
图式 1.
α-羰基重氮化合物的反应活性
Scheme 1.
Reactivity of α-diazocarbonyl compounds

重氮化合物化学性质活泼, 是一类具有重要应用价值的有机合成中间体, 在有机合成、药物合成等领域得到了广泛应用[1].在此类化合物中, α-羰基重氮化合物由于α位羰基的引入, 可分散与重氮基相连的碳上的负电荷, 增加了重氮化合物的稳定性, 使其在实验室里可以用常规的方法分离提纯和保存, 同时又具有很好的反应活性, 因此在涉及底物为重氮化合物的有机合成反应中常作为首选底物.在有机合成中主要用作产生卡宾的前体, 在光或热以及过度金属催化的条件下分别形成自由卡宾(free carbene)和金属卡宾(metal carbene), 形成的卡宾中间体具有非常高的反应活性, 可以发生多种类型的反应, 例如环丙烷化反应[2]、生成叶立德反应[3]、X—H插入反应[4~7] (X=C、O、N、S等)和Wolff重排反应[8].
许多含氮有机化合物, 如α-氨基酸、生物碱和含氮杂环化合物, 都具有广泛的生物活性, 因而发展高效构筑C—N键的方法具有重要的研究意义[9].过渡金属催化卡宾介导的N—H插入反应是构筑C—N键的高效方法之一[10]. 1978年, Cama课题组[11]报道了醋酸铑催化α-羰基重氮化合物发生分子内N—H键插入反应, 实现分子内环化合成β-内酰胺类抗生素, 为合成β-内酰胺类抗生素带来深远影响.随后, Bouffard课题组[12]通过分子内N—H键插入反应合成硫霉素, 进而促使抗生素亚胺培南的发现和工业化制造, 这是目前α-羰基重氮化合物在工业上最为成功的应用.自此以后, 过渡金属、生物大分子以及有机小分子等催化剂相继被开发并运用在N—H键插入反应, 以构筑各类C—N键产物. α-羰基重氮化合物对N—H键的插入反应可以合成各类具有生物活性的化合物, 在现代药物合成方法中被广泛应用于合成抗生素[13]、具有光学活性的α-氨基酸衍生物[14]以及含氮杂环化合物[15]等(Scheme 2).详细介绍了以α-羰基重氮化合物为原料, 过渡金属催化、有机小分子催化、生物大分子催化及光和热条件下, 实现α-羰基重氮化合物对N—H键的插入反应, 阐明反应机理, 介绍反应合成应用价值.
自1952年Yates[16]使用Cu催化N—H插入反应以来, 各类过渡金属[17]催化N—H插入反应得到了广泛开发, 使利用过渡金属催化α-羰基重氮化合物对胺类化合物N—H键的插入反应成为目前最常用的方法.近二十年来, 利用过渡金属配合物催化α-羰基重氮化合物对N—H键的不对称插入反应取得了重大突破, 使过渡金属催化α-羰基重氮化合物对N—H键的不对称插入反应已成为高效、高对映选择性获得手性α-氨基酸及其衍生物的重要方法.
铜及其配合物作为催化剂广泛应用于N—H插入反应中, 例如Saegusa等[18]以CuCN为催化剂, 将重氮乙酸乙酯对六氢吡啶N—H键的插入反应收率提升至72%, Sivasankar等[19]使用铜配合物为催化剂, 将苯基重氮乙酸乙酯对苯胺N—H键的插入反应收率提升至97%. 2015年, Sivasankar课题组[20]报道了一种含膦配体的Cu(Ⅰ)配合物催化剂1, 此催化剂在空气中能稳定存在, 催化剂结构通过X射线晶体学表征证实.反应以芳基重氮酯类化合物和氨基苯酚类化合物作为起始原料, 反应条件温和, 收率良好至优秀, 底物的普适性广.值得一提的是, 反应能够避免O—H插入产物和重氮酯的二聚副反应产物生成, 从而高选择性地得到N—H插入产物, 目标产物结构通过X射线晶体学表征证实(Scheme 3).

2017年, 祖连锁课题组[21]报道了利用醋酸铜催化2-炔基-苯胺类化合物2和α-羰基重氮类化合物3的串联反应, 合成了吲哚酮类化合物4.反应过程可能经历N—H插入反应、分子内迈克尔加成和碎片化反应, 最后经过环化得到终产物, 反应收率中等至良好, 官能团兼容性良好以及底物普适性广.值得一提的是, 作者用2-重氮基-1, 3-环己二酮类化合物5作为底物时发现并没有经过N—H插入最终得到吲哚酮类化合物, 而是经过wolff重排、分子内迈克尔加成和碎片化反应, 最终得到2-喹啉酮类化合物6, 反应效果良好, 底物普适性广(Scheme 4).
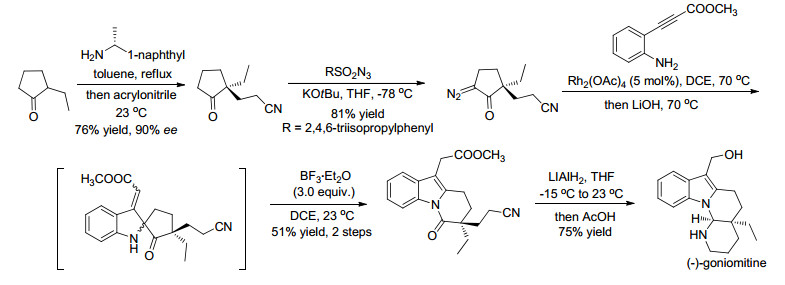
作者对此类含N—H插入反应过程的串联反应进行了合成应用, 通过5步化学反应就可以实现天然产物(-)-goniomitine的不对称全合成, 该合成路线是迄今最为简短的(-)-goniomitine合成策略, 进一步推动了该天然产物的合成研究, 如Scheme 5所示.
2012年, Gillingham课题组[22]报道了在2-吗啉乙磺酸(MES)缓冲液中用Rh2(OAc)4催化α-重氮酯类化合物对天然核苷酸的N—H键的插入反应, 实现了天然核苷酸的烷基化反应, 如Scheme 6所示.值得一提的是, 实验中发现核苷酸的二级结构可以诱导发生选择性的烷基化反应.此类方法, 对于核苷酸衍生物的结构修饰和标记具有重大的意义, 可以应用于生物学领域研究或临床治疗过程中对核苷酸衍生物进行标记和修饰.

近几年, 过渡金属钌在生物分子偶联反应[23]中同样取得相应优秀成果, 对生物大分子(核苷酸和肽链等)的烷基化修饰效果很好. 2014年, 支志明课题组[23a]合成了一系列含氮杂环卡宾(NHC)配体的钌卟啉配合物7, [Ru(Por)(NHC)2] (7)能够催化苯基重氮乙酸甲酯对芳香胺和脂肪胺N—H键的插入反应, 具有良好的催化效果.值得一提的是, 在乙腈和水作为溶剂下, 该催化剂同样能够催化苯基重氮乙酸乙酯与肽链的N-末端反应, 实现在温和条件下对肽链的N端修饰(Scheme 7).

2006年, Gross课题组[24]报道了利用铁卟啉催化剂8首次实现了重氮乙酸乙酯对氨气N—H键的插入反应, 合成了甘氨酸乙酯, 这为α-氨基酸酯的制备提供了新的有效合成方法.值得一提的是, 如果以醋酸铵作为氮源, 反应可以高选择性地得到三次插入产物(Scheme 8).

2009年, Mangion课题组[25]首次报道利了用过渡金属铱催化α-羰基硫叶立德化合物对N—H键的插入反应, 此后, 过渡金属铱催化α-羰基重氮化合物对N—H键插入反应同样得到科研工作者的关注, 并取得了相应的成果. 2013年, Woo课题组[26]首次报道了Ir(TTP)CH3催化重氮乙酸乙酯(EDA)或苯基重氮乙酸甲酯(MPDA)对胺类化合物N—H键的插入反应. EDA与伯胺反应往往会得到一次插入和二次插入反应产物, ,但EDA对大多数苯胺类化合物N—H键插入反应主要得到一次插入产物, 收率大部分在80%以上. MPDA和芳香胺类化合物反应得到一次插入产物, 产率中等至良好, 但与脂肪胺类化合物反应效果却很差(Scheme 9a).
2017年, Sivasankar课题组[27]报道了[(COD)IrCl]2催化重氮类化合物9对苯胺类化合物N—H键的插入反应, 相对于Woo课题组开发的Ir(TTP)CH3催化剂, [(COD)IrCl]2催化反应效果更好, 反应的产物单一, 收率可达98%, 值得一提的是, 反应以水为溶剂, 但反应过程中并不会得到O—H键插入反应产物(Scheme 9b).
2004年, Jørgensen等[28]首次利用铜配合物实现不对称N—H键插入反应, 随后许多课题组相继开发出不同的手性配体实现铜配合物催化N—H键的不对称插入反应, 其中包括周其林[29]、Fu[30]、冯小明[31]等课题组, 如Scheme 10所示.

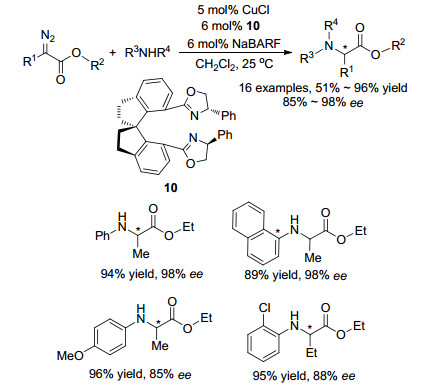
2007年, 周其林课题组[29]报道了使用CuCl和手性螺环双噁唑啉配体10作为催化剂, 催化烷基重氮酯类化合物对芳香胺类化合物N—H键的高对映选择性插入反应, 反应条件温和, 在室温下即可反应, 反应收率可达95%, ee值可达98%.底物普适性研究表明:大多数的苯胺类化合物的反应效果较好; 当苯胺类化合物苯环上的对位取代基为供电子基团或邻位为卤素基团取代时, 会使产物ee值略微降低, 底物的普适性广泛.这是首例高对映选择性的催化不对称N—H键插入反应, 为手性α-氨基酸类化合物的合成提供了新的有效方法(Scheme 11).
过渡金属催化 α-羰基重氮化合物对胺类化合物N—H键的不对称插入反应的机理未被明确证实, 目前机理主要认为形成叶立德中间体和通过1, 2-质子迁移过程完成插入反应[32].为探究反应的机理[33], 作者研究了氢动力学同位素效应, 竞争实验结果观测到一级动力学同位素效应(kH/kD=4), 说明质子迁移过程可能为限速步骤.从而提出反应可能的机理(Scheme 12).首先, Cu配合物催化α-重氮酯失去一分子氮气形成铜卡宾中间体Ⅰ, 胺类化合物的氮原子上的孤对电子进攻缺电子的铜卡宾进而形成Cu配合物相连的叶立德中间体Ⅱ, 然后同时发生质子迁移和Cu配合物离去, 以达到控制立体选择性的效果, 最终得到高对映选择性产物.质子迁移的详细过程仍然未被证实, 可能包括Ⅲ、Ⅳ两种转移方式, 主要区别在于胺类化合物是否参与质子转移过程.

2007年, Fu课题组[30]报道了CuBr和双联吡啶配体11催化芳基重氮酯类化合物对氨基甲酸叔丁酯N—H键的高对映选择性插入反应.反应需要加入AgSbF6, 室温下即可反应, 反应收率可达89%, ee值可达95%.底物普适性研究表明:苯基重氮酯苯环上的取代基电子效应对反应效果影响不明显, 苯基重氮酯类化合物对氨基甲酸苄酯N—H键的插入反应同样能够获得高对映选择性的产物, 如Scheme 13所示.

2010年, 冯小明课题组[31]报道了CuCl和联萘酚衍生物配体12催化α-重氮酯对芳香胺N—H键的高对映选择性N—H插入反应.反应条件温和, 反应收率可达99%, ee值可达98%, 在反应中加入适量分子筛会提高反应的对映选择性(Scheme 14).底物普适性研究表明: 2-重氮基丙酸叔丁酯对芳香族伯胺类化合物N—H键插入反应具有优秀的收率和ee值; 苯基重氮酯对芳香族仲胺类化合物具有良好收率但ee值中等至良好.

金属Pd很早被应用于形成金属钯卡宾[34], 但较少被应用在催化X—H插入反应(X=N、O、S等杂原子).近年来, 过渡金属钯在不对称N—H插入反应中得到了很好开发[35]. 2014年, 冯小明课题组[35b]利用[Pd2(dba)3]和手性胍衍生物13催化α-重氮酯对苯胺类化合物高对映选择性N—H键插入反应, 获得了高达99%的收率和达到94% ee的对映选择性.底物普适性研究表明:苯胺类化合物的苯环上取代基的电子效应和位阻效应对于ee值影响不大, 底物普适性广.作者为探究反应可能的机理, 研究了氢动力学同位素效应, 未观测到动力学同位素效应(kH/kD=1), 表明反应过程中质子迁移过程不是限速步骤, 重氮化合物分解或胺类化合物的氮原子发生亲核加成过程可能为限速步骤, 产物具有高对映选择性的原因可能由于质子迁移和金属催化剂同时离去达到控制立体选择性的效果(Scheme 15a).
2017年, Van Vranken课题组[35c]报道了Pd(Ⅱ)与手性双噁唑啉配体14催化苯基重氮乙酸甲酯对芳香含氮杂环化合物高对映选择性N—H键的插入反应.实现了首例钯催化α-重氮酯对咔唑或C(3)-取代吲哚的高对映选择性N—H键插入反应.在温和反应条件下, 合成一系列具有药效基团的吲哚类衍生物和具有生物活性的咔唑类衍生物, 获得了高达99%的分离收率和99% ee的对映选择性.底物普适性研究表明:在苯基重氮酯类化合物对咔唑类化合物N—H键插入反应中, 苯基重氮酯类化合物苯环上的对位取代基为供电子基团反应效果良好, 吸电子取代基团反应效果很差; 吲哚类化合物的C(3)位置被取代时, N—H插入产物收率会得到明显的提高, 且C(3)位取代基团的位阻越大收率越高, 官能团兼容性良好, 底物普适性广.作者同时探究了此反应合成应用价值, 高对映选择性合成了5-HT6受体阻断剂15, 反应ee值可达92% (Scheme 15b).
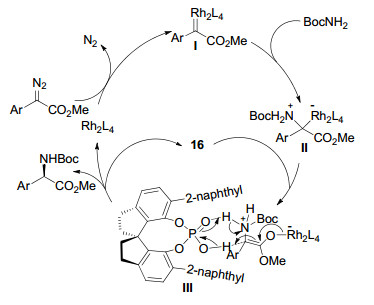
2011年, 周其林课题组[36]报道了Rh2(TPA)4和手性螺环磷酸16协同催化实现α-重氮酯对BoC—NH2的高对映选择性不对称N—H插入反应.手性螺环磷酸作为质子梭催化剂, 成功实现α-重氮酯对N—H键插入反应中的质子转移过程的手性控制.反应以苯基重氮酯类化合物17和氨基甲酸叔丁酯为原料, 在室温条件下, 对映选择性合成一系列产物18, 反应催化剂用量低, 反应时间仅需1 min, 收率可达99%, ee值可达95% (Scheme 16).

可能的反应机理如Scheme 17所示.首先, Rh2(TPA)4催化α-重氮酯脱去一份子氮气, 形成铑卡宾中间体Ⅰ, 然后与氨基甲酸叔丁酯反应形成叶立德中间体Ⅱ, 手性螺环磷酸与叶立德中间体可能通过形成七元环中间体Ⅲ控制质子转移过程, 最后得到高对映选择性的产物.该研究显示了在含有质子转移步骤的有机反应中, 手性质子梭催化剂对于手性控制的独特优势, 凸显其在不对称合成中的广阔应用前景.
2016年, 周其林课题组[37]进一步使用Rh2(TPA)4和手性螺环磷酸19协同催化N—H插入反应, 实现了首例α-烯基重氮酯类化合物20对氨基甲酸叔丁酯N—H键的高对映选择性插入反应, 反应过程中避免了重氮烯基部分发生环丙烷化和重排等副反应.手性质子梭催化剂通过叶立德中间体质子转移过程的直接控制实现了高效的不对称诱导, 显著提高了收率.该反应催化剂用量低活性高(转化频率>600/h), 副反应少, 底物普适性广, 收率可达99%, ee值可达98%, 成为合成α-烯基-α-氨基酸及其衍生物最高效的方法之一.作者对此方法进行了简单的应用研究, 在Pd/C催化条件下, 产物21与氢气发生加成, 然后在盐酸条件下水解, 高收率获得了具有光学活性的α-氨基酸22 (Scheme 18).

2003年, 胡文浩和Doyle课题组[38]合作报道了利用亚胺作为亲电试剂, 成功地得到了铵基叶立德对亚胺的亲核加成产物, 证明铵基叶立德是金属卡宾介导的N—H插入反应的活性中间体, 验证了金属卡宾N—H插入反应的分步反应机理.随后, 胡文浩课题组[39]基于亲电试剂捕捉活性铵基叶立德多组分反应取得一系列显著成果.
2014年, 胡文浩课题组[39d]报道了高效构建手性γ-硝基-α-氨基琥珀酸酯类化合物的三组分反应, 该方法使用Rh[(C2H4)2Cl]2与手性双烯配体23为催化剂, 以芳基重氮酯、芳香胺和β-硝基丙烯酸酯类化合物为底物, 在温和的反应条件下, 高非对映选择性和高对映选择性制备具有两个连续的手性中心的γ-硝基-α-氨基琥珀酸酯类化合物, 反应的收率中等至良好, 相对于传统的合成方法[40]此方法具有原子经济性和步骤经济性的特点, 作者对此反应进行了简单的合成应用的研究, 合成了具有药物活性基团的3-氨基吡咯烷酮类衍生物24 (Scheme 19).

2016年, Moody课题组[41]报道了过渡金属Rh(Ⅱ)、Cu(Ⅰ)、Fe(Ⅲ)催化卡宾介导的N—H键插入反应, 进而发生分子内的Aldol反应, 高非对映选择性合成多取代吡咯烷.反应以α-重氮酯类化合物和β-氨基酮类化合物为原料, 二氯甲烷为溶剂, 过渡金属Fe(TPP)Cl、Rh(esp)2或(CuOTf)2·toluene为催化剂, 在加热回流条件下, 高非对映选择性合成多取代吡咯烷.终产物中未监测到N—H键插入产物, 反应收率中等至优秀, 催化剂用量低, 官能团兼容性良好以及底物普适性广泛(Scheme 20).
在过去的十几年中, 氢键供体型有机小分子催化剂如脲和硫脲类化合物, 在反应中可以通过形成氢键来活化反应底物或稳定过渡态的阴离子, 进而起到催化作用, 越来越多的人将氢键供体型有机小分子催化剂用于各类有机合成反应中.目前, 使用有机小分子脲类化合物[42]催化N—H插入反应已经实现.
2012年, Mattson课题组[42a]通过利用小分子脲类化合物催化剂25形成的氢键作用对α-硝基重氮化合物26起到活化作用, 促进重氮酯的分解和实现N—H键的插入反应.利用氢键供体小分子催化剂实现了α-硝基重氮酯类化合物、苯胺衍生物和亲核试剂三组分反应, 在温和反应条件下, 可以高收率合成α-氨基酸酯类衍生物27 (Scheme 21).

反应可能的机理如Scheme 22所示.首先, 小分子脲催化剂与α-硝基重氮酯通过非共价键作用形成中间体Ⅰ, 然后脱去一分子氮气形成氢键供体稳定的卡宾中间体Ⅱ, 与苯胺发生N—H插入反应形成叶立德中间体Ⅲ, 硝基基团离去形成亚胺离子中间体Ⅳ, 亚胺离子最后和亲核试剂发生反应, 得到终产物.

利用环境友好的生物大分子如酶和蛋白质等催化α-羰基重氮化合物对N—H键的插入反应具有反应条件温和, 可以利用蛋白的空间构型控制反应的立体选择性和减少副反应和产物中可避免重金属残余等优点, 因此得到科研工作者的青睐.
2014年, Arnold课题组[43]报道了将来源于Bacillus megaterium的细胞色素P450酶(P450-BM3)进行定点突变后应用于催化卡宾介导的N—H插入反应中, 这是首例酶催化N—H键插入反应, 为N—H插入反应的绿色合成开启了新的天地.反应以水作为溶剂, 在还原剂Na2S2O4和无氧条件下进行反应, 反应收率中等至良好. α-重氮酯不需要采用缓慢加料的方式以避免α-重氮酯的二聚副反应, 与伯胺N—H键的插入反应的产物高选择性得到一次插入产物.反应产物单一的原因可能是酶蛋白的空间构型控制了反应的选择性, 同时又限制了胺类化合物的底物结构类型(吗啉、苄胺等不反应).反应可能的机理如Scheme 23所示.
2015年, Fasan课题组[44]对来源于抹香鲸(sperm whale)的肌红蛋白进行定向突变, 得到的突变肌红蛋白能够催化重氮乙酸乙酯与芳香胺发生分子间N—H插入反应, 相比Arnold课题组报道的细胞色素P450-BM3, 突变后的肌红蛋白的催化效率更高, 反应产物单一, 收率可达到99%, 并没有观察到二次插入产物和α-重氮酯的二聚副反应(Scheme 24).底物普适性研究表明:苯胺类化合物的苯环上的取代基电子效应对反应收率影响不大, 但与脂肪胺的反应效果很差.这是首例肌红蛋白突变体应用于催化N—H键的插入反应.
2015年, 潘远江课题组[45]开发了在水中血红素-环糊精体系催化α-重氮酯对芳香胺N—H键的插入反应, 反应条件温和, 收率中等至优秀(Scheme 25).底物普适性研究发现对于绝大多数的脂肪胺和水溶性胺都不反应, 因此作者提出了一个猜想:环糊精在该反应体系中相当于一个微反应器, 将水溶性差的反应物容纳在疏水性的腔体中, 为N—H插入反应提供条件.作者同时考察了环糊精腔体大小(α-环糊精、β-环糊精、γ-环糊精)对反应的影响, 发现α-环糊精更倾向于产生一次插入产物, 疏水腔体更大的环糊精一次插入产物产率减低, 二次插入产物产率升高, 由此证明了环糊精腔体大小会对反应产生影响.

反应可能的机理如Scheme 25所示.首先, 重氮化合物在氯化血红素的催化作用下, 脱去一分子氮气, 亲电加成到血红素中的铁原子上, 形成血红素卡宾复合物中间体, 苯胺的氮原子发生亲核进攻, 形成相应的叶立德, 继而发生叶立德重排得到终产物.
α-羰基重氮化合物在光、热等条件下可以脱去氮气, 进而发生N—H插入反应, 促使光和热条件下实现α-羰基重氮化合物对N—H键的插入反应成为研究热点, 近年来也取得一些优秀成果.
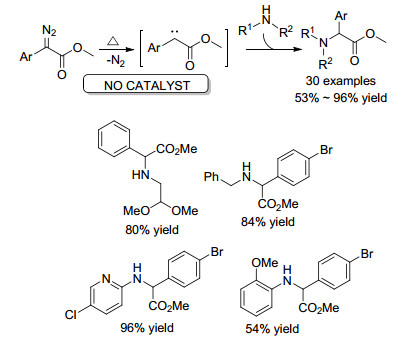
2012年, Davies课题组[46]报道了直接在加热回流条件下, 芳基重氮酯会失去一分子氮气, 形成给体/受体卡宾, 与胺类化合物进行N—H插入反应, 得到各种α-氨基酸酯衍生物.反应溶剂选择了对高反应活性的卡宾中间体表现为惰性的三氟甲苯, 收率可达96%, 底物普适性广, 对脂肪胺和芳香胺N—H键的插入反应效果都较好(Scheme 26).
2018年, Davies课题组[47]发现蓝光(460~490nm)可以促进芳基重氮酯发生光解失去一分子氮气, 从而实现芳基重氮酯对胺类化合物N—H键的插入反应(Scheme 27).反应条件温和, 无需惰性气体保护, 反应收率中等至优秀, 底物普适性广泛.

可能的反应机理如Scheme 28所示, 首先, 芳基重氮酯在光的激发下形成高能的单线态中间体Ⅰ, 然后失去氮气形成单线态的卡宾中间体Ⅱ, 与胺类化合物反应得到终产物, 其中也无法排除高能单线态中间体直接与胺类化合物反应, 失去氮气得到终产物.

主要对近十年来过渡金属催化、有机小分子催化、生物大分子催化以及光和热条件下实现α-羰基重氮化合物对N—H键的插入反应所取得的重要成果进行简单综述.目前α-羰基重氮化合物对N—H键的插入反应可以很好实现高对映选择性α-氨基酸衍生物制备、天然核苷酸的烷基化修饰, 具有生物活性的含氮杂环化合物的合成及其它有价值的应用.此类反应同时存在一些局限之处, 主要包括: (1)目前应用最为广泛的催化剂为价格昂贵的过渡金属催化剂, 环境友好、价格较便宜的非金属催化剂有待进一步开发; (2)胺类底物比较受限, 对胺类化合物N—H键的插入反应, 底物普适性主要适用于芳香胺类化合物; (3)应用于高对映选择性N—H键插入反应的手性配体较少和对生物大分子(核苷酸和肽链等)N—H键的插入反应研究报道较少.总体而言, α-羰基重氮化合物对N—H键的插入反应的研究领域充满活力, 未来仍然会得到重要发展, 并用于构筑各类具有应用价值的C—N键化合物.
(a) Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley-Inter- science, New York, 1998.
(b) Zhao, X.; Zhang, Y.; Wang, J. Chem. Commun. 2012, 48, 10162.
(c) Zhu, S.-F.; Zhou, Q.-L. Nat. Sci. Rev. 2014, 1, 580.
(d) Gillingham, D.; Fei, N. Chem. Soc. Rev. 2013, 42, 4918.
(e) Ford, A.; Miel, H.; Ring, A.; Slattery, C. N.; Maguire, A. R.; Mckervey, M. A. Chem. Rev. 2015, 115, 9981.
(f) Zhang, Z.; Wang, J. Chem. Commun. 2009, 5350.
(g) Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704.
(h) Zhu, S.-F.; Zhou, Q.-L. Acc. Chem. Res. 2012, 45, 1365.
(i) Candeias, N.; Paterna, R.; Gois, P. M. P. Chem. Rev. 2016, 116, 2937.
(j) Ren, Y.-Y.; Zhu, S.-F.; Zhou, Q.-L. Org. Biomol. Chem. 2018, 16, 3087.
(k) Zhang, D.; Hu, W. H. Chem. Rec. 2017, 17, 739.
(l) Wang, J. Chin. J. Org. Chem. 2001, 21, 980 (in Chinese).
(王剑波, 有机化学, 2001, 21, 980.)
(a) Pelphrey, P.; Hansen, J.; Davies, H. M. L. Chem. Sci. 2010, 1, 254.
(b) Sambasivanand, R.; Ball, Z. T. Angew. Chem., Int. Ed. 2012, 51, 8568.
(c) Adly, F. G.; Gardiner, M. G.; Ghanem, A. Chem.-Eur. J. 2016, 22, 3447.
(d) Qin, C.; Boyarskikh, V.; Hansen, J. H.; Hardcastle, K. I.; Musaev, D. G.; Davies, H. M. L. J. Am. Chem. Soc. 2011, 133, 19198.
(e) Xu, H.; Li, Y.-P.; Cai, Y.; Wang, G.-P.; Zhu, S. F.; Zhou, Q.-L., J. Am. Chem. Soc. 2017, 139, 7697.
(f) Maas, G. Chem. Soc. Rev. 2004, 33, 183.
(a) Padwa, A.; Weingarten, M. D. Chem. Rev. 1996, 96, 223.
(b) Padwa, A.; Hornbuckle, S. F. Chem. Rev. 1991, 91, 263.
(c) Yakura, T.; Ozono, A.; Matsui, K.; Yamashita, M.; Fujiwara, T. Synlett 2013, 24, 65.
(d) Moody, C. J.; Taylor, R. J. Tetrahedron 1990, 46, 6525.
(e) Roberts, E.; Sançon, J. P.; Sweeney, J. B. Org. Lett. 2005, 7, 2075.
(a) Liao, K.; Negretti, S.; Musaev, D. G.; Bacsa, J.; Davies, H. M. L. Nature 2016, 533, 230.
(b) Liao, K.; Pickel, T. C.; Boyarskikh, V.; Bacsa, J.; Musaev D. G.; Davies, H. M. L. Nature 2017, 551, 609.
(c) Qin, C.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 9792.
(a) Wang, J.; Hou, Y.; Wu, P. J. Chem. Soc., Perkin Trans. 1 1999, 2277.
(b) Clapham, B.; Spanka, C.; Janda, K. D. Org. Lett. 2001, 3, 2173.
(c) Matsushita, H.; Lee, S.-H.; Yoshida, K.; Clapham, B.; Koch, G.; Zimmermann, J.; Janda, K. D.Org. Lett. 2004, 6, 4627.
(d) Pavlyuk, O.; Teller, H.; McMills, M. C. Tetrahedron Lett. 2009, 50, 2716.
(a) Tan, F.; Liu, X.; Hao, X.; Tang, Y.; Lin, L.; Feng, X. ACS Catal. 2016, 6, 6930.
(b) Zhang, Y.; Yao, Y.; He, L.; Liu, Y.; Shi, L. Adv. Synth. Catal. 2017, 359, 2754.
(c) Zhu, S.-F.; Cai, Y.; Mao, H.-X.; Xie, J.-H.; Zhou, Q.-L. Nat. Chem. 2010, 2, 546.
(a) Zhang, Y.-Z.; Zhu, S.-F.; Cai, Y.; Mao, H.-X.; Zhou, Q.-L. Chem. Commun. 2009, 5362.
(b) Brunner, H.; Wutz, K.; Doyle, M. P. Monatsh. Chem. 1990, 121, 755.
(a) Neupane, P.; Li, X.; Jung, J. H.; Lee, Y. R.; Kim, S. H. Tetrahedron 2012, 68, 2496.
(b) Zrig, S.; Andrioletti, B.; Rose, E.; Colin, J. Tetrahedron Lett. 2005, 46, 1103.
(c) Dyer, J.; Jockusch, S.; Balsanek, V.; Sames, D.; Turro, N. J. Org. Chem. 2005, 70, 2143.
(d) Dussault, P. H.; Xu, C. Tetrahedron Lett. 2004, 45, 7455.
Yudin, A. K. Catalyzed Carbon—Heteroatom Bond Formation, Wiley-VCH, Weinheim, 2011.
Burtoloso, A. C. B.; Dias, R. M. P.; Bernardim, B. Acc. Chem. Res. 2015, 48, 921. doi: 10.1021/ar500433t
Cama, L. D.; Christensen, B. G. Tetrahedron Lett. 1978, 19, 4233. doi: 10.1016/S0040-4039(01)95189-5
Salzmann, T. N.; Ratcliffe, R. W.; Christensen, B. G.; Bouffard, F. A. J. Am. Chem. Soc. 1980, 102, 6161. doi: 10.1021/ja00539a040
Ratcliffe, R. W.; Salzmann, T. N.; Christensen, B. G. Tetrahedron Lett. 1980, 21, 31. doi: 10.1016/S0040-4039(00)93616-5
Xu, B.; Zhu, S. F.; Zuo, X. D.; Zhang, Z. C.; Zhou, Q. L. Angew. Chem., Int. Ed. 2014, 126, 3994. doi: 10.1002/ange.201400236
(a) Davies, J. R.; Kane, P. D.; Moody, C. J. J. Org. Chem. 2005, 70, 7305.
(b) Bagley, M. C.; Bashford, K. E.; Hesketh, C. L.; Moody, C. J. J. Am. Chem. Soc. 2000, 122, 3301.
(c) Garcia, C. F.; McKervey, M. A.; Ye, T. Chem. Commun. 1996, 1465.
(d) Lee, S.-H.; Yoshida, K.; Matsushita, H.; Clapham, B.; Koch, G.; Zimmermann, J.; Janda, K. D. J. Org. Chem. 2004, 69, 8829.
(e) Liu, K.; Zhu, C.; Min, J.; Peng, S.; Xu, G.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 12962.
(f) Shi, B.; Blake, A. J.; Lewis, W.; Campbell, I. B.; Judkins, B. D.; Moody, C. J. J. Org. Chem. 2010, 75, 152.
Yates, P. J. Am. Chem. Soc. 1952, 74, 5376. doi: 10.1021/ja01141a047
(a) Zhang, X.; Sui, Z. Tetrahedron Lett. 2006, 47, 5953.
(b) Livant, P.; Jie, Y.; Wang, X. Tetrahedron Lett. 205, 46, 2113.
(c) Chanthamath, S.; Thongjareun, S.; Shibatomi, K.; Iwasa, S. Tetrahedron Lett. 2012, 53, 4862.
(d) Wang, Y.; Zhu, S. Org. Lett. 2003, 5, 745.
(e) Vyavahare, V. P.; Chattopadhyay, S.; Puranik, V. G.; Dhavale, D. D. Synlett 2007, 559.
(f) Saito, H.; Uchiyama, T.; Miyake, M.; Anada, M.; Hashimoto, S.; Takabatake, T.; Miyairi, S. Heterocycles 2010, 81, 1149.
(g) Lian, X.; Meng, J.; Han, Z. Org. Lett. 2016, 18, 4270.
(h) Huang, H.; Wang, Y.; Chen, Z.; Hu, W. Adv. Synth. Catal. 2005, 347, 531.
(i) Medvedev, J. J; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640.
(j) Xu, X. F.; Zavalij, P. Y.; Doyle, M. P. Angew. Chem., Int. Ed. 2012, 51, 9829.
Saegusa, T.; Ito, Y.; Kobayashi, S.; Hirota, K.; Jhimizu, T. Tetrahedron Lett. 1966, 7, 6131. doi: 10.1016/S0040-4039(00)70153-5
(a) Ramakrishna, K.; Sivasankar, C. J. Organomet. Chem. 2016, 805, 122.
(b) Ramakrishna, K.; Sivasankar, C. J. Org. Chem. 2016, 81, 6609.
Tishinov, K.; Schmidt, K.; Häussinger, D.; Gillingham, D. G. Angew. Chem., Int. Ed. 2012, 51, 1200 http://www.ncbi.nlm.nih.gov/pubmed/23090851
Li, H.; Cheng, P.; Jiang, L.; Yang, J. L.; Zu, L. S. Angew. Chem., Int. Ed. 2017, 56, 2754. doi: 10.1002/anie.201611830
Tishinov, K.; Schmidt, K.; Häussinger, D.; Gillingham, D. G. Angew. Chem., Int. Ed. 2012, 51, 1200 http://www.ncbi.nlm.nih.gov/pubmed/23090851
(a) Chan, K. H.; Guan, X.; Lo, V. K.; Che, C. M. Angew. Chem., Int. Ed. 2014, 53, 2982.
(b) Ho, C.-M.; Zhang, J.-L.; Zhou, C.-Y.; Chan, O.-Y.; Yan, J. J.; Zhang, F.-Y.; Huang, J.-S.; Che, C.-M. J. Am. Chem. Soc. 2010, 132, 1886.
Aviv, I.; Gross, Z. Chem. Commun. 2006, 4477. http://www.ncbi.nlm.nih.gov/pubmed/17283790
Mangion, I. K.; Nwamba, I. K.; Shevlin, M.; Huffman, M. A. Org. Lett. 2009, 11, 3566. doi: 10.1021/ol901298p
Anding, B. J.; Woo, L. K. Organometallics 2013, 32, 2599. doi: 10.1021/om400098v
Ramakrishna, K.; Sivasankar, C. Org. Biomol. Chem. 2017, 15, 2392 doi: 10.1039/C7OB00177K
Bachmann, S.; Fielenbach, D.; Jørgensen, K. A. Org. Biomol. Chem. 2004, 2, 3044 doi: 10.1039/B412053A
Liu, B.; Zhu, S. F.; Zhang, W.; Chen, C.; Zhou, Q.-L. J. Am. Chem. Soc. 2007, 129, 5834. doi: 10.1021/ja0711765
Lee, E. C.; Fu, G. C. J. Am. Chem. Soc. 2007, 129, 12066. doi: 10.1021/ja074483j
Hou, Z.-R.; Wang, J.; He, P.; Wang, J.; Qin, B.; Liu, X.-H.; Lin, L.-L.; Feng, X.-M. Angew. Chem., Int. Ed. 2010, 49, 4763. doi: 10.1002/anie.201001686
Zhang, Z.-H.; Wang, J.-B. Tetrahedron 2008, 64, 6577. doi: 10.1016/j.tet.2008.04.074
Zhu, S.-F.; Xu, B.; Wang, G.-P.; Zhou, Q.-L. J. Am. Chem. Soc. 2012, 134, 436. doi: 10.1021/ja2084493
(a) Zhang, Y.; Wang, J. Eur. J. Org. Chem. 2011, 1015.
(b) Xiao, Q.; Zhang, Y.; Wang, J. Acc. Chem. Res. 2013, 46, 236.
(c) Devine, S. K. J.; Van Vranken, D. L. Org. Lett. 2007, 9, 2047.
(d) Kudirka, R.; Devine, S. K. J.; Adams, C. S.; Van Vranken, D. L. Angew. Chem., Int. Ed. 2009, 48, 3677.
(a) Liu, G.; Li, J.; Qiu, L.; Liu, L.; Xu, G. Y.; Ma, B.; Sun, J. T. Org. Biomol. Chem. 2013, 11, 5998.
(b) Zhu, Y.; Liu, X.; Dong, S.; Zhou, Y.; Li, W.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2014, 53, 1636.
(c) Arredondo, V.; Hiew, S. C.; Gutman, E. S.; Premachandra, I. D. U. A.; Van Vranken, D. L. Angew. Chem., Int. Ed. 2017, 56, 4156.
Xu, B.; Zhu, S. F.; Xie, X. L.; Shen, J. J.; Zhou, Q. L. Angew. Chem., Int. Ed. 2011, 50, 11483. doi: 10.1002/anie.201105485
Guo, J. X.; Zhou, T.; Xu, B.; Zhu, S. F.; Zhou, Q. L. Chem. Sci. 2016, 7, 1104. doi: 10.1039/C5SC03558A
Wang, Y. H.; Zhu, Y. X.; Chen, Z. Y.; Mi, A. Q.; Hu, W. H.; Doyle, M. P. Org. Lett. 2003, 5, 3923. doi: 10.1021/ol035490p
(a) Jiang, J.; Xu, H.-D.; Xi, J.-B.; Ren, B.-Y.; Lü, F.-P.; Xin, G.; Jiang, L.-Q.; Zhang, Z.-Y.; Hu, W.-H. J. Am. Chem. Soc. 2011, 133, 8428.
(b) Jiang, J.; Ma, X.-C.; Liu, S.-Y.; Qian, Y.; Lü, F.-P.; Qiu, L.; Wu, X.; Hu, W.-H. Chem. Commun. 2013, 49, 4238.
(c) Jiang, L.-Q.; Zhang, D.; Wang, Z.-Q.; Hu, W.-H. Synthesis 2013, 45, 452.
(d) Ma, X.-C.; Jiang, J.; Lü, S.-Y.; Yao, W.-F.; Yang, Y.; Liu, S.-Y.; Xia, F.; Hu, W.-H. Angew. Chem., Int. Ed. 2014, 53, 13136.
(a) Ooi, T.; Kameda, M.; Fuji, J.; Maruoka, K. Org. Lett. 2004, 6, 2397.
(b) Knudsen, K. R.; Jørgensen, K. A. Org. Biomol. Chem. 2005, 3, 1362.
(c) Puglisi, A.; Raimondi, L.; Benaglia, M.; Bonsignore, M.; Rossi, S. Tetrahedron Lett. 2009, 50, 4340.
Nicolle, S. M.; William, L.; Hayes, C. J.; Moody, C. J. Angew. Chem., Int. Ed. 2016, 55, 3749. doi: 10.1002/anie.201511433
(a) So, S. S.; Mattson, A. E. J. Am. Chem. Soc. 2012, 134, 8798.
(b) Auvil, T. J.; So, S. S.; Mattson, A. E. Angew. Chem., Int. Ed. 2013, 52, 11317.
(c) So, S. S.; Oottikkal, S.; Badjić, J. D.; Hadad, C. M.; Mattson, A. E. J. Org. Chem. 2014, 79, 4832.
Wang, Z. J.; Peck, N. E.; Renata, H.; Arnold, F. H. Chem. Sci. 2014, 5, 598. doi: 10.1039/C3SC52535J
Sreenilayam, G.; Fasan, R. Chem. Commun. 2015, 51, 1532. doi: 10.1039/C4CC08753D
Xu, X.; Li, C.; Tao, Z.; Pan, Y. J. Adv. Synth. Catal. 2015, 357, 3341. doi: 10.1002/adsc.201500418
Hansen, S. R.; Spangler, J. E.; Hansen, J. H.; Davies, H. M. L. Org. Lett. 2012, 14, 4626. doi: 10.1021/ol3020754
Jurberg, I. D.; Davies, H. M. L. Chem. Sci. 2018, 9, 5112. doi: 10.1039/C8SC01165F

图式 3 Cu(Ⅰ)-磷配体催化N—H插入反应
Scheme 3 Cu(Ⅰ)-phosphine complexes catalyzed N—H insertion reaction

图式 5 5步反应不对称全合成(-)-goniomitine
Scheme 5 Five-step asymmetric total synthesis of (-)-goniomitine

图式 6 Rh2(OAc)4催化四种脱氧核苷酸的N—H插入反应
Scheme 6 Rh2(OAc)4-catalyzed N—H insertion reaction of a tetradeoxynucleotide

图式 7 [Ru(Por)(NHC)2)]配合物催化N—H插入反应
Scheme 7 [Ru(Por)(NHC)2)] complexes catalyzed N—H insertion reaction

图式 8 铁卟啉催化氨气和醋酸铵N—H键的插入反应
Scheme 8 Iron porphyrin catalyzed N—H insertion of ammonia and ammonium acetate

图式 10 铜配合物催化不对称N—H插入反应
Scheme 10 Copper complexes catalyzed asymmetric N—H insertion reaction

图式 11 CuCl和手性螺环双噁唑啉催化不对称N—H插入反应
Scheme 11 Asymmetric N—H insertion reaction catalyzed by CuCl and chiral spiro bisoxazolines

图式 12 铜催化不对称N—H插入反应可能的机理
Scheme 12 Proposed mechanism for copper-catalyzed asymmetric N—H insertion reaction

图式 13 CuBr和双联吡啶催化不对称N—H插入反应
Scheme 13 Asymmetric N—H insertion reaction catalyzed by CuCl and bipyridine

图式 14 CuCl和联萘酚衍生物催化不对称N—H插入反应
Scheme 14 Asymmetric N—H insertion reaction catalyzed by CuCl and binol derivative

图式 16 Rh2(TPA)4和手性螺环磷酸催化不对称N—H插入反应
Scheme 16 Asymmetric N—H insertion reaction catalyzed by Rh2(TPA)4 and chiral spiro phosphoric acid

图式 17 Rh2(TPA)4和手性螺环磷酸协同催化N—H插入反应的可能机理
Scheme 17 Proposed mechanism for Rh2(TPA)4 and chiral spiro phosphoric acid co-catalyzed N—H insertion reaction

图式 18 N—H插入反应合成高对映选择性的α-烯基-α-氨基酸
Scheme 18 Enantioselective synthesis of α-alkenyl α-amino acids via N—H insertion reaction

图式 19 合成γ-硝基-α-氨基琥珀酸酯类化合物和3-氨基吡咯烷酮类衍生物
Scheme 19 Synthesis of γ-nitro-α-amino-succinates and 3-aminopyrrolidinone derivatives

图式 21 有机小分子催化N—H插入反应
Scheme 21 N—H insertion reaction catalyzed by organic small molecules

图式 22 脲类化合物催化N—H插入反应可能的机理
Scheme 22 Proposed mechanism for Urea-catalyzed N—H insertion reaction

图式 25 血红素催化N—H插入反应可能的机理
Scheme 25 Proposed mechanism for Hemin-catalyzed N—H insertion reaction

图式 27 蓝光促进芳基重氮酯光解实现N—H插入反应
Scheme 27 Blue light-promoted photolysis of aryldiazoacetates to achieve N—H insertion reaction

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:








 下载:
下载:









 下载:
下载: