同, 作者使用了手性螺环双异噁唑啉氮为配体.体系在室温下即可高效进行, 产率高达98%, 产物的ee值在15%~82%之间.
2005年, Rybak-Akimova小组[50 采用1 equiv.苯甲酸, 2 equiv.活性的非血红素亚铁配合物以及3 equiv.的H2 O2 , 室温下, 在乙腈中高效地实现了苯甲酸邻位的羟基化反应(Eq. 35).该反应使用了廉价与低毒的H2 O2 和亚铁配合物.
2009年, 余金权小组[51 以氧气为羟基中氧的来源实现了苯甲酸邻位羟基化反应(Eq. 36).反应以Pd(OAc)2 为催化剂, 以KOAc为碱, 以苯醌为氧化剂, 以101 kPa O2 为氧源, 在N , N -二甲基乙酰胺中实现了苯甲酸的邻位羟基化.对于含有吸电子基团的苯甲酸则需要506 kPa O2 才能有效发生反应.同位素原子示踪也证实了羟基中的氧来自于氧气.
2013年, 余金权小组[52 又报道了Pd(OAc)2 催化苄位含取代基的苯乙酸发生分子内C—O偶联生成苯并呋喃酮的反应(Eqs. 37, 38).体系加入PhI(OAc)2 为氧化剂, KOAc为碱.根据底物的不同催化量的氨基酸类配体对反应具有关键性的促进作用.当苯乙酸苄位的两个取代基不同时, 在40 mol%的N -叔丁氧羰基异亮氨酸作配体的条件下, 催化体系能高选择性地得到手性的苯并呋喃酮类化合物, ee 值高达95%.
几乎同时, 施章杰小组[53 也报道了该反应(Eq. 39).体系虽然没有使用氨基酸作为配体, 但加入了AgOAc作为添加剂, CsOAc/NaOAc作为碱, 同时使用了PhCl/t -BuOH混合溶剂, 因此该催化体系条件稍复杂.
2013年, 王细胜小组[54 报道了Pd(OAc)2 催化邻苯基苯甲酸分子内C—O偶联关环生成联芳基内酯的反应(Scheme 18 2 为氧化剂, KOAc为碱.这说明这类条件对Pd(OAc)2 催化芳香羧酸C—H活化和分子内C—O偶联关环反应具有良好的促进作用.同时, 作者用该策略实现了天然产物大麻醇的全合成.
图式 18
同年, Martin小组[55 在Cu(OAc)2 催化体系中对该反应进行了研究(Scheme 19 2 为催化剂, 过氧化苯甲酰为氧化剂, 避免了贵金属催化剂的使用, 且反应条件简单.反应结束后向体系中加入LiOH和MeOH, 能够有效地解离产物中的酯键, 从而间接得到底物分子中邻位苯基羟基化的产物.作者推测了可能的机理, 首先底物分子与Cu(OAc)2 和过氧化苯甲酰作用得到邻苯基苯甲酸Cu(Ⅲ)的环状配合物Ⅰ 和苯甲酰氧自由基Ⅱ 以及苯基自由基Ⅲ , 然后自由基Ⅱ 或Ⅲ 从配合物Ⅰ 上夺取一个氢原子得到邻苯基苯甲酸Cu(Ⅲ)的自由基配合物Ⅳ , 最后通过分子内自由基的C—O偶联得到内酯产物,并释放出Cu(Ⅱ).同时, 作者认为配合物Ⅰ 经历金属协同脱氢(CMD)过程得到七元环的Cu(Ⅲ)-C配合物Ⅴ , 然后经过还原消除得到内酯产物的机理也是不能排除的.
图式 19
2017年, Baidya小组[56 报道了[Ru(p- cymene)Cl2 ]2 催化苯甲酸与二苯基二硫醚生成邻位硫苯基苯甲酸的反应(Eq. 40).体系反应条件简单, 无需惰性气体的保护, 对含吸电子基团的苯甲酸底物同样兼容.而且将二苯基二硫醚换为二苯基二硒醚后, 体系也可高效得到苯甲酸邻位硒苯基化的产物.
1.4
其它类型的偶联
2008年, 余金权小组[57 报道了Pd(OAc)2 催化的苯甲酸邻位碘化和溴化的反应(Scheme 20 N , N -二甲基甲酰胺(DMF)可以夺取羧酸的质子促进羧基与Pd(Ⅱ)的配位, 从而促进C—H活化过程.同时, 当使用IOAc为碘源时, 羧基邻位的两边都会被碘化; 当使用IOAc和Bu4 NI为碘源, 碘化则发生在单边; 使用IOAc与Bu4 NBr为溴源, 溴化也发生在单边.作者认为其中原因可能是季铵盐促使了Pd(Ⅱ)-Ⅰ的形成, 而Pd(Ⅱ)-Ⅰ不利于进一步的C—H活化.
图式 20
该小组在2010年对该体系进行了进一步的研究, 研究发现在避光的条件下体系能对苯乙酸及其衍生物进行高效的邻位碘化[58 .避光能够有效地避免苯乙酸底物的分解, 进而避免其它的副反应.同时作者采用该策略合成了药物分子双氯酚酸和罗美昔布.
2018年, Martin-Matute小组[59 采用Ir(Ⅲ)为催化剂, 以N -碘代琥珀酰亚胺为碘源, 在温和条件下实现了苯甲酸邻位的碘化反应(Eq. 41).体系无需其他任何酸、碱和添加剂的介入, 反应条件温和.
N -碘代琥珀酰亚胺扩大到2~3倍且延长反应时间, 可选择性地得到双边碘化的产物.
2014年, 余金权小组[60 报道了首例苯乙酸导向邻位氘代的反应(Eq. 42).反应以Pd(OAc)2 为催化剂, 以Na2 CO3 为碱, 以全氘代乙酸为氘的来源, 苯乙酸和苯甲酸均能在该反应条件下高效的实现双边的氘代.作者认为反应机理可能是D+ 对苯乙酸邻位C—H活化后形成的Pd(Ⅱ)-C五元环中间体进行的亲电取代.
2.
羧基无痕导向的C—H偶联
2.1
C—C偶联
2.1.1
芳基化偶联
羧基作为无痕导向基团是以羧基为导向的重要优越性表现所在.早在2007年, Daugulis[19 报道了一例苯甲酸邻位芳基化后, 芳基化产物进一步脱羧的反应.脱羧反应以氧化亚铜为催化剂, 邻菲啰啉为配体, N -甲基吡咯烷酮和喹啉为溶剂(Scheme 21
图式 21
Larrosa小组[61 在2011年首次报道了苯甲酸邻位导向芳基化与脱羧一步进行的反应(Eq. 43).体系在Pd(OAc)2 为催化剂, Ag2 CO3 为碱, AcOH同时为添加剂和溶剂的条件下, 反应16 h即可实现苯甲酸邻位芳基化和脱羧的一步进行, 高效得到间位芳基化的产物.
2014年, 该小组[62 又报道了Pd(OAc)2 催化水杨酸邻位芳基化后脱羧一步得到间芳基化苯酚的反应(Scheme 22 2 的作用下得到水杨酸, 然后向反应后的体系中加入碘苯, 在氮杂卡宾钯为催化剂, Ag2 CO3 为碱的条件下实现了水杨酸邻位芳基化和脱羧过程, 直接一步得到了苯酚邻位芳基化的产物.整个过程中羧基的引入、芳基化和脱羧“一锅”进行, 充分体现出了羧基无痕导向的优势.
图式 22
2018年, 他们[63 又用该策略实现了氟苯间位的芳基化反应(Eq. 44).体系巧妙地用“一锅法”实现了氟苯邻位引入羧基、Pd(Ⅱ)催化苯甲酸邻位导向芳基化和芳基化产物脱羧的三步反应, 首次直接实现了氟苯的间位芳基化, 并成功将该方法用于γ 分泌酶抑制剂的全合成.
Larrosa小组[64 在2016年又将体系扩展到了吡啶甲酸类底物(Eq. 45).该体系的难点是克服了吡啶甲酸底物上强配位的氮原子对羧基导向的干扰作用, 在Pd(Ⅱ)作为催化剂, Cs2 CO3 为碱的条件下, 催化体系对氯苯和溴苯均有较好的效果.芳基化反应完毕后, 用三氟甲磺酸使体系酸化, 最后加入Cu2 O和邻菲啰啉在145 ℃条件下实现脱羧, 从而“一锅两步”得到吡啶对位芳基化的产物.
2015年, 苏伟平小组[65 报道了Rh(Ⅲ)催化的苯甲酸邻位导向与噻吩C—H/C—H偶联脱羧的反应(Eq. 46).体系使用K2 HPO4 为碱, Ag2 CO3 为氧化剂, 2, 2, 6, 6-四甲基哌啶-氮-氧化物(TEMPO)为助氧化剂, 反应条件温和且具有较好的官能团兼容性和底物适应性.体系主要应用于富电子的苯甲酸和取代的噻吩环.机理研究表明, 苯甲酸银盐的形成对反应后的脱羧具有重要作用.几乎同时, 游劲松小组[66 也报道了类似的工作, 反应以[(RhCp*Cl2 )2 ]为催化剂, 以AgSbF6 为助催化剂, 以K2 HPO4 为碱, 以Ag2 CO3 为氧化剂, 虽然需要在150 ℃的高温才能完成反应, 但体系将底物的适应范围扩展到了强吸电子基的苯甲酸.
2018年, 石先莹小组[67 报道了Pd(OAc)2 催化的苯甲酸邻位导向自偶联和脱羧为联苯化合物的反应(Scheme 23 2 HPO4 为碱, 以Ag2 CO3 为氧化剂, 在150 ℃的条件下能有效实现苯甲酸邻位导向自偶联和脱羧的多米诺反应过程.值得注意的是, 当不同的苯甲酸以1:1的比例混合时, 体系能选择性地得到交叉偶联和脱羧的产物.机理研究表明, 两个苯甲酸的羧基是分两次脱去的, 先是双五元环的Pd-C配合物中一个羧基脱CO2 后还原消除得到邻苯基苯甲酸, 然后在Pd(Ⅱ)的作用下另一个羧基再脱去CO2 得到联苯类化合物.
图式 23
同年, 杨宇东[68 报道了Ir(Ⅲ)催化的苯乙酮酸邻位导向与噻吩杂环偶联后脱羧关环的反应(Scheme 24 2 ]2 为催化剂, 以AgNTf2 为助催化剂, 以Ag2 O为氧化剂, 以金刚烷甲酸为添加剂.机理研究表明, 反应可能是苯乙酮酸先邻位导向芳基化后再脱CO2 转化为邻芳基苯甲酸; 或者是苯乙酮酸首先脱羧转化为苯甲酸, 然后苯甲酸邻位导向芳基化生成邻芳基苯甲酸; 最后经历还原消除得到含噻吩杂环的苯并吡喃酮类化合物.
图式 24
2.1.2
烯基化偶联
2008年, Miura小组[69 报道了吲哚-2-甲酸类底物与丙烯酸酯邻位导向烯基化后脱羧的反应(Eq. 54).反应以Pd(OAc)2 为催化剂, 以Cu(OAc)2 为氧化剂, 以LiOAc为碱, 一步实现了杂环上羧基邻位导向的烯基化和脱羧的反应, 得到了杂环3号位烯基化的产物.但体系对呋喃和噻吩类底物的选择性稍差. 2011年, 该课题组[70 又用[RhCp*Cl2 ]2 为催化剂, 在类似的条件下研究了该反应.在[RhCp*Cl2 ]2 的催化作用下, 体系克服了呋喃和噻吩类底物选择性差的缺点, 对苯甲酸和杂环羧酸底物均具有较好兼容性, 同时对苯乙烯底物也具有较好效果.
2014年, 谢作伟小组[71 以碳硼烷羧酸为底物, 二苯乙炔为烯基来源, 首次实现了羧基导向的碳硼烷骨架邻位B—H活化烯基化与脱羧的反应(Eq. 48).反应以[IrCp*Cl2 ]2 为催化剂, Cu(OAc)2 与AgOAc为碱, 在甲苯溶剂中130 ℃的条件下一步实现了羧基导向碳硼烷骨架邻位烯基化与脱羧过程.反应对各种对称的二芳基乙炔均具有较好的效果, 但对脂肪族炔烃效果稍差.
2016年, Ackermann小组[72 在Ru(Ⅱ)催化体系中, 分别以丙烯酸酯和二苯乙炔一步实现了苯甲酸导向邻位烯基化和脱羧反应(Eqs. 49, 50).在[Ru(MesCO2 )2 (p -cymene)]的催化作用下, 体系在相对温和的条件下即可实现烯基化和脱羧过程, 且无需铜盐和银盐的介入, 条件简单.当以丙烯酸酯为烯基来源时, 反应经过了烯烃的插入和β 氢的消除, 故需要加入V2 O5 为氧化剂.几乎同时, 在Gooßen小组[10 也报道了10例以二苯基乙炔为烯基来源的苯甲酸邻位导向烯基化反应.反应以[RuCl2 (p -cymene)]2 为催化剂, 以碳酸胍与2-甲基吡啶为碱, 以醋酸为添加剂, 同样在没有铜盐和银盐介入的条件下实现了苯甲酸邻位的烯基化和脱羧.
同年, 赵品晶小组[73 以Ru(OAc)2 (p -cymene)为催化剂, 以1, 4-二氧六环、均三甲苯和庚烷为混合溶剂, 在温和条件下实现了苯甲酸与二芳基炔的烯基化和脱羧反应(Scheme 25
图式 25
2016年, Hong小组[74 报道了Pd(OAc)2 催化的2-苯基苯乙酸与丙烯酸酯的邻位导向烯基化、Heck偶联关环和脱羧的多米诺反应(Scheme 26 N -乙酰-L -异亮氨酸为配体, 以K2 CO3 为碱, 以O2 和苯醌为氧化剂的条件下一步实现了2-苯基苯乙酸邻位导向的烯基化、苯环与烯基的分子内Heck偶联关环以及关环产物的脱羧.体系反应条件温和, 对各种官能团具有良好的兼容性, 同时对稠环芳烃和杂环芳烃均具有较好的效果.
图式 26
2018年, Studer小组[75 报道了环己二烯基甲酸与苯乙烯的邻位导向烯基化和脱羧芳构化反应(Scheme 27 2 的催化作用下一步完成了羧基邻位导向烯基化、脱羧和芳构化的多米诺反应.机理研究表明, TEMPO能夺取β 氢消除后Pd(Ⅱ)—H键中的氢原子, 同时将生成的Pd(Ⅰ)的烯烃配合物氧化成为Pd(Ⅱ)的烯烃配合物, 最后Pd(Ⅱ)的烯烃配合物发生脱羧和芳构化得到目标产物.
图式 27
2.1.3
烷基化偶联
2017年, Ackermann小组[76 报道了Ru(Ⅱ)催化的苯甲酸邻位导向与琥珀酰亚胺Heck偶联脱羧的反应(Eq. 51).体系仅需以[Ru(MesCO2 )2 (p -cymene)]为催化剂, 无需铜盐和银盐的介入, 在温和条件下即实现偶联和脱羧过程.同时作者通过量子化学方法对不同溶剂中反应能量的变化情况分别进行了计算, 为在温和条件下探索羧基导向偶联和脱羧反应提供重要理论参考.
2.1.4
羰基化偶联
2015年, 李朝军小组[77 报道了以异氰酸酯为酰胺来源, Ru(Ⅲ)催化苯甲酸邻位C—H酰胺化后脱羧制备N -苯基苯甲酰胺的反应(Scheme 28 2 O作为助催化剂, 更换了K2 HPO4 为碱, 在150 ℃的高温条件下, 实现了邻位酰胺化后的脱羧反应.机理研究表明, 异氰酸酯的亚胺键迁移插入到五环配合物的C—Rh(Ⅲ)中得到苯甲酸邻位苯甲酰胺化产物, 进一步羧基在碱和Cu2 O的作用下脱去CO2 得到N -苯基苯甲酰胺.
图式 28
2018年, Swamy小组[78 采用异硫氰酸酯为酰胺源, 实现了3-甲酸基吲哚邻位导向酰胺化脱羧的反应(Eq. 52).与上述工作相比, 作者使用了Pd(Ⅱ)催化体系, 实现了吲哚环2号位的酰胺化以及温和条件下的脱羧.值得一提的是, 作者本想实现硫代酰胺化, 但体系中并未分离出硫代酰胺产物, 而是直接得到氧化后的酰胺产物.
2.2
C—O和C—S偶联
Gooßen小组[79 在2013年报道了Cu(OAc)2 催化的苯甲酸钾与三甲氧基硼邻位导向甲氧基化后脱羧基的反应(Scheme 29 2 为催化剂, 碳酸银和O2 为氧化剂, 反应对杂环羧酸底物和手性的烷氧基硼均有较好的兼容性.机理研究表明, Cu(Ⅱ)歧化为Cu(Ⅲ)后, 苯甲酸在Cu(Ⅲ)作用下发生邻位C—H活化形成五元环的C-Cu(Ⅲ)配合物, 由三甲氧基硼与Ag(Ⅰ)生成的甲氧基银经过转金属作用后与五元环的C-Cu(Ⅲ)配合物配位, 然后经历还原消除得到邻位甲氧基化的产物, 最后在Ag(Ⅰ)的作用下羧基脱去CO2 得到甲氧基苯类化合物.
图式 29
2.3
其它类型的偶联
2019年, 蔡琥小组[80 以Pd(OAc)2 为催化剂, 首次采用NaX为卤素来源, 实现了苯甲酸邻位的碘化/溴化与脱羧的多米诺反应(Eq. 53).用低毒与价廉的NaX来替代有机的卤素源极大地提高了反应的经济性和实用性, 但反应仅对邻位含有强吸电子基硝基的苯甲酸有较好效果.机理研究表明, 2 equiv.的Bi(NO3 )3 作为添加剂对反应发生具有重要意义, 同时1 equiv.的Cu2 O能在高温下促进卤化后的苯甲酸脱羧也是促进反应高效进行的因素.
3.
结论与展望
近10年来, 随着导向基团辅助下过渡金属催化特定位置的C—H活化与官能团化反应研究的不断深入, 羧基作为一种极富吸引力的导向基团越来越受到广大C—H活化工作者的重视, 许多新的和深入的工作不断的呈现出来.
虽然过渡金属催化羧酸导向的C—H活化反应已经取得了重大的研究进展, 但仍然存在一些亟待改进和探索的科学问题: (1)羧酸导向C—H官能团化的工作主要集中于C—C偶联领域, C—X偶联的报道还很不充分, 需要进一步的充实和完善; (2)羧酸导向C—H官能团化尤其是作为无痕导向基团的反应, 大都需要在100 ℃以上的高温下才能完成, 探索更加温和的反应体系仍然是未来急需解决的难题; (3)苯甲酸导向的邻位C—H官能团化反应仍然存在如何调控单边官能团化和双边官能团化的问题, 而目前解决的主要措施是在邻位引入取代基来调控, 寻找更加有效的方法来调控单边官能团化和双边官能团化的选择性也是急需解决的问题; (4)脂肪族羧酸类化合物广泛存在于自然界, 但直接对脂肪族羧酸β 位C—H进行官能团化的工作仅限于末端甲基的芳基化, 更多其他类型官能团化的工作急需开展; (5)羧基作为无痕导向基团应用充分发挥了其重要的优势, 但反应体系和类型相对单一, 更多的新型反应体系也急需开发.相信在不久的将来, 过渡金属催化羧酸导向的C—H活化反应会得到更大的突破和改善, 在C—H活化领域发挥更重要的作用.

 下载:
下载:

















































































 下载:
下载:






























 下载:
下载:
