
图 1.
含芳香醚结构的重要分子
Figure 1.
Important molecules containing aryl ethers

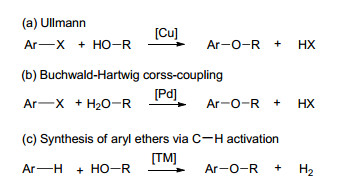
芳香醚骨架广泛存在于天然产物和药物分子中, 比如BMS-777607 (muti-kinase inhibitor, 多激醚抑制剂)、antitumor riccardin C(riccardin C靶向抗癌药)、antithyroid levothyroxine(抗甲状腺左甲状腺素)、Gefitinib (EGFR inhibitor, EGFR抑制剂)以及Vitamin E(维生素E)(图 1)[1].另外, 芳基醚还是重要的有机合成中间体, 通过切断芳基C—O键, 可以引入不同的官能团, 从而实现不同功能有机化合物分子的合成[2].传统的芳香醚类化合物的合成是通过Ullmann反应(Scheme 1, a)实现的, 但是该反应需要等物质的量或过量的铜作为催化剂, 且需要较高的反应温度(200~350 ℃)[3]. Chan-Evans-Lam反应改进了Ullmann反应, 它使用芳基硼酸代替卤代物, 利用催化剂量的铜即可实现芳香醚化合物的制备[4].相对而言, Buchwald-Hartwig交叉偶联反应提供了更为有效的策略, 其通过钯催化的芳基卤代烃与醇或酚的偶联实现芳香醚化合物的制备(Scheme 1, b)[5].最近, MacMillan利用光催化氧化还原的方法, 发展了镍和铱的双金属催化策略, 实现了芳基卤代烃与醇的偶联反应[6].
上述方法中均以含离去基团的芳香烃(芳基卤代烃或金属试剂)作为反应底物, 其需要对芳香烃进行预官能团化, 因此大大降低了芳基醚的合成效率; 另一方面, 离去基团的引入导致了副产物的增加, 且大多数情况下会生成毒性较大的含卤副产物或金属废弃物. C—H键的直接官能化, 即用芳香烃直接与醇或酚偶联, 提供了另一种更为高效绿色的芳香醚合成方法.该方法无需预官能团化引入离去基团, 具有步骤经济性; 且理论上副产物只有氢气, 因此具有较强的原子经济性(Scheme 1, c).近年来尽管有不少关于C—H活化/C—O键偶联方法的进展报道[7], 但基于脱氢偶联制备芳香醚的方法尚未报道.本综述对近几年报道的利用C—H活化策略合成芳香醚的方法进行了总结, 相信这对芳基醚类化合物绿色高效合成方法的发展具有一定的帮助.
目前, 钯金属催化惰性键活化是发展最为成熟的C—H键官能团化方法.为了提高反应的选择性, 往往需要引入一个导向基团, 其配位基团可与钯催化剂络合, 诱导Pd催化剂活化其邻位的C—H键形成环钯中间体(通常为五元或六元环中间体), 环钯化合物经氧化加成、还原消除可得到官能团化产物(Scheme 2).
2006, Sanford课题组[8]首次应用了该策略, 以肟醚或吡咯烷酮作为导向基团, 在Pd催化下实现了邻位C—H甲氧基化, 完成了茴香醚衍生物的制备(Eqs. 1, 2).该课题组设计了四价钯金属催化氧化体系, 使用廉价、安全、环保的硫酸氢钾复合物作为氧化剂.另外甲醇作为甲氧基源, 同时作为反应溶剂, 因此提高了反应效率.
|
|
(1) |
|
|
(2) |
王官武课题组[9]以N-甲氧基酰胺基(CONHOMe)作为导向基团, 在类似的反应条件, 分别以甲醇、乙醇和异丙醇作为偶联试剂实现了其邻位C—H键的烷氧基化(Eq. 3), 扩大了当前方法的潜在合成应用.作者在相同反应条件下尝试了乙酰苯胺邻位C—H甲氧基化反应, 发现其反应效率大幅降低, 仅以30%的收率得到官能化产物.随后, 该课题组[10]通过向反应体系中添加催化量的甲磺酸实现了乙酰苯胺衍生物邻位C—H键的高效烷氧基化反应.反应在室温下即可进行(Eq. 4), 且大量不同的醇包括甲醇、乙醇、正丁醇、仲丁醇、正丙醇、异丙醇、2-甲氧基乙醇、2-氯乙醇、环己醇和环戊醇等均可作为偶联试剂.作者推测甲磺酸在反应中不仅促进了C—H键的活化, 同时也扮演离去基团的角色, 从而提高了反应活性.在该策略的启发下, 匡春香课题组[11]利用相似的反应条件, 以1, 2, 3-三氮唑作为导向基团实现了其邻位的C—H烷氧基化反应.
|
|
(3) |
|
|
(4) |
孙培培课题组[12]以氰基作为导向基团, 在甲醇或乙醇存在下, 实现邻位C—H键的甲氧基化或乙氧基化(Eq. 5).当底物无其他取代基或对位取代时反应发生双C—H官能团化, 而当底物邻位或间位取代时则发生单C—H官能团化.与其他导向基团相比, 氰基则可以更多地衍生化, 比如可还原成胺基或醛基、水解成羧基或环加成得到四氮唑.作者认为氰基作为导向基团是通过π键与钯金属配位形成环钯中间体19.乙酰基参与去质子活化C—H键, 进一步氧化加成、还原消除得到邻位甲氧基化产物.
|
|
(5) |
随后, 孙培培课题使用偶氮基[13]、2-吡啶氧基[14]和N-烷基-N-亚硝基胺[15]为导向基团, 以Pd金属作为催化剂, 二乙酸碘苯(PhI(OAc)2)为氧化剂, 烷基醇作为烷氧化剂和溶剂, 通过邻位C—H键活化构建芳基醚衍生物(Eq. 6).上述的底物均具有较好的官能团耐受性.底物N-烷基-N-亚硝基苯胺可通过铁粉简单处理, 还原生成苯胺衍生物.
|
|
(6) |
Fabis课题组[16]采用N-甲苯磺酰甲酰胺基团作为导向基团, 在类似的反应条件下, 实现邻位C—H烷氧基化. N-卤代琥珀酰亚胺(NXS)代替二乙酸碘苯作为氧化剂时, 醚化反应并没有顺利进行, 而是在导向基团的邻位发生了卤代反应.
史炳锋课题组[17]发展了新型的双齿导向基团2-(2-吡啶基)-2-胺基丙烷基, 通过其邻位C—H键的活化实现苯甲酸衍生物的烷氧基化(Eq. 7).该导向基团具有易制备和易脱除等优良特点.值得注意的是作者利用该导向基团实现了大量脂肪族羧酸衍生物β位的C(sp3)—H烷氧基化.作者通过密度泛函理论(DFT)计算推测C—H键的活化是通过协同金属去质子化过程(CMD)实现的.
|
|
(7) |
2, 3-二氢苯并呋喃广泛存在天然产物和药物分子中[18].通过芳香烃C—H活化并与醇羟基发生分子内偶联, 可快速得到二氢苯并呋喃衍生物. 2010年, 余金权课题组[19]报道了一种钯催化苯乙醇邻位C—H键活化进而实现分子内C—O键构建的方法(Eq. 8), 该方法可快速实现二氢苯并呋喃衍生物的制备.碳酸锂(Li2CO3)在反应中具有重要的作用, 作者认为可能的原因是C—H键活化的前体钯配合物在非碱性条件下不稳定.可能的催化循环如Scheme 3所示:首先羟基导向Pd催化邻位C—H键活化形成六元环钯中间体24, 进而氧化剂将其氧化得到四价钯中间体25, 最后中间体25发生还原消除反应得到二氢苯并呋喃衍生物, 并重新生成钯催化剂完成催化循环.
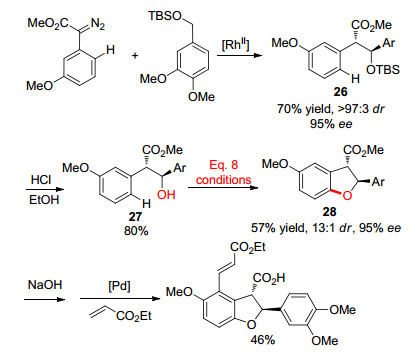
随后, 该课题组[20]通过连续的C—H官能团化反应制备了具有对映选择性的二氢苯并呋喃衍生物.该方法首先是叔丁基二甲基甲硅烷基醚(TBS)保护的苄醇在铑催化下与重氮化合物发生不对称偶联, 生成化合物26.酸性条件下化合物26中的TBS脱保护得到27, 其在与Eq. 8相同的条件下发生分子内环化得到二氢苯并呋喃衍生物28, 产物保留原有的对映选择性(Scheme 4).产物28中的酯基水解成羧基后还可诱导芳香环上邻位的C—H烯基化反应.同年, 余金权课题组[21]将该策略拓展到苯乙酸衍生物的邻位C—H活化反应, 通过分子内环化反应实现了苯并呋喃酮的高效合成.
铜作为一种常见的过渡金属, 具有良好的兼容性, 且价格相对低廉, 因而受到广泛关注, 其催化脱氢偶联构建芳基醚的方法亦有不少报道.王梅祥课题组[22]合成了Azacalixaromatics的铜化合物29, 该化合物在碱1, 8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)存在下可与醇或苯酚发生偶联生成芳基醚衍生物(Eq. 9).
2014年, 宋毛平课题组[23]报道了N, O-二齿导向基团促进的邻位C—H烷氧基化反应, 需要在等物质的量的氯化亚铜(CuCl)存在的条件下进行(Eq. 10).反应具有很好的底物官能团耐受性, 卤素、硝基、醚、烷氧基、酯和磺酰等多种官能团的底物均可顺利发生反应.作者推测铜和氧气作用后与醇反应可产生烷氧自由基EtO•和超氧二价铜ClCuOOH. ClCuOOH和二齿导向基团生成二价铜化合物33, 33和EtO•作用生成三价铜化合物34, 最后34还原消除生成芳基醚产物(Scheme 5).
相比于介导的方法, 金属催化更加绿色环保, 且极大降低反应成本. 2013年Gooβen课题组[24]使用催化量的醋酸铜(Cu(OAc)2)实现了含N导向基团的邻位C—H烷氧基化(Eq. 11).作者认为可能的反应催化循环如Scheme 6所示:三氟甲磺酸银(AgOTf)作为醇的活化试剂, 与醇发生作用生成瞬态烷氧化银38. 38与环铜化合物37发生单电子转移生成三价铜化合物39, 并发生还原消除过程得到芳基醚产物和一价铜Cu(Ⅰ)Ln, 一价铜被氧气氧化重新生成二价铜完成催化循环.
|
|
(11) |
同年, Daugulis等[25]以8-氨基喹啉作为导向基团, 实现了铜催化苯甲酸衍生物邻位的C—H烷氧基化反应(Eq. 12).不同于之前的工作, 该策略以吡啶作为反应溶剂, 而醇仅仅作为偶联试剂, 实现了芳环与多类型醇的偶联.但是该反应策略仅对对三氟甲基取代的苯甲酸衍生物有效.
|
|
(12) |
2015年, 史炳锋课题组[26]利用2-(2-吡啶基)-丙胺(PIP)作为导向基团, 将上述策略拓展应用于更多的芳香底物(Eq. 13).反应不仅适用于不同官能团取代的苯环, 同时也适用于杂环化合物, 如吡啶、哒嗪和噻吩.反应可能的机理如Scheme 7所示:二价铜Cu(Ⅱ)X2首先与导向基团配位生成二价铜化合物42, 接下来42与二价铜催化剂发生歧化反应生成三价铜化合物43和一价铜Cu(Ⅰ)X. 43还原消除生成一价铜化合物44.最后44质子化得到芳香醚产物和Cu(Ⅰ)X. Cu(Ⅰ)X被氧化重新生成Cu(Ⅱ)X2催化剂完成催化循环.
|
|
(13) |
2015年, 宋毛平课题组[27]首次报道了钴催化的芳香烃C—H键烷氧基化反应(Eq. 14).作者对不同的导向基团筛选后发现, 2-氨基-吡啶-N-氧化物作为双齿导向基团时, 反应效率最高.尽管反应的机理尚不明确, 但是作者并未发现反应有动力学同位素效应(KIE, kH/kD≈1.0), 表明芳烃的C—H键断裂不是限速步骤.另一方面, 添加自由基捕捉剂2, 2, 6, 6-四甲基哌啶氮氧化物(TEMPO)会阻止反应.因此作者推测该反应可能经历了自由基过程.
|
|
(14) |
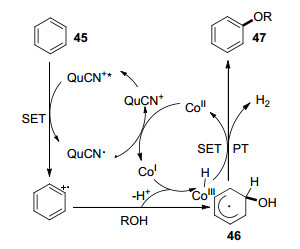
2017年, 佟振合团队[28]发现以醇作溶剂, 苯在光和钴的协同催化下可以发生脱氢偶联生成芳香醚化合物(Eq. 15).该反应在室温下即可发生, 无需额外氧化剂, 氢气为唯一的副产物.在这之前的报道中反应没有钴催化剂存在时效率则很低[29].反应可能经历的过程如Scheme 8所示, 光照下3-氰基-N-甲基喹啉盐(QuCN+)被激活形成单线态QuCN+*, 苯环上单电子转移至QuCN+*生成苯环正离子自由基和QuCN•.同时, 苯环正离子自由基与醇在一价钴催化剂Co(Ⅰ)作用下生成二烯自由基47与三价钴氢化合物Co(Ⅲ)—H.最后47与Co(Ⅲ)—H作用脱氢生成芳香醚产物和二价钴Co(Ⅱ). Co(Ⅱ)可与QuCN•作用重新生成光催化剂QuCN+和Co(Ⅰ)催化剂完成催化循环.
|
|
(15) |
2011年, 赵康和杜云飞等[30]发现廉价金属铁也可促进芳香环C—H键的醚化反应(Eq. 16).该方法可快速制备一系列的香豆素类似物.氯化铁(FeCl3)可能与底物作用, 首先通过单电子转移(SET)生成自由基中间体, 进而再次单电子转移至FeCl3生成正离子中间体, 因此该反应需要在2 equiv.以上的FeCl3存在下方可进行.
|
|
(16) |
酚羟基较易被氧化, 因此苯酚衍生物作为偶联试剂的报道相对较少, 且多数反应为分子内偶联. 2011年, 刘磊课题组[31]报道了Pd催化的2-羟基联苯的分子内偶联反应, 实现了二芳基醚结构二苯并呋喃(Eq. 17)的合成.酚羟基在反应中既作为导向基团, 促进其邻位C—H键的活化; 同时本身作为偶联组分, 用于构建新的C—O键.作者将2-羟基联苯与等物质的量的的Pd催化剂和卡宾配体混合得到环钯化合物52, 该化合物在120 ℃下以58%的产率转化为二氢苯并呋喃.同位素标记实验发现C—H键和C—D键的活化得到相同的产率.因此, 作者认为该反应的决速步骤是二价钯中间体C—O键的还原消除步骤, 反应经历Pd(0)/Pd(Ⅱ)催化循环.
|
|
(17) |
同年, Yoshikai课题组[32]也报道了2-羟基联苯的分子内偶联反应(Eq. 18).反应以3-硝基吡啶作为配体, 过氧化苯甲酸叔丁酯(tert-butyl peroxybenzoate, BzOOtBu)作为氧化剂.不同于刘磊教授发展的反应体系, 作者通过动力学同位素效应(KIE)发现在该条件下, 反应经历Pd(Ⅱ)/Pd(Ⅳ)催化循环, 而C—H键断裂则为反应的决速步骤.
|
|
(18) |
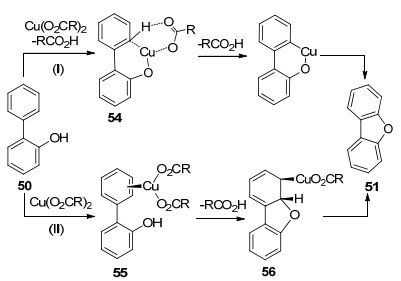
2012年, 朱强课题组[33]发展了铜催化活化C(sp2)—H键构建C—O键的策略(Eq. 19), 该方法可快速制备二苯并呋喃衍生物.该反应成功的关键在于酚羟基对位强吸电子取代基的存在, 可能的原因是强吸电子基团的存在可防止苯酚结构的氧化.作者推测了两种可能的反应机理: (Ⅰ)铜催化剂通过协同金属去质子化(CMD)的过程活化邻位C—H键生成环金属化合物54, 还原消除得到二苯并呋喃衍生物; (Ⅱ)铜催化剂与芳环作用生成η2络合物55, 酚羟基亲核进攻铜络合的双键生成56, 56经过顺式β-H消除得到二苯并呋喃(Scheme 9).
同年, 该课题组[34]研究了含酰胺导向基团的邻芳基苯酚的分子内C—H醚化反应(Eq. 20).该反应中, 酰胺导向基团不仅控制反应的区域选择性, 同时也增加了反应的活性.另外, 当过量碘化亚铜(CuI)存在时, 苯酚环首先发生碘代反应, 然后铜催化分子内C—H醚化反应环生成碘代二苯并呋喃(Eq. 21).
|
|
(20) |
|
|
(21) |
在苯甲酸衍生物的C—H烷氧基化反应中(Eq. 22), Daugulis等[35]发现当反应的溶剂为N, N-二甲基甲酰胺(DMF), 碱为碳酸钾(K2CO3)时, 8-氨基喹啉保护的苯甲酸衍生物可与苯酚衍生物偶联, 实现其邻位C—H键的苯氧基化, 生成二芳基醚产物.酯基、胺基、硝基、腈基和卤素等官能团取代的苯甲酸衍生物均能顺利进行反应, 高效得到目标产物. 2014年, 宋毛平课题组[36]利用2-氨基吡啶-N-氧化物双齿导向基团, 实现了铜介导的邻位C—H苯氧基化反应(Eq. 23).
|
|
(22) |
|
|
(23) |
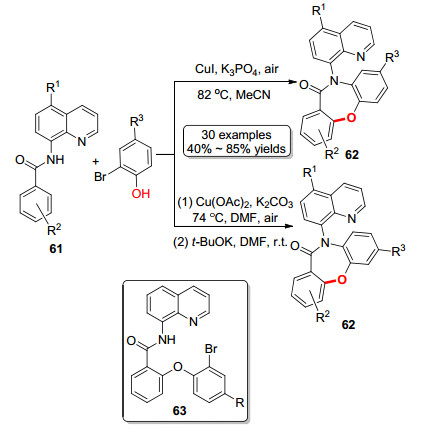
2016年, 朱维良课题组[37]报道了一种二苯氧氮杂䓬酮的合成方法(Scheme 10).首先是8-氨基喹啉保护的羧酸衍生物与邻溴苯酚偶联生成二芳基醚化合物63, 卤代芳环进而发生胺化反应生成二苯氧氮杂䓬酮衍生物.作者认为芳基醚化合物63通过Goldberg胺化生成非重排产物, 而在碱存在发生Smile重排从而胺化生成重排产物.
与C(sp2)—H键和醇或酚偶联反应相比, 通过活化C(sp3)—H键制备芳香醚的方法报道仍然较少. Kappe等[38]报道了一种醚α-位的C—H苯氧基化反应(Eq. 24).反应以叔丁基过氧化氢(tert-butyl hydroperoxide, TBHP)作为氧化剂, 在铜的催化下进行.该反应中的苯酚底物受限于酚羟基邻位含羰基的化合物, 可能的原因是铜催化剂需要与酚羟基氧原子以及羰基氧原子形成双配位的中间体才能促进反应进行. 2015年, Chang和Wang等[39]在类似的条件下, 实现了对氧化剂敏感的水杨醛与醚的偶联反应(Eq. 25).另外该反应还可以在铁的催化下进行[40].
|
|
(24) |
|
|
(25) |
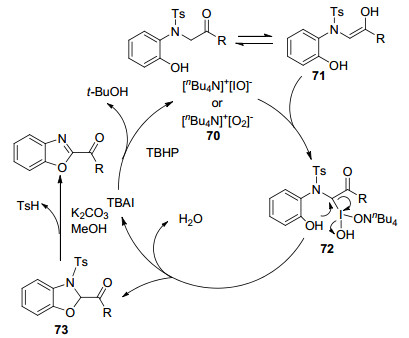
2014年, Wang课题组[41]报道了四正丁基碘化铵(TBAI)催化的分子内C(sp3)—H键苯氧基化反应, 实现了α-酮苯并噁唑衍生物的合成(Eq. 26).反应在室温下即可进行, 且具有较好的官能团耐受性.其可能的机理如Scheme 11所示:催化剂TBAI与氧化剂TBHP作用生成次碘酸盐70.该中间体与烯醇71作用生成中间体72.酚羟基分子内亲核进攻得到环化产物73, 并重新生成催化剂TBAI, 最终73在碱性条件下脱磺酰化得到α-酮苯并噁唑.
|
|
(26) |
近年来基于脱氢偶联策略制备芳香醚衍生物的方法已经取得了快速发展.目前, 芳香烃与醇的脱氢偶联研究相对较为成熟, 其主要在Pd或Cu催化下进行.但是此类方法一般需要用醇作为反应溶剂, 这对芳烃与复杂醇的反应造成了较大限制.与此同时, 也有不少文献报道了芳香烃与酚偶联制备二芳基醚化合物的方法, 但是反应往往是在分子内发生, 而分子间的芳香烃与苯酚偶联的方法报道仍然较少.利用C(sp3)—H键的活化制备芳基烷基醚的方法亦有报道, 但是方法单一, 并且具有较大的局限性.
综上所述, 尽管芳香烃与醇或酚之间的脱氢偶联为芳香醚的制备提供了绿色经济的途径, 但是已报道的方法大多数需要额外的氧化剂, 真正以氢气为唯一副产物的方法极少.因此, 设计新型的氧化还原中性的催化剂体系, 发展条件温和、普适性高的C—H烷氧基化或苯氧基化反应来制备芳基醚化合物, 仍是该领域研究的长期目标.另外, 通过丰富反应底物类型, 使相关策略能应用于复杂的天然产物和药物分子的合成中, 这无疑具有极其重要的学术和应用价值.
(a) Jabran, K.; Ehsanullah; Hussain, M.; Farooq, M.; Babar, M.; Doǧan M.-N.; Lee, D.-J. Weed Biol. Manage. 2012, 12, 136.
(b) Negro, R.; Formoso, G.; Mangieri, T.; Pezzarossa, A.; Dazzi, D.; Hassan, H. J. Clin. Endocrinol. Metab. 2006, 91, 2587.
(c) Kosenkova, Y.-S.; Polovinka, M.; Komarova, N.; Korchagina, D.; Kurochkina, N. Y.; Cheremushkina, V.; Salakhutdinov, N. Chem. Nat. Compd. 2007, 43, 712.
(d) Deng, H.; Jung, J.-K.; Liu, T.; Kuntz, K. W.; Snapper, M. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2003, 125, 9032.
(a) Chen, G.; Du, J. Chin. J. Org. Chem. 2014, 34, 65(in Chinese). (陈国军, 杜建时, 有机化学, 2014, 34, 65.)
(b) Tobisu, M.; Chatani, N. Acc. Chem. Res. 2015, 48, 1717.
(a) Ullmann, F.; Sponagel, P. Ber. Dtsch. Chem. Ges. 1905, 38, 2211.
(b) Xiao, S.; Zhu, J.; Mu, X.; Li, Z. Chin. J. Org. Chem. 2013, 33, 1668(in Chinese). (肖尚友, 朱俊, 穆小静, 李正华, 有机化学, 2013, 33, 1668.)
(a) Chan, D. M. T.; Monaco, K. L.; Wang, R.-P.; Winters, M. P. Tetrahedron Lett. 1998, 39, 2933.
(b) Evans, D. A.; Katz, J. L.; West, T. R. Tetrahedron Lett. 1998, 39, 2937.
(c) Lam, P. Y. S.; Vincent, G.; Clark, C. G.; Deudon, S.; Jadhav, P. K. Tetrahedron Lett. 2001, 42, 3415.
(a) Aranyos, A.; Old, D. W.; Kiyomori, A.; Wolfe, J. P.; Sadighi, J. P.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 4369.
(b) Torraca, K. E.; Huang, X.; Parrish, C. A.; Buchwald, S. L. J. Am. Chem. Soc. 2001, 123, 10770.
(c) Mann, G.; Hartwig, J. F. J. Am. Chem. Soc. 1996, 118, 13109.
(b) Mann, G.; Incarvito, C.; Rheingold, A. L.; Hartwig, J. F. J. Am. Chem. Soc. 1999, 121, 3224.
(c) Hartwig, J. F. Nature 2008, 455, 314.
Terrett, J. A.; Cuthbertson, J. D.; Shurtleff, V. W.; MacMillan, D. W. C. Nature 2015, 524, 330. doi: 10.1038/nature14875
(a) Yang, F.; Zhang, H.; Liu, X.; Wang, B.; Ackermann, L. Chin. J. Org. Chem. 2019, 39, 59(in Chinese). (杨帆致, 张晗, 刘旭日, 王博, Lutz A., 有机化学, 2019, 39, 59.)
(b) Leila, R.; Asieh, Y.; Mehdi, S. ChemCatChem 2018, 10, 20.
(c) Kamellia, N.; Sheida, A.; Mohammad, N.; Parvaneh, D. K. N.; Esmail, V. RSC Adv. 2018, 8, 19125.
(d) Bhunia, S.; Pawar, G. G.; Kumar, S. V.; Jiang, Y.; Ma, D. Angew. Chem., Int. Ed. 2017, 56, 16136.
(e) Ding, H.; Li, J.; Guo, Q.; Xiao, Y. Chin. J. Org. Chem. 2017, 37, 3112(in Chinese). (丁怀伟, 李娟, 郭庆辉, 肖琰, 有机化学, 2017, 37, 3112.)
Desai, L. V.; Malik, H. A.; Sanford, M. S. Org. Lett. 2006, 8, 1141. doi: 10.1021/ol0530272
Wang, G.-W.; Yuan, T.-T. J. Org. Chem. 2010, 75, 476. doi: 10.1021/jo902139b
Jiang, T.-S.; Wang, G.-W. J. Org. Chem. 2012, 77, 9504 doi: 10.1021/jo301964m
Shi, S.-P.; Kuang, C.-X. J. Org. Chem. 2014, 79, 6105. doi: 10.1021/jo5008306
Li, W.; Sun, P.-P. J. Org. Chem. 2012, 77, 8362 doi: 10.1021/jo301384r
Yin, Z.-W.; Jiang, X.-Q.; Sun, P.-P. J. Org. Chem. 2013, 78, 10002 doi: 10.1021/jo401623j
Zhang, C.; Sun, P.-P. J. Org. Chem. 2014, 79, 8457. doi: 10.1021/jo5014146
Gao, T.-T.; Sun, P.-P. J. Org. Chem. 2014, 79, 9888. doi: 10.1021/jo501902d
Peron, F.; Fossey, C.; Santos, J. O. S.; Cailly, T.; Fabis, F. Chem.-Eur. J. 2014, 20, 1. doi: 10.1002/chem.201390210
Chen, F.-J.; Zhao, S.; Hu, F.; Chen, K.; Zhang, Q.; Zhang, S.-Q.; Shi, B.-F. Chem. Sci. 2013, 4, 4187. doi: 10.1039/c3sc51993g
(a) Shen, T.; Wang, X.-N.; Lou, H.-X. Nat. Prod. Rep. 2009, 26, 916.
(b) Veitch, N. C. Nat. Prod. Rep. 2007, 24, 417.
(c) Watzke, A.; O'Malley, S. J.; Bergman, R. G.; Ellman, J. A. J. Nat. Prod. 2006, 69, 1231.
(d) Tsui, G. C.; Tsoung, J.; Dougan, P.; Lautens, M. Org. Lett. 2012, 14, 5542.
Wang, X.-S.; Lu, Y.; Dai, H.-X.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 12203. doi: 10.1021/ja105366u
Wang, H.-B.; Li, G.; Engle, K. M.; Yu, J.-Q.; Davies, H. M. L. J. Am. Chem. Soc. 2013, 135, 6774. doi: 10.1021/ja401731d
Cheng, X.-F.; Li, Y.; Su, Y.-M.; Yin, F.; Wang, J.-Y.; Sheng, J.; Vora, H. U.; Wang, X.-S.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 1236. doi: 10.1021/ja311259x
Wang, Z.-L.; Zhao, L.; Wang, M.-X. Org. Lett. 2011, 13, 6560. doi: 10.1021/ol202874n
Zhang, L.-B.; Hao, X.-Q.; Zhang, S.-K.; Liu, K.; Ren, B.-Z.; Gong, J.-F.; Niu, J.-L.; Song, M.-P. J. Org. Chem. 2014, 79, 10399. doi: 10.1021/jo502005j
Bhadra, S.; Matheis, C.; Katayev, D.; Gooßen, L. J. Angew. Chem. 2013, 125, 9449. doi: 10.1002/ange.201303702
Roane, J.; Daugulis, O. Org. Lett. 2013, 15, 5842. doi: 10.1021/ol402904d
Yin, X.-S.; Li, Y.-C.; Yuan, J.; Gua, W.-J.; Shi, B.-F. Org. Chem. Front. 2015, 2, 119. doi: 10.1039/C4QO00276H
Zhang, L.-B.; Hao, X.-Q.; Zhang, S.-K.; Liu, Z.-J.; Zheng, X.-X.; Gong, J.-F.; Niu, J.-F.; Song, M.-P. Angew. Chem., Int. Ed. 2015, 54, 272. doi: 10.1002/anie.201409751
Zheng, Y.-W.; Ye, P.; Chen, B.; Meng, Q.-Y.; Feng, K.; Wang, W.-G.; Wu, L.-Z.; Tung, C.-H. Org. Lett. 2017, 19, 2206. doi: 10.1021/acs.orglett.7b00463
Ohkubo, K.; Kobayashi, T.; Fukuzumi, S. Opt. Express 2012, 20, A360. doi: 10.1364/OE.20.00A360
Tang, L.; Pang, Y.; Yan, Q.; Shi, L.-Q.; Huang, J.-H.; Du, Y.-F.; Zhao, K. J. Org. Chem. 2011, 76, 2744. doi: 10.1021/jo2000644
Xiao, B.; Gong, T.-J.; Liu, Z.-J.; Liu, J.-H.; Luo, D.-F.; Xu, J.; Liu L. J. Am. Chem. Soc. 2011, 133, 9250. doi: 10.1021/ja203335u
Wei, Y.; Yoshikai, N. J. Org. Chem. 2011, 13, 5504.
Zhao, J.-J.; Wang, Y.; He, Y.-M.; Liu, L.-Y.; Zhu, Q. Org. Lett. 2012, 14, 1078. doi: 10.1021/ol203442a
(a) Zhao, J.-J.; Wang, Y.; Zhu, Q. Synthesis 2012, 44, 1551.
(b) Zhao, J.-J.; Zhang, Q.; Liu, L.-Y.; He, Y.-M.; Li, J.; Li, J.; Zhu, Q. Org. Lett. 2012, 14, 5362.
Roane, J.; Daugulis, O. Org. Lett. 2013, 15, 5842. doi: 10.1021/ol402904d
Hao, X.-Q.; Chen, L.-J.; Ren, B.; Li, L.-Y.; Yang, X.-Y.; Gong, J.-F.; Niu, J.-L.; Song, M.-P. Org. Lett. 2014, 16, 1104. doi: 10.1021/ol500166d
Zhou, Y.-F.; Zhu, J.-M.; Li, B.; Zhang, Y.; Feng, J.; Hall, A.; Shi, J.-Y.; Zhu, W.-L. Org. Lett. 2016, 18, 3803
Kumar, G. S.; Pieber, B.; Reddy, K. R.; Kappe, C. O. Chem.-Eur. J. 2012, 18, 6124. doi: 10.1002/chem.201200815
Barve, B. D.; Wu, Y.-C.; El-Shazly, M.; Korinek, M.; Cheng, Y.-B.; Wang, J.-J.; Chang, F.-R. Tetrahedron 2015, 71, 2290 doi: 10.1016/j.tet.2015.02.035
Barve, B. D.; Wu, Y.-C.; El-Shazly, M.; Korinek, M.; Cheng, Y.-B.; Wang, J.-J.; Chang, F.-R. Org. Lett. 2014, 16, 1912. doi: 10.1021/ol5004115
Boominathan, S. S. K.; Hu, W.-P.; Senadi, G. C.; Vandavasia, J. K.; Wanga, J.-J. Chem. Commun. 2014, 50, 6726. doi: 10.1039/C4CC02425G

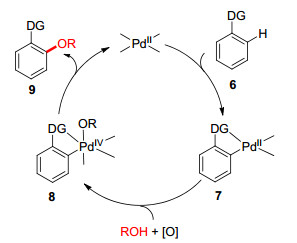
图式 2 Pd催化C—H活化制备芳基醚机理
Scheme 2 Mechanism for the synthesis of aryl ethers via Pd- catalyzed C—H activation

图式 4 C—H官能化反应合成二氢苯并呋喃衍生物
Scheme 4 C—H functionalization for the synthesis of dihdro- benzofuran derivatives

图式 9 铜催化分子内C—H芳氧基化机理图
Scheme 9 Mechanism for Cu-catalyzed intramolecular aryloxylation.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


























 下载:
下载:








 下载:
下载: