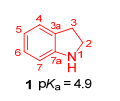
图 1.
二氢吲哚结构图
Figure 1.
Indoline nucleus

二氢吲哚1, 也称为苯并吡咯烷(或2, 3-二氢-1H-吲哚或1-氮杂茚二酮), 是具有双环结构的芳香杂环骨架, 双环结构由五元含氮杂环稠合的六元苯环组成, pKa值为4.9(图 1)[1].
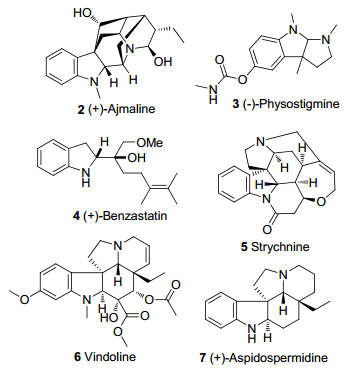
吲哚类化合物广泛存在于自然界中, 很多天然产物和药物分子都含有二氢吲哚的结构骨架(图 2), 二氢吲哚的衍生物具有较好的药理活性和生理活性, 例如在抗菌、抗惊厥、抗肿瘤和抗炎等方面具有较好的活性等[2, 3].具有二氢吲哚骨架的化合物在医药、农药、染料等方面具有良好活性[4].在吲哚化学中, 制备二氢吲哚骨架比合成其他衍生物如吲哚酮更具挑战性[5~7].获得二氢吲哚骨架的主要方法: (1)吲哚的脱芳构化.然而使用非环状化合物制备二氢吲哚类衍生物的实例很少[8]; (2)利用吲哚酮还原二氢吲哚.此方法需要严苛的条件[9].因此对二氢吲哚及其衍生物的合成研究仍具备重要意义.
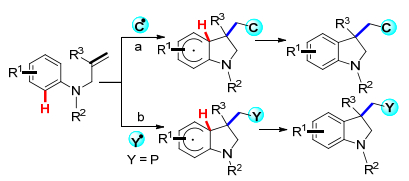
烯烃的双官能化是合成具有生物活性的杂环非常重要的方法[10].特别是, 活性烯烃的邻位双官能化反应是过去几十年中获得分子复杂性的有力手段, 而在未活化的烯烃中同时引入两种官能团的意义同样重要[11].最近, 多个课题组研究使用未活化的烯烃作为自由基受体, 使用铁或银等金属催化剂或在无金属条件下, 通过自由基加成/环化反应, 使未活化的烯烃双官能化来构建二氢吲哚.此方法可以将多种官能团如三氟甲基、多氯甲基、氰基、羰基和磷酰基等引入到二氢吲哚骨架中, 来构建具有各种官能团的二氢吲哚类衍生物.根据烯烃双官能化转化类型, 主要分为两种: (a)在碳-碳双键上加入碳自由基, 然后进行芳基化, 通过烯烃1, 2-碳碳官能化合成二氢吲哚; (b)在碳-碳双键上加入杂原子和碳自由基, 然后进行芳基化, 通过烯烃的1, 2-碳杂官能化合成二氢吲哚(Scheme 1).本综述按照上述烯烃的双官能化的转化类型, 结合各种自由基、反应机理介绍不同的C(3)取代的二氢吲哚分子骨架的构建, 对近年来未活化烯烃通过自由基加成/环化反应合成二氢吲哚的研究进展进行综述.
三氟甲基已被认为是可以增强母体分子的代谢稳定性、亲脂性和生物利用度的重要药效团[12].虽然研究开发将三氟甲基结合到生物活性分子中的方法受到广泛关注, 然而成功的例子却很少见.活化烯烃的三氟甲基化邻位双官能化已被证明是获得分子复杂性的有力策略[13~16], 而3-(2, 2, 2-三氟乙基)二氢吲哚的合成仍然是一项重大挑战, 可能因为未活化的烯烃易于聚合和双键转移.
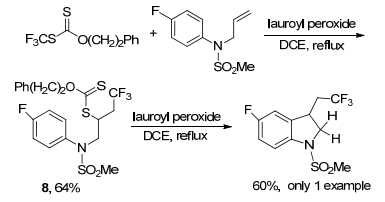
2001年, Zard及其同事[17]报道了在过氧化月桂酰作为引发剂存在下, 在回流的1, 2-二氯乙烷(DCE)中加热黄原酸酯和受保护的N-烯丙基对氟苯胺, 得到中间产物8, 产率为64%.将8暴露在过氧化月桂酰中, 最终得到二氢吲哚的产率为60% (Scheme 2).
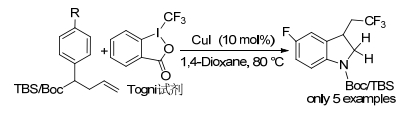
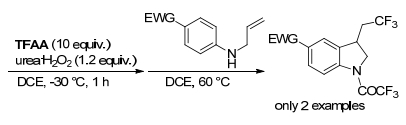
2013年, Sodeoka课题组[18]报道了使用CuI/Togni试剂催化的未活化烯烃的三氟甲基化, 以及独特的1, 6-氧三氟甲基化反应.通过烯烃和芳基之间的轨道相互作用加速反应, 有利于三氟甲基化反应以及分子内C—C键形成.该反应体系被应用于N-Boc-烯丙基苯胺和TBS保护的烯丙基苯胺, 得到对应的三氟甲基化二氢吲哚, 产率分别为85%和87% (Scheme 3). 2017年, 该课题组[19]报道了使用全氟酸酐(TFAA)作为全氟烷基源, 用于N-芳基烯丙胺的三氟烷基化, 得到三氟烷基化的二氢吲哚(Scheme 4).
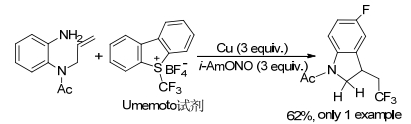
2013年, Fu及其同事[20]研究报道了以铜粉作为催化剂, 使用Umemoto试剂作为CF3源, 使用亚硝酸异戊酯(i-AmONO)进行原位重氮化反应, 得到三氟甲基化的二氢吲哚, 产率为62% (Scheme 5).此反应条件苛刻, 需使用大量的的铜才能顺利完成反应, 而且底物的范围比较窄, 成本较高.
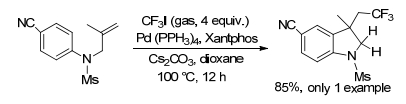
2017年, Cheng课题组[21]描述了有效且高度实用的钯催化未活化的烯烃的芳基全氟烷基化, 得到三氟甲基化二氢吲哚的产率为85% (Scheme 6).该反应温和地进行且没有芳基的电子活化, 具有高通用性, 低成本的氟烷基化来源和良好的官能团相容性.但该反应涉及到有毒气体以及苛刻的反应条件.
尽管利用未活化烯烃为起始物成功地构建了二氢吲哚类化合物, 但是往往反应程序繁琐, 底物范围窄, 成本高, 或反应条件苛刻、毒性等条件.这样不仅导致在底物的兼容性方面有诸多限制, 而且也降低了这类方法的实用性.
2018年, Liang及其同事[22]报道了未活化的烯烃的无金属串联三氟甲基化/芳基化, 以CF3SO2Na作为三氟甲基化剂和N-烯丙基苯胺的未活化烯烃作为自由基受体, 发生三氟甲基烷基化/环化级联反应, 反应以高收率获得含CF3CH2的二氢吲哚和1, 2, 3, 4-四氢异喹啉(Scheme 7).使用廉价且稳定的固体CF3SO2Na (Lang- lois’试剂)作为三氟甲基化剂.该反应具有条件温和, 操作简便, 成本低和底物范围广的特点.
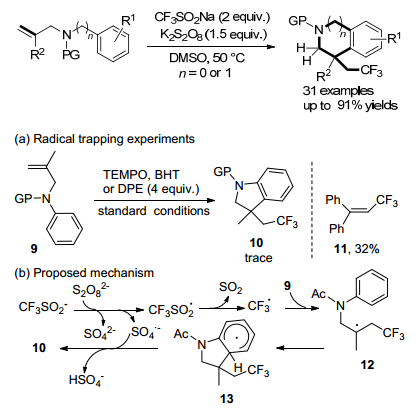
自由基捕获实验表明, 加入4 equiv.的2, 2, 6, 6-四甲基哌啶-1-氧基(TEMPO)和2, 6-二叔丁基-4-甲基苯酚(BHT), 完全抑制标准条件下的模型反应.在DPE实验中, 1, 1-二苯基乙烯(DPE)作为自由基清除剂, DPE-CF3加合物11以32%的产率分离.根据上述结果和文献报告[15, 23], 提出了一种可能机制.最初, CF3SO2Na与K2S2O8反应释放出硫酸盐二价阴离子, 硫酸根阴离子和三氟甲磺酰自由基反应, 释放出一个二氧化硫分子得到三氟甲基自由基, 与N-烯丙基苯胺9的未活化双键反应得到烷基12. 12通过分子内自由基进行环化产生脱芳基化的芳基13.随后13脱氢转移到硫酸根阴离子释放出二氢吲哚10以及硫酸氢根阴离子.
腈类物质在材料、药物和有机合成方面的应用日益增多, 并且已经致力于制备含氰基的吲哚衍生物. 2012年, Liu及其同事[24]使用Pd(OAc)2/PhI(O2Ct-Bu)2/AgF系统将未活化的N-烯丙基苯胺烯烃进行氰基烷基化/环化反应, 以乙腈作为氰化物源生成氰化二氢吲哚(Scheme 8).腈溶剂是理想的氰基烷基化试剂, 但为了激活它们的C—H键, 使用了贵金属钯催化剂和超化学计量的AgF, 以及高价碘试剂.此外, 这种极性反应对腈分子中的空间位阻敏感, 并且不能将二级或三级腈部分引入二氢吲哚骨架.
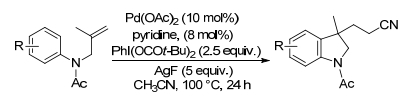
偶氮二异丁腈(AIBN)是聚合物化学[25]和自由基有机合成[26]中最广泛使用的自由基引发剂之一.传统上, 它仅引发自由基过程, 不参与反应, 因为源自AIBN的异丁腈基团通常被认为是无活性的, 部分原因是空间位阻[27].然而, 过去几年, AIBN作为三级腈源可以构建一个α-氰基季碳中心[28]. Li课题组[29]将N-烯丙基苯胺的未活化烯烃发生氰基烷基化/环化级联, 生成3-氰基烷基二氢吲哚(Scheme 9).
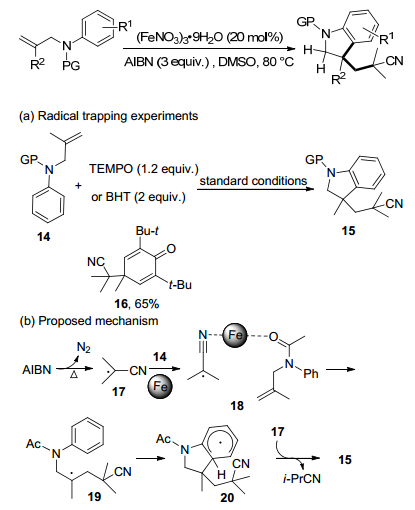
通过对照实验以证实该环化的自由基性质.以1.2 equiv.的2, 2, 6, 6-四甲基哌啶-1-氧基(TEMPO)或2 equiv.的2, 6-二叔丁基-4-甲基苯酚(BHT)作为自由基抑制剂, 几乎完全抑制了模型反应.此外, 氰基异丙基-BHT加合物16以65%的产率分离, 作为BHT实验中的唯一产物.
根据上述结果和以前的报告[28], 提出了一种可能的机制.最初, AIBN的热分解释放出一个氮分子, 得到异丁腈自由基17. 17进攻N-烯丙基苯胺14的双键, 与18和铁络合形成中间产物19, 随后19闭环得到中间体20. 20的氢原子转移到17, 释放出二氢吲哚产物15和异丁腈.
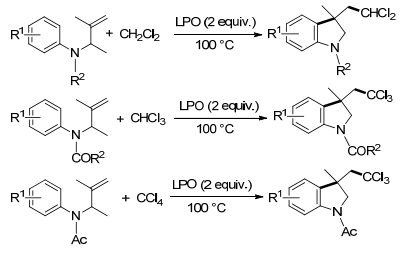
含卤素的化合物在天然产物和合成的有机化合物中广泛存在, 并表现出多种生物活性[30].其中, 具有二氯或三氯甲基的多卤化合物存在于许多具有生物活性的天然产物, 、药物以及农药中[31]. Pan课题组[32]报道了使用二氯甲烷、氯仿和四氯甲烷作为多氯甲基源, 在无金属和无添加剂条件下, 利用双官能化未活化的烯烃来合成二氯和三氯甲基化二氢吲哚(Scheme 10).
通过对照实验来研究反应机理(Scheme 11).分子内和分子间竞争动力学同位素效应(KIE)实验表明. CH2Cl2与CD2Cl2的分子间KIE (kH/kD=6.7)差异显著, 在CH2Cl2和CD2Cl2的单独反应中KIE kH/kD=5.2.结果表明, 该反应的速率决定步骤是DCM中sp3-C—H键的裂解, 而不是芳基中sp2-C—H键的裂解[33].
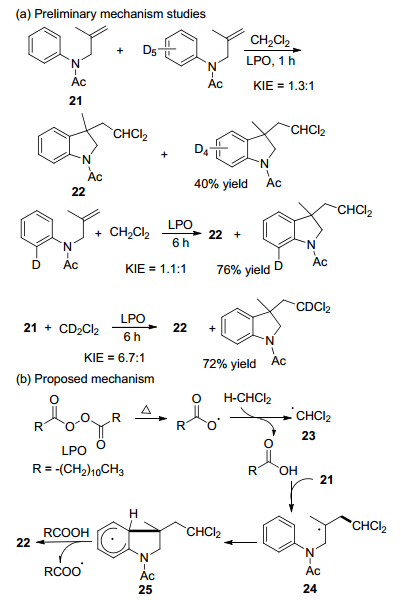
基于以上结果, 提出了反应机理.首先LPO均裂解产生月桂酰基, 然后, 月桂酰基从CH2Cl2中夺去氢原子产生二氯甲基自由基23, 23进攻N-烯丙基苯胺21的双键得到自由基中间体24, 24分子内环化产生自由基中间体25.最后, 夺氢产生二氯甲基化的二氢吲哚产物22.
羰基C(sp2)—H键是高度反应性的化学键, 广泛用于合成[34], 含羰基的二氢吲哚是药物和天然产物中的常见结构骨架, 也是有机合成中的通用中间体.
2018年, Pan课题组[35]研究报道了不含金属和酸的N-烯丙基苯胺与丙酮的氧化加成/环化, 产生一系列3-(3-氧代丁基)官能化的二氢吲哚.该反应的特征在于容易产生与未活化的双键反应的酮基, 得到3-(3-氧代丁基)二氢吲哚, 是天然产物和药物的重要结构骨架(Scheme 12).
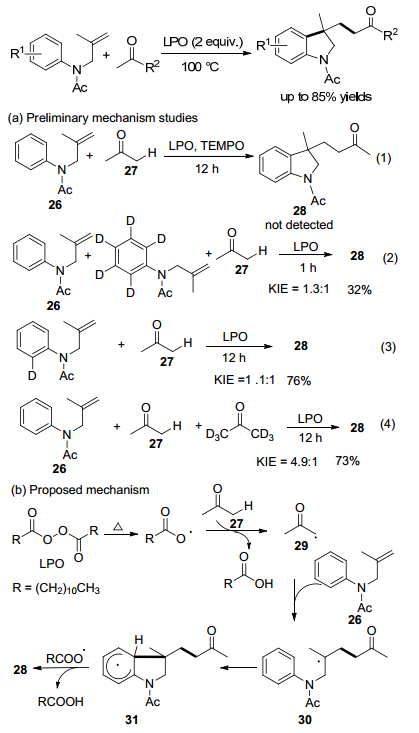
通过对照实验以研究反应机理, 添加自由基清除剂TEMPO成功地抑制了模型反应.分子间动力学同位素实验表明, 芳烃C—H键的kH/kD为1.3和1.1, 说明自由基中间体参与反应, 并且芳烃C—H键的裂解不参与速率确定步骤.另一方面, 在该过程中观察到丙酮的大的动力学同位素效应, 表明丙酮中sp3-C—H键的裂解参与该转化的速率确定步骤.
可能的机理如Scheme 12所示.首先, 29均裂产生的月桂酰自由基, 与丙酮27的α-H形成丙酮自由基29.然后, 29与N-烯丙基苯胺26发生分子间加成产生自由基中间体30.分子内自由基环化得到中间体31, 其经历氧化脱氢得到产物28.
含磷有机化合物广泛存在于药物和农用化学品中; 吲哚基和磷基的结合产生了一系列具有抗癌活性[36]、PET显像[37]、抗艾滋病毒[38]、抗病毒[39]、抗分枝杆菌[40]等化合物[41]和线粒体靶向制剂[42]等.这些益处推动了生成含磷吲哚啉的新方法的发展.
自由基亲核取代或SRN1反应是具有自由基和自由基阴离子作为中间体的链过程, 该链式过程需要引发剂.最常用的引发方法是液氨中的碱金属化学引发, 阴极的电化学引发和光引发. Rossi研究小组[43]于2002年报道了使用Ph2P-作为亲核试剂, 通过自由基亲核取代机理, 由邻位官能化的卤代芳基化合物成功制备3-位被磷酰基取代的二氢吲哚(Scheme 13).
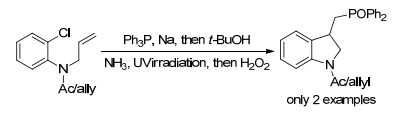
2003年, Zard及其同事[44]报道了黄原酸的自由基交换机理.用少量过氧化物作为引发剂, 黄原酸酯与未活化烯烃直接合成官能化的偕二膦酸酯.在回流条件下于1, 2-二氯乙烷中加入偕二膦酸酯与过氧化物, 在过氧化物的诱导作用下发生自由基加成/环化反应, 得到相应的二氢吲哚, 产率62% (Scheme 14).
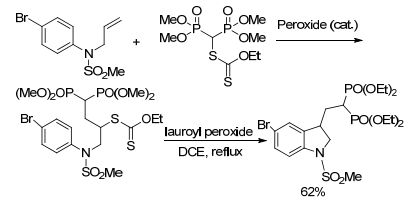
2010年, Curran小组[45]报道了邻碘苯胺在自由基条件下与Me3SnPPh2反应, 经历芳基自由基磷酰化、氧化得到磷酰基化二氢吲哚衍生物, 收率为70%. 2011年, 该课题组[46]用Me3SnPPh2和引发剂V-40在苯中处理二碘酰苯胺.为了简化分析和产物分离, 将反应混合物用S8处理, 得到了双硫化膦二氢吲哚衍生物(Scheme 15).
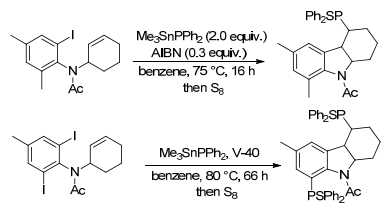
随着对未活化烯烃双官能化的深入研究, Liang课题组[47]开发了一种银催化的N-烯丙基苯胺的磷酸化自由基加成/环化反应, 直接获得3-膦酰基烷基二氢吲哚, 未活化的烯烃用作自由基受体, H-膦酸盐或H-膦氧化物用作自由基前体.该方案操作简单、底物范围广、外部选择性高、很容易进行大规模合成(Scheme 16).
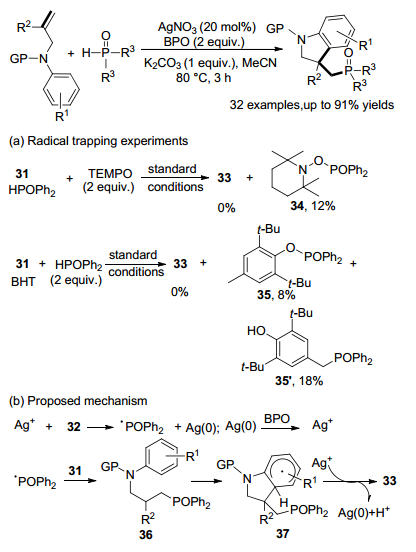
通过一些对照实验以探测反应机理.在标准条件下, 通过加入TEMPO或BHT作为自由基清除剂, 模型反应被完全抑制.基于上述观察和先前的报告[48], 提出了可能的机制.开始, 膦酸盐或膦氧化物32与AgNO3反应得到磷酰自由基, 磷酰自由基与N-烯丙基苯胺31双键反应, 形成具有C—P键的自由基中间体36, 36的苯环通过分子内自由基进行环化得到闭环中间体37, 37与Ag+发生单电子转移(SET)释放出二氢吲哚产物33、质子和Ag(0).最后通过BPO将Ag(0)氧化成Ag+以完成催化性循环.
在过去的几年中, 使用未活化的烯烃作为自由基受体, 通过自由基加成/环化反应合成二氢吲哚及其衍生物的反应策略取得了很大进展.这些新方法由碳或杂原子基团引发, 容易将许多官能团(包括烷基、羰基、三氟甲基、二氯甲基、氰基和磷酰基)引入到二氢吲哚骨架中.鉴于反应底物N-芳基丙烯酰胺具有廉价、易于制备和储存等特点, 该类型的反应必将在医药中间体、功能性有机材料等的合成中得到更加广泛的应用.
尽管在使用自由基加成/环化反应合成二氢吲哚方面取得了重大进展, 但在未活化烯烃的双官能化转化过程中, 对非绿色氧化剂的依赖性非常突出(如过氧化物、过硫酸钾、高价碘或叔丁基).因此, 开发涉及使用绿色氧化剂(例如O2过氧化氢或其他可持续氧化剂)的新的有效自由基加成/环化反应策略变得尤为重要.该领域的重要机遇和挑战还将涉及开发更加绿色有效的催化剂和发现更多新的功能基团, 以便进一步研究未开发的自由基加成/环化反应.
Silva, T. S.; Rodrigues, M. T.; Santos, H.; Zeoly, L. A.; Almeida, W. P.; Barcelos, R. C.; Gomes, R. C.; Fernandes, F. S.; Coelho, F. Tetrahedron 2019, 75, 2553. doi: 10.1016/j.tet.2019.03.032
Collins, M. A.; Hudak, V.; Bender, R.; Fensome, A.; Zhang, P.; Miller, L. Med. Chem. Lett. 2004, 14, 2185. doi: 10.1016/j.bmcl.2004.02.054
Tokunaga, T.; Hume, W. E.; Umezone, T.; Okazaki, K.; Ueki, Y.; Kumagai, K. J. Med. Chem. 2001, 44, 4641. doi: 10.1021/jm0103763
(a) Wang, T.; Xu, Q.; Yu, P. Org. Lett. 2001, 3, 345.
(b) Bui, T.; Syed, S.; Barbas, C F. J. Am. Chem. Soc. 2009, 131, 8758.
(c) Toda, N.; Ori, M.; Takami, K.; Tago, K.; Kogen, H. Org. Lett. 2003, 5, 269.
(d) Zhang, H.; Boonsombat, J.; Padwa, A. Org. Lett. 2007, 9, 279.
(e) Langlois, N.; Gueritte, F.; Langlois, Y. J. Am. Chem. Soc. 1976, 98, 7017.
(f) Marino, J. P.; Rubio, M. B.; Cao, G. J. Am. Chem. Soc. 2002, 124, 13398.
(a) Van Order, R. B.; Lindwall, H. G. Chem. Rev. 1942, 30, 69.
(b) Taber, D. F.; Tirunahari, P. K. Tetrahedron 2011, 67, 7195.
(c) Petrini, M. Chem. Eur. J. 2017, 23, 16115.
(d) Leitch, J. A.; Bhonoah, Y.; Frost, C. G. ACS Catal. 2017, 7, 5618.
(a) Kirillova, M. S.; Miloserdov, F. M.; Echavarren, A. M. Org. Chem. Front. 2018, 5, 273.
(b) Song, J.; Chen, D.; Gong, L. Natl. Sci. Rev. 2017, 4, 381.
(c) Corsello, M. A.; Kim, J.; Garg, N. Chem. Sci. 2017, 8, 5836.
(d) Homer, J. A.; Sperry, J. J. Nat. Prod. 2017, 80, 2178.
(a) Singh, A. K.; Raj, V.; Saha, S. Eur. J. Med. Chem. 2017, 142, 244.
(b) Sravanthi, T. V.; Manju, S. L. Eur. J. Pharm. Sci. 2016, 91, 1.
(c) Sugimoto, S.; Naganuma, M.; Kanai, T. J. Gastroenterol. 2016, 51, 853.
(d) Megna, B. W.; Carney, P. R.; Nukaya, M.; Geiger, P.; Kennedy, G. D. J. Surg. Res. 2016, 204, 47.
(a) Zheng, C.; Zheng, S. Y. Chem 2016, 1, 830.
(b) Liang, X.; Zheng, C.; You, S. Chem.-Eur. J. 2016, 22, 11918.
(c) Roche, S. P.; Tendoung, J. Y.; Treguier, B. Tetrahedron 2015, 71, 3549.
(d) Ding, Q.; Zhou, X.; Fan, R. Org. Biomol. Chem. 2014, 12, 4807.
(a) Wang, L.; Li, S.; Blmel, M.; Puttreddy, R.; Peuronen, A.; Rissanen, K.; Enders, D. Angew. Chem., Int. Ed. 2017, 56, 8516.
(b) Zhao, B.; Du, D.; Chem. Commun. 2016, 52, 6162.
(c) Kayal, S.; Mukherjee, S.Org. Biomol. Chem. 2016, 14, 10175.
(d) Du, D.; Jiang, Y.; Xu, Q.; Tang, X.; Shi, M. ChemCatChem 2015, 7, 1366.
For selected reviews on difunctionalization of alkenes, see:
(a) Kolb, H. C.; Van Nieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483.
(b) Beccalli, E. M.; Broggini, G.; Martinelli, M.; Sottocornola, S. Chem. Rev. 2007, 107, 5318.
(c) Jacques, B.; Muniz, K. In Catalyzed Carbon-Heteroatom Bond Formation, Ed.: Yudin, A. K., Wiley-VCH, Weinheim, 2010, 119.
(d) McDonald, R. I.; Liu, G.; Stahl, S. S. Chem. Rev. 2011, 111, 2981.
(e) Chen, J.-R.; Yu, X.-Y.; Xiao, W.-J. Synthesis 2015, 47, 604.
(f) Sun, K.; Luan, B.; Liu, Z.; Zhu, J.; Du, J.; Bai, E.; Fang, Y.; Zhang, B. Org. Bimol. Chem.2019, 17, 4208.
(g) Sun, K.; Wang, S.; Feng, R.; Zhang, Y.; Wang, X.; Zhang, Z.; Zhang, B. Org. Lett. 2019, 21, 2052.
(h) Sun, K.; Shi, Z.; Liu, Z.; Luan, B.; Zhu, J.; Xue, Y. Org. Lett. 2018, 20, 6687.
(a) Lan, X.; Wang, N.; Xing, Y. Eur. J. Org. Chem. 2017, 5821.
(b) Bag, R.; De, P. B.; Pradhan, S.; Punniyamurthy, T. Eur. J. Org. Chem. 2017, 5424.
(c) Romero, R. M.; Wçste, T. H.; MuÇiz, K. Chem. Asian J. 2014, 9, 972.
(d) Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076.
(a) Smart, B. E. Chem. Rev. 1996, 96, 1555.
(b) Shimizu, M.; Hiyama, T. Angew. Chem., Int. Ed. 2005, 44, 214.
(c) Muller, C. K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881.
(d) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320.
(e) Wang, F.; Wang, D.-H.; Mu, X.; Chen, P.-H.; Liu, G.-S. J. Am. Chem. Soc. 2014, 136, 10202.
For recent reviews on trifluoromethylation of alkenes, see:
(a) Merino, E.; Nevado, C. Chem. Soc. Rev. 2014, 43, 6598.
(b) Egami, H.; Sodeoka, M. Angew. Chem. Int. Ed. 2014, 53, 8294.
For selected examples, see:
(a) Li, L.; Gu, Q.; Wang, N.; Song, P.; Li, Z.; Li, X.; Wang, F.; Liu, X. Chem. Commun. 2017, 53, 4038.
(b) Koike, T.; Akita, M. Acc. Chem. Res. 2016, 49, 1937.
(c) Ye, J.; Song, L.; Zhou, W.; Ju, T.; Yin, Z.; Yan, S.; Zhang, Z.; Li, J.; Yu, D. Angew. Chem., Int. Ed. 2016, 55, 10022.
(d) Liu, C.; Lu, Q.; Huang, Z.; Zhang, J.; Liao, F.; Peng, P.; Lei, A. Org. Lett. 2015, 17, 6034.
(e) Yang, B.; Xu, X.; Qing, F. Org. Lett. 2015, 17, 1906.
Recent examples for trifluoromethylative alkene difunctionalization of N-arylacrylamides:
(a) Kawamura, S.; Sodeoka, M. Angew. Chem., Int. Ed. 2016, 55, 8740.
(b) Guo, J.; Wu, R.; Jin, J.; Tian, S. Org. Lett. 2016, 18, 3850.
(c) Liu, C.; Zhao, W.; Huang, Y.; Wang, H.; Zhang, B. Tetrahedron 2015, 71, 4344.
Synthesis of CF3CH2-containing oxindoles from acryl sulfonamides:
(a) Zheng, L.; Yang, C.; Xu, Z.; Gao, F.; Xia, W. J. Org. Chem. 2015, 80, 5730.
(b) Li, L.; Deng, M.; Zheng, S.; Xiong, Y.; Tan, B.; Liu, X. Org. Lett. 2014, 16, 504.
(c) Kong, W.; Casimiro, M.; Merino, E.; Nevado, C. J. Am. Chem. Soc. 2013, 135, 14480.
Bertrand, F.; Pevere, V.; Quiclet-Sire, B.; Zard, S. Z. Org. Lett. 2001, 3, 1069. doi: 10.1021/ol0156446
Egami, H.; Shimizu, R.; Kawamura, S.; Sodeoka, M. Angew. Chem., Int. Ed. 2013, 52, 4000. doi: 10.1002/anie.201210250
Kawamura, S.; Dosei, K.; Valverde, E.; Ushida, K.; Sodeoka, M. J. Org. Chem. 2017, 82, 12539. doi: 10.1021/acs.joc.7b02307
Dai, J.; Fang, C.; Xiao, B.; Yi, J.; Xu, J.; Liu, Z.; Lu, X.; Liu, L.; Fu, Y. J. Am. Chem. Soc. 2013, 135, 8436. doi: 10.1021/ja404217t
Zheng, J.; Chen, P.; Yuan, Y.; Cheng, J. J. Org. Chem. 2017, 82, 5790. doi: 10.1021/acs.joc.7b00598
Liang, D.; Dong, Q.; Xu, P.; Dong, Y.; Li, W.; Ma, Y. J. Org. Chem. 2018, 83, 11978. doi: 10.1021/acs.joc.8b01861
(a) Yang, B.; Xu, X.; Qing, F. Chin. J. Chem. 2016, 34, 465.
(b) Huang, H.; Yan, H.; Gao, G.; Yang, C.; Xia, W. Asian J. Org. Chem. 2015, 4, 674.
(c) Lu, Q.; Liu, C.; Huang, Z.; Ma, Y.; Zhang, J.; Lei, A. Chem. Commun. 2014, 50, 14101.
(a) Wu, T.; Mu, X.; Liu, G. Angew. Chem., Int. Ed. 2011, 50, 12578.
(b) Zhang, H.; Chen, P.; Liu, G. Synlett 2012, 23, 2749.
Krys, P.; Matyjaszewski, K. Eur. Polym. J. 2017, 89, 482. doi: 10.1016/j.eurpolymj.2017.02.034
Studer, A.; Curran, D. P. Angew. Chem. Int. Ed. 2016, 55, 58. doi: 10.1002/anie.201505090
Recent examples see:
(a) Li, Z.; Yu, H.; Bolm, C. Angew. Chem., Int. Ed. 2017, 56, 9532.
(b) Sato, M.; Azuma, H.; Daigaku, A.; Sato, S.; Takasu, K.; Okano, K.; Tokuyama, H. Angew. Chem., Int. Ed. 2017, 56, 1087.
(c) Ren, S.; Zhang, F.; Qi, J.; Huang, Y.; Xu, A.; Yan, H.; Wang, Y. J. Am. Chem. Soc. 2017, 139, 6050.
(d) Gharpure, S. J.; Shelke, Y. G. Org. Lett. 2017, 19, 5022.
(e) Zhang, H.; Ma, S.; Xing, Z.; Liu, L.; Fang, B.; Xie, X.; She, X. Org. Chem. Front. 2017, 4, 2211.
(f) Liu, X.; Luo, X.; Wu, Z.; Cui, X.; He, Y.; Huang, G. J. Org. Chem. 2017, 82, 2107.
(a) Irudayanathan, F. M.; Lee, S. Org. Lett. 2017, 19, 2318.
(b) Liu, B.; Wang, C.; Hu, M.; Song, R.; Chen, F.; Li, J. Chem. Commun. 2017, 53, 1265.
(c) Song, W.; Yan, P.; Shen, D.; Chen, Z.; Zeng, X.; Zhong, G. J. Org. Chem. 2017, 82, 4444.
(d) Lan, X.; Wang, N.; Bai, C.; Lan, C.; Zhang, T.; Chen, S.; Xing, Y. Org. Lett. 2016, 18, 5986.
(e) Deng, Y.; Tang, S.; Ding, G.; Wang, M.; Li, J.; Li, Z.; Yuan, L.; Sheng, R. Org. Biomol. Chem. 2016, 14, 9348.
(f) Gao, B.; Xie, Y.; Yang, L.; Huang, H. Org. Biomol. Chem. 2016, 14, 2399.
(g) Xie, Y.; Guo, S.; Wu, L.; Xia, C.; Huang, H. Angew. Chem., Int. Ed. 2015, 54, 5900.
(h) Rong, G.; Mao, J.; Zheng, Y.; Yao, R.; Xu, X. Chem. Commun. 2015, 51, 13822.
Li, Y.; Chang, Y.; Li, Y.; Cao, C.; Yang, J.; Wang, B.; Liang, D. Adv. Synth. Catal. 2018, 360, 2488. doi: 10.1002/adsc.201800296
(a) Gribble, G. W. Acc. Chem. Res. 1998, 31, 141.
(b) Vaillancourt, F. H.; Yeh, E.; Vosburg, D. A.; Garneau-Tsodikova, S.; Walsh, C. T. Chem. Rev. 2006, 106, 3364.
(c) Wang, J.; Sánchez-Roselló, M.; Acena, J. L.; Pozo, C. del; Sorochinsky, A. E.; Fustero, S.; Soloshonok V. A.; Liu, H. Chem. Rev. 2014, 114, 2432.
(d) Paul C.; Pohnert, G. Nat. Prod. Rep. 2011, 28, 186.
(e) Latham, J.; Brandenburger, E.; Shepherd, S. A.; Menon, B. R. K.; Micklefield, J. Chem. Rev. 2018, 118, 232.
(a) Sitachitta, N.; Rossi, J.; Roberts, M. A.; Gerwick, W. H.; Fletcher M. D.; Willis, C. L. J. Am. Chem. Soc. 1998, 120, 7131.
(b) Owusu-Ansah, E.; Durow, A. C.; Harding, J. R.; Jordan, A. C.; O'Connell S.J.; Willis, C. L. Org. Biomol. Chem. 2011, 9, 265.
(c) Yu, H.-Y.; Bao, L.-J.; Liang, Y.; Zeng, E. Y. Environ. Sci. Technol. 2011, 45, 5245.
(d) Wagner, C.; Omari M. E.; Kö nig, G. M. J. Nat. Prod. 2009, 72, 540.
(e) Ardá, A.; Soengas, R. G.; Nieto, M. I.; Jiménez, C.; Rodríguez, J. Org. Lett. 2008, 10, 2175.
(f) Orjala, J. O.; Gerwick, W. H. J. Nat. Prod. 1996, 59, 427.
(g) Nguyen, V.-A.; Willis, C. L.; Gerwick, W. H. Chem. Commun. 2001, 1934.
(h) Durow, A. C.; Long, G. C.; O'Connell, S. J.; Willis, C. L. Org. Lett. 2006, 8, 5401.
Pan, C.; Gao, D.; Yang, Z.; Wu, C.; Yu, J. Org. Biomol. Chem. 2018, 16, 5752. doi: 10.1039/C8OB01554F
Simmons, E. M.; Hartwig, J. Angew. Chem., Int. Ed. 2012, 51, 3066. doi: 10.1002/anie.201107334
(a) Chan, C.-W.; Zhou, Z.; Chan, A. S. C.; Yu, W.-Y. Org. Lett. 2010, 12, 3296.
(b) Jia, X.; Zhang, S.; Wang, W.; Luo, F.; Cheng, J. Org. Lett. 2009, 11, 3120.
(c) Tang, B.-X.; Song, R.-J.; Li, J.-H. J. Am. Chem. Soc. 2010, 132, 8900.
(d) Wu, Y.; Li, B.; Mao, F.; Li, X.; Kwong, F. Y. Org. Lett. 2011, 13, 3258.
Pan, C.; Yang, Z.; Gao, D.; Yu, J. Org. Biomol. Chem. 2018, 16, 6035. doi: 10.1039/C8OB01329B
(a) Tercel, M.; Lee, H. H.; Mehta, S. Y.; Youte Tendoung, J.-J.; Bai, S. Y.; Liyanage, H. D. S.; Pruijn, F. B. J. Med. Chem. 2017, 60, 5834.
(b) Yan, J.; Chen, J.; Zhang, S.; Hu, J.; Huang, L.; Li, X. J. Med. Chem. 2016, 59, 5264.
(c) Palmerini, C. A.; Tartacca, F.; Mazzoni, M.; Granieri, L.; Goracci, L.; Scrascia, A.; Lepri, S. Eur. J. Med. Chem. 2015, 102, 403.
(d) Na, Z.; Pan, S.; Uttamchandani, M.; Yao, S. Q. Angew. Chem., Int. Ed. 2014, 53, 8421.
(e) Kandekar, S.; Preet, R.; Kashyap, M.; Mu, R. P.; Mohapatra, P.; Das, D.; Satapathy, S. R.; Siddharth, S.; Jain, V.; Choudhuri, M.; Kundu, C. N.; Guchhait, S. K.; Bharatam, P. V. ChemMedChem 2013, 8, 1873.
(f) Zhao, R. Y.; Erickson, H. K.; Leece, B. A.; Reid, E. E.; Goldmacher, V. S.; Lambert, J. M.; Chari, R. V. J. J. Med. Chem. 2012, 55, 766.
(a) Chansaenpak, K.; Wang, M.; Liu, S.; Wu, Z.; Yuan, H.; Conti, P. S.; Li, Z.; Gabbaï, F. P. RSC Adv. 2016, 6, 23126.
(b) Cai, Z.; Ouyang, Q.; Zeng, D.; Nguyen, K. N.; Modi, J.; Wang, L.; White, A. G.; Rogers, B. E.; Xie, X.; Anderson, C. J. J. Med. Chem. 2014, 57, 6019.
(a) Dousson, C.; Alexandre, F.; Amador, A.; Bonaric, S.; Bot, S.; Caillet, C.; Convard, T.; da Costa, D.; Lioure, M.; Roland, A.; Rosinovsky, E.; Maldonado, S.; Parsy, C.; Trochet, C.; Storer, R.; Stewart, A.; Wang, J.; Mayes, B. A.; Musiu, C.; Poddesu, B.; Vargiu, L.; Liuzzi, M.; Moussa, A.; Jakubik, J.; Hubbard, L.; Seifer, M.; Standring, D. J. Med. Chem. 2016, 59, 1891.
(b) Alexandre, F.; Amador, A.; Bot, S.; Caillet, C.; Convard, T.; Jakubik, J.; Musiu, C.; Poddesu, B.; Vargiu, L.; Liuzzi, M.; Roland, A.; Seifer, M.; Standring, D.; Storer, R.; Dousson, C. B. J. Med. Chem. 2011, 54, 392.
Okon, A.; Matos de Souza, M. R.; Shah, R.; Amorim, R.; da Costa, L. J.; Wagner, C. R. ACS Med. Chem. Lett. 2017, 8, 958. doi: 10.1021/acsmedchemlett.7b00277
Li, M.; Nyantakyi, S. A.; Gopal, P.; Aziz, D. B.; Dick, T.; Go, M. ACS Med. Chem. Lett. 2017, 8, 1165. doi: 10.1021/acsmedchemlett.7b00287
(a) Fricke, J.; Blei, F.; Hoffmeister, D. Angew. Chem., Int. Ed. 2017, 56, 12352.
(b) Hofmann, A.; Heim, R.; Brack, A.; Kobel, H.; Frey, A.; Ott, H.; Petrzilka, T.; Troxler, F. Helv. Chim. Acta 1959, 42, 1557.
Wu, S.; Cao, Q.; Wang, X.; Cheng, K.; Cheng, Z. Chem. Commun. 2014, 50, 8919. doi: 10.1039/C4CC03296A
Vaillard, S. E.; Postigo, A.; Rossi, R. A. J. Org. Chem. 2002, 67, 8500. doi: 10.1021/jo026404m
Gagosz, F.; Zard, S. Z. Synlett 2003, 387.
Bruch, A.; Ambrosius, A.; Fröhlich, R.; Studer, A.; Guthrie, D. B.; Zhang, H.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 11452. doi: 10.1021/ja105070k
Bruch, A.; Fröhlich, R.; Grimme, S.; Studer, A.; Curran, D. P. J. Am. Chem. Soc. 2011, 133, 16270. doi: 10.1021/ja2070347
Liang, D.; Ge, D.; Lü, Y.; Huang, W.; Wang, B.; Li, W. J. Org. Chem. 2018, 83, 4681. doi: 10.1021/acs.joc.8b00450
(a) Xu, J.; Yu, X.; Song, Q. Org. Lett. 2017, 19, 980.
(b) Li, Y.; Sun, M.; Wang, H.; Tian, Q.; Yang, S. Angew. Chem., Int. Ed.2013, 52, 3972.
(c) Zheng, J.; Zhang, Y.; Wang, D.; Cui, S. Org. Lett. 2016, 18, 1768.

图 2 具有二氢吲哚结构的代表性天然生物碱
Figure 2 Representative natural alkaloids with the indoline framework

图式 1 未活化的烯烃通过自由基加成/环化反应双官能化
Scheme 1 Difunctionalization of unactivated alkenes through radical addition/cyclization

图式 2 黄原酸酯和N-烯丙基对氟苯胺的多氟烷基化
Scheme 2 Polyfluoroalkylation of xanthate and N-allyl p-fluoroaniline

图式 3 铜催化的未活化烯烃的三氟甲基化
Scheme 3 Copper-catalyzed trifluoromethylation of unactivated alkenes

图式 5 Umemoto和未活化烯烃的多氟烷基化反应
Scheme 5 Polyfluoroalkylation of Umemoto and unactivated alkenes

图式 6 钯催化的未活化的烯烃的芳基全氟烷基化
Scheme 6 Palladium-catalyzed aryl perfluoroalkylation of unactivated alkenes

图式 7 未活化烯烃的分子内三氟甲基化/芳基化串联反应
Scheme 7 Intramolecular trifluoromethylation/arylation cascade reaction of unactiveted alkenes

图式 8 钯催化的未活化烯烃的氧化芳基烷基化
Scheme 8 Palladium-catalyzed oxidative arylalkylation of unactivated alkenes

图式 9 未活化烯烃和偶氮二异丁腈的自由基氰甲基化/芳基化反应
Scheme 9 Radical cyanomethylation/arylation reaction of unactivated alkenes with azobisisobutyronitrile

图式 10 无金属条件下未活化烯烃的自由基级联氯甲基化反应
Scheme 10 Metal-free radical cascade chloromethylation of unactivated alkenes

图式 11 未活化烯烃的自由基级联/氯甲基化反应的可能机制
Scheme 11 Possible mechanism for radical cascade chloromethylation of unactivated alkenes

图式 12 无金属条件下未活化烯烃与丙酮的氧化自由基级联加成反应
Scheme 12 Metal-free oxidative radical cascade addition reaction of unactivated alkenes with acetone

图式 13 串联环化-SRN1反应合成二氢吲哚
Scheme 13 Syntheses of indolines by tandem ring closure-SRN1 reactions

图式 14 黄原酸酯与未活化烯烃合成磷酰基化二氢吲哚
Scheme 14 Synthesis of phosphorylated indolin by xanthates and unactivated alkenes

图式 15 Me3SnPPh 2进行芳基自由基磷酰化氧化得到二氢吲哚
Scheme 15 Indolin was obtained by aryl radical phosphorylation oxidation of Me3SnPPh2

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:


 下载:
下载: