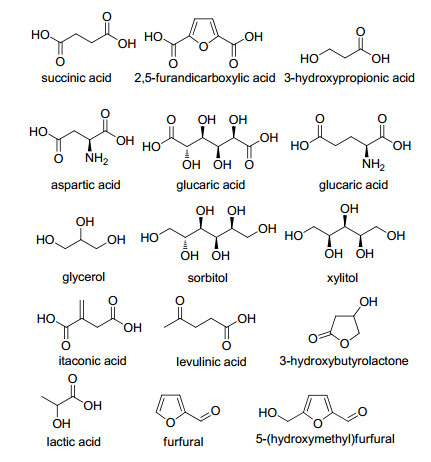
图 1.
“Top10+4”生物质基平台化合物
Figure 1.
"Top10+4" biomass-derived platform compounds

生物质资源因其可再生、低污染、来源广泛等优点成为21世纪最具开发价值的可再生资源之一[1, 2].生物质是地球上唯一的可再生碳源[3, 4].以生物质为资源的生物质炼制是通过化学或生物方法将生物质原料转化为大宗的低附加价值产品或高附加价值的精细和特种化学品[5, 6].现阶段, 生物质炼制产品与传统的石油炼制产品在市场中处于相对竞争地位.因此, 人们尤其关注于以廉价易得的生物质资源为原料制备高附加值的高分子材料和医药化学品的研究, 为生物质炼制产品争取更多的竞争空间[7, 8]. 2004年, 美国能源部首次提出可用于合成高附加价值化学品的12种生物质基平台化合物, 包括丁二酸、2,5-呋喃二甲酸、3-羟基丙酸、天冬氨酸、葡萄糖二酸、谷氨酸、甘油、山梨糖醇、木糖醇、衣康酸、乙酰丙酸和3-羟基丁内酯[9].在此基础上, Bozell和Petersen[10]对其进行了补充, 使其扩展为“Top10+4”生物质基平台化合物, 具体包括醇类(乙醇、甘油、山梨糖醇、木糖醇)、呋喃类(糠醛、5-羟甲基糠醛、2,5-呋喃二甲酸)和羧酸类(乙酰丙酸、琥珀酸、乳酸)等, 其结构式如图 1所示.
多组分反应是以廉价、易得的有机小分子为原料构建复杂分子的有效手段.为了实现多个组分的有序组装, 多官能性化合物经常作为反应的底物分子[11]. 图 1中的生物质基平台化合物大多含有多个官能团, 潜在反应位点较多[12].以廉价、易得的生物质基平台化合物为多组分反应的原料之一, 可在实现复杂分子合成的同时实现生物质高值化转化.受益于生物质基平台化合物分子结构的多样性、多官能性和来源的可持续性, 所实现的多组分反应具有绿色、良好的可持续的特点.借此实现的某些特定结构分子库的构建往往具有传统反应难以具备的独特性质, 这为生物质炼制产品在化学制造业中的进一步应用提供了选择.虽然该研究方向正在逐渐引起绿色化学家的关注, 但是尚缺乏对已报道的成功案例进行阶段性总结的综述论文.为此, 本文介绍了小分子多元醇、二元酸、乙酰丙酸、糠醛衍生物等生物质基平台分子参与的典型多组分反应案例, 并对生物质基平台化合物参与的多组分反应的未来发展进行了展望.
乙醇醛可以由纤维素或者葡萄糖经过水解/逆羟醛缩合反应制备得到, 因此被认为是一种生物质基化学品[13].化学合成的乙醇醛已经被广泛应用于食品、医药、化工等领域[14~16].乙醇醛分子中同时含有醛基和羟基两种官能团, 是构建多组分反应的理想合成子.但是生物质基乙醇醛很难与水分离, 因此以其为原料发展合成方法学必须考虑反应体系的水兼容性问题.本文作者[17]报道了一种低共熔溶剂催化生物质基乙醇醛合成二氢呋喃类化合物的方法(Eq. 1).反应以吲哚、乙酰乙酸乙酯和乙醇醛水溶液为原料, 以预合成的六水合三氯化铁/葡甲胺低共熔溶剂为催化剂.该体系可在80℃条件下实现上述三个组分的选择性组装得到二氢呋喃类化合物.六水合三氯化铁/葡甲胺低共熔溶剂可以在反应完成后回收再用.尽管以乙醇醛水溶液为原料时, 该三组分反应的分离收率只有中等, 但该研究证明生物质基乙醇醛水溶液是极具价值的合成子, 有望用于更多杂环化合物的合成.乙醇醛中亲核性羟基和亲电性醛基同时存在, 确保了环化反应得以发生.
|
|
(1) |
乙二醇是一种大宗化学品.虽然市场中的乙二醇均来自于石油化工路线, 但是以生物质为原料制备乙二醇已经被证明具有很好的经济和技术可行性, 是生物质高值化转化最有前途的路线之一[18].目前, 生物质乙二醇可以通过四种途径合成: (1)生物乙醇经脱水、环氧化、水合制备乙二醇; (2)生物质热解后经合成气制备乙二醇; (3)生物质经糖醇催化转化制备乙二醇; (4)生物质发酵制备乙二醇.其中, 生物质经糖醇催化转化制备乙二醇技术已相对成熟, 正在进行产业化之前的中试[19].
乙二醇可以作为多组分反应的关键组分用于构建具有生物活性和药理功能的吡咯类杂环化合物. 2013年, Beller等[20]借助金属诱导借氢反应实现了多取代吡咯类化合物的合成(Eq. 2).以1,2-二苯乙酮、2-苯乙胺和乙二醇为起始原料, [Ru3(CO)12]为催化剂, Xantphos为配体, 在20 mol% K2CO3作用下, 于叔戊醇中130 ℃条件下反应18 h, 最终以82%的气相收率(73%的分离收率)得到1, 2, 3-三取代吡咯类化合物.该方法对芳香酮类化合物和氨水、芳香胺类和脂肪胺类化合物均有较好的底物适用性, 分离收率为42%~83%.与传统吡咯类化合物的合成方法相比, 该方法反应原料廉价易得, 步骤简洁, 避免了有毒的烷基化试剂的使用.
上述报道的吡咯类化合物的合成方法虽然有效, 但是需要使用价格昂贵的Ru催化剂. Seayad等[21]以一种相对廉价的铜-氮杂环卡宾络合物为催化剂实现了同种类型吡咯类化合物的合成(Eq. 2).以苯乙酮、2-苯乙胺和乙二醇为起始原料, 以CuBr和SIAd(图 2)原位生成的Cu-NHC络合物为催化剂, 在催化量叔丁醇锂存在下, 于叔戊醇中140 ℃条件下反应24 h之后即可以合成得到1,2-二取代吡咯类化合物, 分离收率高达86%.该反应本质上是多步串联过程, 具体包括: (ⅰ)金属诱导乙二醇原位脱氢生成乙醇醛; (ⅱ)后者与胺发生缩合反应生成亚胺, 或与活泼亚甲基化合物反应生成烯醇; (ⅲ)得到的中间体经过类Paal-Knorr反应合成多取代吡咯类化合物.整个串联反应过程包括两个C—N键的生成、一个C—C键的生成、三个脱水反应.上述反应得以发生的关键在于乙二醇氧化后的乙醇醛中间体可以和胺或者烯醇化的羰基化合物发生反应, 而在缩合反应后, 未被氧化的羟基可以经过烯醇互变过程转化为另一个亲电中心, 进而实现环化反应.因为该方法合成的产物吡咯类化合物的C(4)、C(5)位未被取代, 进而有别于传统吡咯类化合物的合成方法, 也便于以其为中间体开展吡咯类化合物的下游转化和应用研究.
|
|
(2) |
丙三醇是一种重要的三碳生物质基平台化合物, 主要以动植物油为原料经过皂化反应合成得到.第一代生物柴油技术在欧洲的产业化导致丙三醇一时供过于求.因此, 催化和合成化学家发展了多种方法利用和转化丙三醇[22].丙三醇可以通过催化氢解、选择氧化、脱水等反应合成丙二醇(1,2-丙二醇、1,3-丙二醇)、羟基丙酮、甘油醛、甘油酸和丙烯醛等重要的精细化学品[23], 也可作为可降解脂肪族聚碳酸酯材料的起始原料[24].
丙三醇含有三个相邻的羟基, 因此可以和二氧化碳反应合成环状碳酸甘油酯类化合物, 但是丙三醇的三个羟基同时和三分子二氧化碳反应合成线性三碳酸甘油酯类化合物却鲜有报道.该反应的难点之一在于, 在二氧化碳存在条件下, 如何有效抑制丙三醇经过分子内反应生成碳酸甘油酯类化合物. 2018年, Mihara等[25]以丙三醇、二氧化碳和正丁基溴为起始原料, 2-叔丁基-1,1,3,3-四甲基胍为碱, 于N-甲基吡咯烷酮(NMP)中室温条件下反应24 h, 即可以高产率地合成得到线性三碳酸酯类化合物(Scheme 1).该反应对多种取代的烷基卤化物均有较好的适用性, 分离收率为21%~92%.当反应温度升高至50 ℃, CO2压力降至0.1 MPa时, 可以得到环状碳酸酯类化合物(最高分离产率为86%).

丁二酸是一种廉价易得的生物质基二元羧酸, 可由糖、淀粉或纤维素等原料经发酵过程制备得到[26].因为生物质基丁二酸产量较大, 以其为平台分子发展有价值下游产物的合成是绿色化学家较为关注的研究方向之一[27].丁二酸分子中同时含有羧基和活性亚甲基片段, 具有较为丰富的反应性. 2017年, Halimehjani等[28]利用丁二酸为反应原料之一, 通过其与芳香醛、芳胺、异腈的四组分Ugi反应得到双酰胺类化合物(Scheme 2).该反应在甲醇中进行, 于室温条件下反应24 h即可完成.双酰胺类化合物的产率为49%~78%.底物拓展实验表明, 该方法不适用于强拉电子基取代的苯胺类化合物.因为产物分子中包含有未反应的羧基, 其与胺、酰胺等片段构成的分子内氢键在一定程度上确保了反应产物具有较低能量, 进而提高了反应产率.作者还以丁二酸酐或顺丁烯二酸酐作为反应底物, 在相同条件下增加1 equiv.的胺, 得到一系列三酰胺类化合物, 其中具有生物活性的两种2,5-二酮哌嗪类化合物也可通过UgiMichael反应得到(Eq. 3).
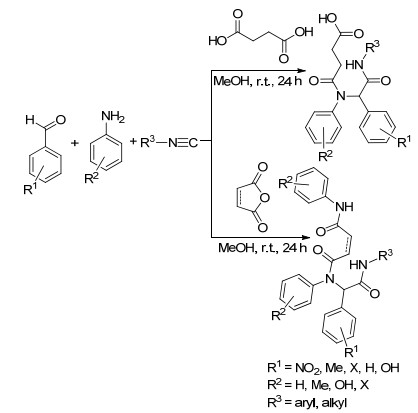
以纤维素为原料通过酸催化水解反应可以大量地制备乙酰丙酸, 因此乙酰丙酸是一种重要的生物质基平台化合物[29].其分子中同时含有酮羰基和羧羰基, 能够发生成盐、酯化、还原等多种反应, 合成乙酰丙酸钙盐、乙酰丙酸酯、γ-戊内酯等重要的有机化工中间体[30].因为乙酰丙酸的两个亲电中心(即1,4-亲电中心)距离适中, 其可以和一级胺经过串联环化反应形成内酰胺类化合物. 1998年, Ugi等[31]利用乙酰丙酸为反应原料之一, 通过其与胺和异腈的三组分反应合成得到了γ-内酰胺类化合物(Scheme 3).反应在甲醇溶剂中进行, 分离收率为64%~100%.当以氨基酸酯作为底物时, 得到的γ-内酰胺还可进一步环化为2, 6-哌嗪二酮类衍生物.但是该反应分离收率仅为5%~33%.这种现象与γ-内酰胺类化合物在三维空间的结构有关, 可能只有一种非对映异构体可以转化为2, 6-哌嗪二酮衍生物, 导致产物分离收率低.
|
|
(3) |
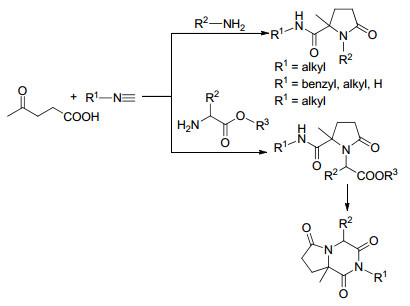
乙酰丙酸还可用作合成聚合物的反应原料. Becer等[32]报道了一种以乙酰丙酸、1,6-二异氰基己烷和乙二胺为起始原料合成聚酰胺类化合物的方法(Eq. 4).在初始研究阶段, 作者对该反应温度、时间、溶剂、加热源等一系列参数进行了优化, 发现以甲醇作为反应溶剂, 于100 ℃微波条件下反应30 min即可以较高产率得到高分子量、高分散度的聚酰胺类化合物.利用一维、二维核磁共振波谱、SEC和MALDI-ToF MS对聚合物的结构进行了表征, 表明聚合物具有良好的分散度和分子量, 分子量可根据所含二胺的链长进行调节, 从而使聚酰胺类化合物具有明显的玻璃转化状态和热分解温度.
|
|
(4) |
糠醛和5-羟甲基糠醛(HMF)是生物质转化过程中两种重要的平台化合物, 前者已在大规模生产, 后者虽已成为易得的市售化学品, 但是其大规模生产尚需在产品质量、下游产品种类和规模上真正实现突破[33].糠醛和5-羟甲基糠醛可通过加氢还原、氧化、酯化和缩合反应合成丁二酸、糠醇、乙酰丙酸和1,6-己二醇等重要的化学品[34].因其分子中含有不可烯醇化的醛基, 为此芳香醛参与的多组分反应自然成为糠醛和HMF下游转化的重要途径.
Skowronski等[35]首次利用HMF为起始原料通过两步法合成α-氨基膦酸酯类化合物(Scheme 4).但是该反应是分步进行的.首先由HMF和伯胺形成亚胺, 经分离后再以三氟乙酸作为催化剂与亚磷酸酯反应, 两步反应后的分离收率为42%~90%.为了简化合成过程, 作者[36]利用单质碘为催化剂在2-甲基四氢呋喃(2-MeTHF)中实现了HMF、伯胺和亚磷酸酯三组分KabachnikFields反应, 以“一锅法”合成得到α-氨基膦酸酯类化合物(Eq. 5).该反应对各种取代的胺类化合物和亚磷酸酯类化合物均适用.值得注意的是, 制备得到的α-氨基膦酸酯类化合物含有呋喃片段, 有望作为底物参与到呋喃衍生物的转化反应中, 为其下游产品的设计和合成提供了契机.
|
|
(5) |
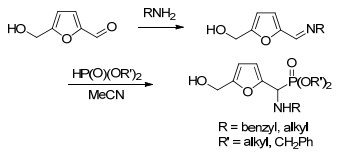
2018年Fernandes等[37]将木糖/木聚糖水解制备糠醛的反应与Kabachnik-Fields反应串联, 以生物质资源为原料通过一锅法合成了α-氨基膦酸酯类化合物(Eq. 6).作者首先以木糖/木聚糖为起始原料, 高铼酸(HReO4)为催化剂, 于1,4-二氧六环中140 ℃条件下反应2 h.生成的糠醛不经分离提纯, 继续与亚磷酸酯、苯胺反应合成得到α-氨基膦酸酯类化合物.该串联反应分离收率为30%~67%.相比于以纯糠醛为原料构建的合成体系, 该方法底物普适性略差, 反应产率也只有中等, 昂贵高铼酸催化剂的使用也限制了其实际应用价值.但该方法首次将糖类化合物制备糠醛和糠醛的转化反应串联为连续反应.这种构建“一锅两步法”而简化合成操作步骤的策略在将来可能被多次使用.
构建一锅连续反应的策略还被用于邻苯二甲酰亚胺类化合物的合成中, 邻苯二甲酰亚胺类化合物具有良好的生物活性而受到合成和药物化学家的关注[38].早在1984年, Potts等[39]以2-糠醛二甲基腙和N-乙基马来酰亚胺为反应原料, 氯仿为溶剂, 通过串联的DielsAlder环加成/芳构化反应合成得到一系列邻苯二甲酰亚胺类衍生物(Eq. 7).因为2-糠醛二甲基腙价格昂贵(AlfaAesar牌价: 720元/10 g), Hailes等[40]以廉价易得的糠醛为起始原料, 通过一锅两步法合成得到邻苯二甲酰亚胺类衍生物(Eq. 8). N,N-二甲基肼中NH2官能团与电负性较强的N原子相连, 使其与糠醛亲核加成反应活性较高.二者混合物在水中即可反应生成2-糠醛二甲基腙, 后者无需分离即可在水中继续与N-乙基马来酰亚胺在温和条件下(<80 ℃)反应生成邻苯二甲酰亚胺类衍生物.该水相中的一锅两步反应分离收率为41%~97%.考虑到作者并未使用外加催化剂, 仅仅以水为溶剂, 该结果较为难得.该反应不但对各种取代的糠醛(包括HMF)和马来酰亚胺底物都有较好的适用性, 还适用于富马酸腈、马来酸二甲酯等亲双烯体.在大部分情况下, 产物均可直接过滤得到, 无需使用色谱法分离产物.糠醛分子中醛羰基的亲电性和呋喃片段作为DielsAlder环加成双烯体的反应性在上述反应中得以应用, 是构建该一锅两步反应的化学基础.
|
|
(6) |
|
|
(7) |
|
|
(8) |
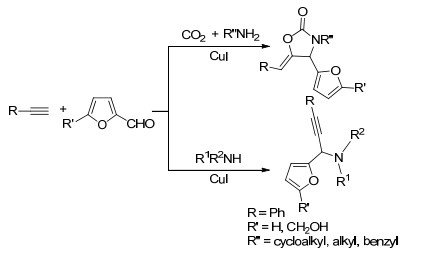
2018年徐益升等[41]报道一种以糠醛或HMF为反应原料合成噁唑烷酮类化合物的方法(Scheme 5).作者发现, 糠醛或HMF可在乙醇溶剂中, 于80 ℃、催化量碘化亚铜(CuI)存在条件下与苯乙炔、环己胺和二氧化碳发生四组分反应得到一系列噁唑烷酮类化合物.反应的中间体应该是糠醛亚胺和炔铜作用得到的丙炔胺, 后者可以和二氧化碳进一步发生缩合反应, 形成环合产物.该反应对芳炔类底物和脂肪族伯胺均有较好的适用性, 但反应分离收率不高.进一步研究表明, 当以苯胺作为反应底物时, 二氧化碳则不能参与反应.糠醛或HMF、苯乙炔和苯胺可经过三组分反应合成得到炔丙胺类化合物.显然, 胺类的碱性对该反应具有举足轻重的作用.当以碱性较弱的苯胺为原料时, 得到的炔丙基胺亲核性不够强, 难以实现对二氧化碳(CO2)的亲核加成过程.
需要指出的是, 上述反应虽然只用了糠醛分子中醛羰基的反应性, 而呋喃片段并未参与反应, 但是得到的产物有呋喃片段, 有望为产物的下游转化提供反应位点.基于这种思想, 本文作者[42]以HMF为起始原料, 通过Biginelli反应合成了一系列3,4-二氢嘧啶酮类化合物(Eq. 9).虽然Biginelli反应制备得到的3,4-二氢嘧啶酮类化合物因为具有抗病毒性、抗肿瘤性、抗菌性和抗炎性等生物活性而被人熟知[43, 44], 但是到目前为止HMF作为一种生物质基平台化合物并未被作为底物用于该反应.我们以HMF、1,3-二羰基化合物和尿素为反应原料, 氯化锌(ZnCl2)为催化剂, 在无溶剂条件下合成得到一系列3,4-二氢嘧啶酮类化合物, 反应收率为30%~86%.得到的3,4-二氢嘧啶酮类产物保留了HMF的羟甲基, 为该类化合物的后续修饰提供了可能的反应位点.
|
|
(9) |
糖类化合物是木质纤维素中碳水化合物的主要组成部分, 也是重要的生物质基化合物.葡萄糖和果糖是代表性的单糖, 可通过脱水、酯化、氢化裂解、氧化等反应合成HMF、乙酰丙酸、丙三醇、山梨醇等重要的化学品.还可以作为多组分反应理想的合成子.
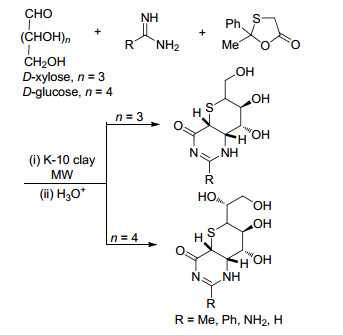
部分硫糖衍生物被发现具有良好的α-糖苷酶抑制性能, 因此该类化合物广泛用作抗血栓、抗肿瘤药物等药物研究中[45~49]. Rai等[50]报道了一种新型K-10黏土催化的三组分反应合成具有重要药用价值的嘧啶基硫代糖类衍生物的方法(Scheme 6).该方法以葡萄糖或者木糖、脒/胍和2-甲基-2-苯基-1,3-氧硫-5-环戊酮作为起始原料, K-10黏土作为催化剂, 无溶剂条件下微波合成具有较高的顺式非对映体选择性(>94%)目标产物, 反应分离收率为72%~93%.该反应对不同取代的脒/胍类底物进行了拓展, 丰富了硫代糖类衍生物的多样性.该工艺实现了可再生的碳水化合物向具有药用价值的精细化学品转化.
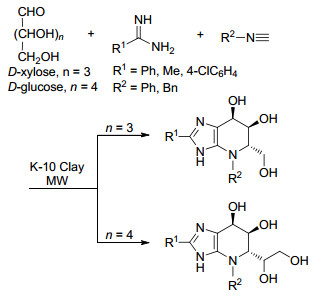
Rai课题组[51]还报道了一种以葡萄糖或木糖作为起始原料, 通过Ugi反应合成咪唑基亚胺糖类衍生物的方法(Scheme 7).该反应同样以K-10黏土为催化剂, 无溶剂、微波辐射条件下合成得到一系列带有不同取代基的目标产物, 分离收率为86%~95%.需要指出的是, 亚胺糖类衍生物对糖苷酶和糖基转移酶具有潜在的抑制作用, 能够有效治疗艾滋病感染、癌症、糖尿病、其他遗传性和代谢性疾病[52, 53].传统的Ugi反应主要以芳香醛/酮或脂肪醛/酮为原料合成, 而涉及到以醛糖等单糖为原料的案例未见报道.因此, 该方法的创新之处在于以醛糖为起始原料实现具有潜在生理和药理活性亚胺糖类衍生物的合成.
在此基础, Rai课题组[54]还报道了一种以葡萄糖或木糖作为起始原料, 通过一锅四组分缩合反应构建亚胺糖环化1,3-噻嗪类衍生物的方法(Eq. 10).该反应以1-丁基-3-甲基咪唑硫氰酸盐离子液体([BMIm]SCN)同时作为介质和硫氰酸根阴离子供体, 在室温条件下以较高的顺式非对映体选择性(>94%)得到一系列目标产物, 反应分离收率为82%~95%.反应结束后生成的氢氧化1-丁基-3-甲基咪唑盐离子液体([BMIm]OH)可以经离子交换后再循环使用, 循环五次后催化活性没有明显下降.
2018年, Khalafi-Nezhad等[55]利用葡萄糖等醛糖、1,3-二羰基化合物和丙二腈作为起始原料, 在对甲基苯磺酸(PTSA)的催化作用下, 乙醇溶剂中于50 ℃条件下反应12 h, 合成得到了一系列含有多元醇尾链的杂环化合物, 反应分离收率为80%~90% (Eq. 11).该反应对单糖类化合物(葡萄糖、半乳糖、阿拉伯糖)和二糖类化合物(麦芽糖、乳糖)都有较好的适用性.抗氧化容量分析发现, 所有杂环化合物都表现出明显的抗氧化活性, 其中化合物A(图 3)表现出最大的抗氧化活性.该方法操作简单、产物分离收率高、纯度高且产物具有良好的抗氧化活性, 因此在合成一些生物活性分子和药物化学相关化合物方面具有很好的应用前景.
|
|
(11) |
综上所述, 以廉价易得的可再生生物质基平台化合物为原料, 通过与其他组分之间发生多组分反应合成结构复杂的高附加价值产物是生物质化学品高值化转化的策略之一.生物质基平台分子的多样性和多功能性恰好满足了多组分反应中关键合成子的基本要求, 这使得立足于生物质基平台分子的反应性来设计多组分反应成为可能.对文献报道的多元醇、二元羧酸、乙酰丙酸、糠醛衍生物、糖类等生物质基平台化合物参与的多组分反应进行了综述.以生物质基乙二醇、乙酰丙酸和糠醛衍生物作为起始原料构建的杂环化合物合成方法, 充分利用了多官能性生物质基平台分子的独特反应性.借助该策略制备得到的化合物往往带有可持续的特点, 这对于日化和医药产品来说具有额外的“绿色标签效应”, 有利于后期的产品宣传和市场开发.
虽然前人已经在该方向取得了可喜的进展, 但借助多组分反应策略实现生物质高值化转化的研究依然存在诸多挑战和不足.生物质基平台化合物参与多组分反应的案例还较少, 这是多种因素造成的.首先, 生物炼制过程尚未充分渗透到化学品制造产业链之中, 所能提供的多官能性底物分子不够丰富.其次, 主流有机化学家长期以来并未对该方向给予足够关注; 随着绿色化学理念在有机化学研究中的不断渗透和接纳, 相信不远的将来这一局面将会有所改变.第三, 糖、多元醇等相对更为上游的生物质基平台分子大多是多羟基化合物, 与简单的有机小分子不同, 其参与的反应受控因素更多, 产物的结构表征也更为复杂, 这也提高了以该类生物质基平台分子作为底物来设计和探索多组分反应的难度.
尽管有很多挑战, 但是利用多组分反应实现生物质基平台分子的高值化转化这一主题依然具有很好的研究价值和挖掘空间.在未来, 人们需要尤其关注于充分利用和挖掘现有生物质基平台分子反应性的研究, “因地制宜”地设计与之相契合的多组分反应, 丰富生物质高值化转化的途径.为此, 从事生物质基平台化合物参与多组分反应研究的科研工作者还需要注意以下几个方面: (ⅰ)尽可能地使用靠近上游的生物质基为原料, 借助催化体系和催化剂的合理设计、反应偶联或其他辅助手段提高生物质高值化转化反应的步骤和原子经济性. (ⅱ)关注所得产物的有用性, 因为借助多组分反应实现生物质高值化转化制备得到的大多是结构复杂的未知分子, 为了避免漫无目的地投入研究精力, 研究者最好以此前已经被多次证明确实具有较高潜在用途的分子或者分子骨架为目标, 尽可能地发展有科学贡献和学科必要性的多组分转化路径. (ⅲ)生物质基平台分子来源于生物炼制过程, 初级生物质加工提质采用加氢脱氧反应, 必然导致水的生成; 而很多生物质基平台分子均含有亲水性的羟基、羧基等官能团, 其去水纯化难度很大.因此研究者需要尽可能地关注所发展反应体系的水兼容性.
Li, H.; Riisager, A.; Saravanamurugan, S.; Pandey, A.; Sangwan, R. S.; Yang, S.; Luque, R. ACS Catal. 2018, 8, 148. doi: 10.1021/acscatal.7b02577
Lai, L.; Zhang, Y. ChemSusChem 2010, 3, 1257. doi: 10.1002/cssc.201000201
Geilen, F. M. A.; Engendahl, B.; Harwardt, A.; Marquardt, W.; Klankermayer, J.; Leitner, W. Angew. Chem., Int. Ed. 2010, 49, 5510. doi: 10.1002/anie.201002060
Wang, N.; Yao, Y.; Li, W.; Yang, Y.; Song, Z.; Liu, W.; Wang, H.; Xia, X.; Gao, H. RSC Adv. 2014, 4, 57164. doi: 10.1039/C4RA09585E
Mascal, M.; Nikitin, E. B. Green Chem. 2010, 12, 370. doi: 10.1039/B918922J
Breeden, S. W.; Clark, J. H.; Farmer, T. J.; Macquarrie, D. J.; Meimoun, J. S.; Nonne, Y.; Reid, J. E. S. J. Green Chem. 2013, 15, 72. doi: 10.1039/C2GC36290B
Huber, G. W.; Iborra, S.; Corma, A. Chem. Rev. 2006, 106, 4044. doi: 10.1021/cr068360d
Corma, A.; Iborra, S.; Velty, A. Chem. Rev. 2007, 107, 2411. doi: 10.1021/cr050989d
Werpy, T.; Petersen, G.; Aden, A; Bozell, J.; Holladay, J.; White, J.; Manheim, A; Eliot, D.; Lasure, L.; Jones, S. Top Value Added Chemicals from Biomass, Vol. 1, Richland Washingto, Pacific Northwest National Laboratory, 2004.
Bozell, J. J.; Petersen, G. R. Green Chem. 2010, 12, 539. doi: 10.1039/b922014c
Ghashghaei, O.; Seghetti, F.; Lavilla, R. Beilstein J. Org. Chem. 2019, 15, 521. doi: 10.3762/bjoc.15.46
Ravichandiran, P.; Lai, B.; Gu, Y. Chem. Rec. 2017, 17, 142. doi: 10.1002/tcr.201600042
Xu, G.; Wang, A.; Pang, J.; Zheng, M.; Yin, J.; Zhang, T. Appl. Catal. A 2015, 502, 65. doi: 10.1016/j.apcata.2015.05.038
辛坤, 李青松, 贾冰, 于记生, 天然气化工, 2016, 41, 88. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=trqhg201605023Xin, K.; Li, Q.; Jia, B.; Yu, J. Nat. Gas Chem. Ind. 2016, 41, 88(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=trqhg201605023
Liang, G.; Wang, A.; Li, L.; Xu, G.; Yan, N.; Zhang, T. Angew. Chem., In. Ed. 2017, 56, 3050. doi: 10.1002/anie.201610964
Flamini, R.; Vedova, A. D. J. Agric. Food Chem. 2003, 51, 2300. doi: 10.1021/jf020639h
Xu, J.; Huang, W.; Bai, R.; Queneau, Y.; Jerome, F.; Gu, Y. Green Chem. 2019, 21, 2061. doi: 10.1039/C8GC04000A
杨学萍, 董丽, 陈璐, 胡云光, 化工进展, 2015, 34, 3609. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hgjz201510015Yang, X.; Dong, L.; Chen, L.; Hu, Y. Chem. Ind. Eng. Proc. 2015, 34, 3609(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hgjz201510015
Zheng, M.; Pang, J.; Sun, R.; Wang, A.; Zhang, T. ACS Catal. 2017, 7, 1939. doi: 10.1021/acscatal.6b03469
Zhang, M.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 597. doi: 10.1002/anie.201206082
Dang, T. T.; Seayad, A. M. Chem. Asian J. 2017, 12, 2383. doi: 10.1002/asia.201701045
乐传俊, 王丽, 苏杨, 朱少萍, 应用化学, 2014, 31, 367. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=yyhx201404001Le, C.; Wang, L.; Su, Y.; Zhu, S. Chin. J. Appl. Chem. 2014, 31, 367(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=yyhx201404001
朱林, 艾珍, 工业催化, 2013, 21, 19. doi: 10.3969/j.issn.1008-1143.2013.12.004Zhu, L.; Ai, Z. Ind. Catal. 2013, 21, 19(in Chinese). doi: 10.3969/j.issn.1008-1143.2013.12.004
Lyadov, A. S.; Khadzhiev, S. N. Russ. J. Appl. Chem. 2017, 90, 1727. doi: 10.1134/S1070427217110015
Mihara, M.; Morogab, K.; Iwasawab, T.; Nakaia, T.; Ito, T.; Ohno, T.; Mizuno, T. Synlett 2018, 29, 1759. doi: 10.1055/s-0037-1610027
王庆昭, 吴巍, 赵学明, 化工进展, 2004, 23, 794. doi: 10.3321/j.issn:1000-6613.2004.07.026Wang, Q.; Wu, W.; Zhao, X. Chem. Ind. Eng. Proc. 2004, 23, 794(in Chinese). doi: 10.3321/j.issn:1000-6613.2004.07.026
Zeikus, J. G.; Jain, M. K.; Elankovan, P. Appl. Microbiol. Biot. 1999, 51, 545. doi: 10.1007/s002530051431
Halimehjani, A. Z.; Sharifi, M. Tetrahedron 2017, 73, 5778. doi: 10.1016/j.tet.2017.08.028
Yan, L.; Yao, Q.; Fu, Y. Green Chem. 2017, 19, 5527. doi: 10.1039/C7GC02503C
Kang, S.; Fu, J.; Zhang, G. Renewable Sustainable Energy Rev. 2018, 94, 340. doi: 10.1016/j.rser.2018.06.016
Hanusch-Kompa, C.; Ugi, I. Tetrahedron Lett. 1998, 39, 2725. doi: 10.1016/S0040-4039(98)00428-6
Hartweg, M.; Becer, C. R. Green Chem. 2016, 18, 3272. doi: 10.1039/C6GC00372A
Rosatella, A. A.; Simeonov, S. P.; Frade, R. F. M.; Afonso, C. A. M. Green Chem. 2011, 13, 754. doi: 10.1039/c0gc00401d
王军, 张春鹏, 欧阳平凯, 化工进展, 2008, 27, 702. doi: 10.3321/j.issn:1000-6613.2008.05.013Wang, J.; Zhang, C.; Ouyang, P. Chem. Ind. Eng. Proc. 2008, 27, 702(in Chinese). doi: 10.3321/j.issn:1000-6613.2008.05.013
Cottier, L.; Descotes, G.; Lewkowski, J.; Skowronski, R. Phosphorus, Sulfur Silicon Relat. Elem. 1996, 116, 93. doi: 10.1080/10426509608040472
Fan, W.; Queneau, Y.; Popowycz, F. RSC Adv. 2018, 8, 31496. doi: 10.1039/C8RA05983G
Isca, V. S.; Fernandes, A. C. Green Chem. 2018, 20, 3242. doi: 10.1039/C8GC01343H
张文娟, 罗浩, 周孟, 卢陆飞, 武峰, 李锐, 中国药物化学杂志, 2012, 23, 181. http://www.cnki.com.cn/Article/CJFDTotal-ZGYH201303003.htmZhang, W.; Luo, H.; Meng, M.; Lu, L.; Wu, F., Li, R. Chin. J. Med. Chem. 2012, 23, 181(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-ZGYH201303003.htm
Potts, K. T.; Walsh, E. B. J. Org. Chem. 1984, 49, 4099. doi: 10.1021/jo00195a058
Higson, S.; Subrizi, F.; Sheppard, T. D.; Hailes, H. C. Green Chem. 2016, 18, 1855. doi: 10.1039/C5GC02935J
Wu, Q.; Chen, J.; Guo, X.; Xu, Y. Eur. J. Org. Chem. 2018, 24, 3105.
Fan, W.; Queneau, Y.; Popowycz, F. Green Chem. 2018, 20, 485. doi: 10.1039/C7GC03425C
权正军, 张彰, 达玉霞, 王喜存, 有机化学, 2009, 29, 876. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract328201.shtmlQuan, Z.; Zhang, B.; Da, Y.; Wang, C. Chin. J. Org. Chem. 2009, 29, 876(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract328201.shtml
Kappe, C. O. Eur. J. Med. Chem. 2000, 35, 1043. doi: 10.1016/S0223-5234(00)01189-2
Hellman, B.; Lernmark, A.; Sehlin, J.; Taljedal, J. B.; Whistler, R. L. Biochem. Pharmacol. 1973, 22, 29. doi: 10.1016/0006-2952(73)90251-7
Zysk, J. R.; Bushway, A. A.; Whistler, R. L.; Carlton, W. W. J. Reprod. Fertil. 1975, 45, 69. doi: 10.1530/jrf.0.0450069
Kim, J. H.; Kim, S. H.; Hahn, E. W.; Song, C. W. Science 1978, 200, 206. doi: 10.1126/science.635582
Hashimoto, H.; Fujimori, T.; Yuasa, H. J. Carbohydr. Chem. 1990, 9, 683. doi: 10.1080/07328309008543863
Johnston, B. D.; Pinto, B. M. J. Org. Chem. 1998, 63, 5797. doi: 10.1021/jo980168r
Yadav, L. S.; Rai, A. Carbohydr. Res. 2009, 344, 2329. doi: 10.1016/j.carres.2009.06.017
Rai, V. K.; Singh, S.; Singh, P.; Yadav, L. D. S. Synthesis 2010, 23, 4051. https://www.mendeley.com/catalogue/expeditious-synthesis-functionalized-piperidines-regioselective-ring-opening-aziridines-enals-cataly/
Goss, P. E.; Baker, M. A.; Carver, J. P.; Dennis, J. W. Clin. Cancer Res. 1995, 1, 935. https://www.ncbi.nlm.nih.gov/pubmed/9816064
Robinson, K. M.; Begovic, M. E.; Rhinehart, B. L.; Heineke, E. W.; Ducep, J. B.; Kastner, P. R.; Marshall, F. N.; Danzin, C. Diabetes 1991, 40, 825. doi: 10.2337/diab.40.7.825
Rai, V. K.; Scharrof, V. R. J. Heterocycl. Chem. 2017, 54, 1178. doi: 10.1002/jhet.2690
Dangolani, S. K.; Panahi, F.; Tavaf, Z.; Nourisefat, M.; Yousefi, R. Khalafi-Nezhad, A. ACS Omega 2018, 3, 10341. doi: 10.1021/acsomega.8b01124

图式 1 三组分反应合成碳酸酯类化合物
Scheme 1 Synthesis of carbonate compounds via three-component reaction

图式 2 四组分反应合成双酰胺类化合物
Scheme 2 Synthesis of bisamide derivatives via four-component reaction

图式 3 三组分反应合成γ-内酰胺类化合物
Scheme 3 Synthesis of γ-lactam compounds via three-component reaction

图式 4 CF3CO2H催化合成α-氨基膦酸酯类化合物
Scheme 4 CF3CO2H-catalyzed synthesis of α-amino phosphonate compounds

图式 6 K-10黏土催化合成硫代糖类衍生物
Scheme 6 K-10 clay-catalyzed synthesis of thiosugar derivatives

图式 7 K-10黏土催化合成亚胺糖类衍生物
Scheme 7 K-10 clay-catalyzed synthesis of iminosugar derivatives

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:











 下载:
下载:



 下载:
下载: