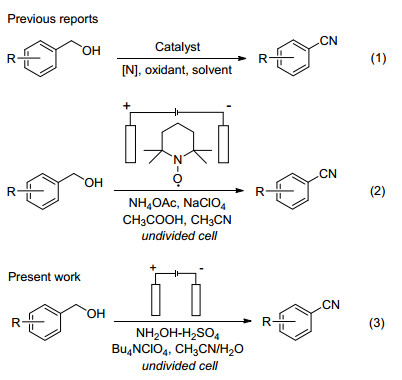
图式 1.
芳香伯醇到芳腈的合成路线
Scheme 1.
Synthesis of aryl nitriles from aromatic benzyl alcohols

芳腈化合物作为一种重要的化工原料和有机合成中间体, 在农药、医药、染料和功能材料等领域具有广泛的应用价值[1~5], 其合成方法一直受到相关研究者的广泛关注.传统的氰化方法包括经典的Sandmeyer反应[6]和Rosenmund-vonBraun反应[7], 以及过渡金属催化卤代芳烃与氰基化试剂的偶联反应[8~12], 这些反应大都涉及毒性较高的氰化试剂, 反应条件苛刻、不易操作.通过酰氨/醛肟的脱水反应[13, 14]是合成腈类化合物常用的方法, 此类反应大都需要在强酸或强碱的条件下进行, 并需使用大量的脱水试剂.芳烃的气相氨氧化法是工业上合成芳腈化合物应用最广泛的方法之一[16~18], 该方法不需要特殊溶剂参与, 但是反应过程中高温(>350 ℃)、高压及气体的强腐蚀性仍是需待解决的问题.过渡金属催化C—H键活化氰基化反应合成腈类化合物受到了广泛研究[19~26], 反应底物无需预活化, 具有原子经济性等优点, 但贵金属催化剂的循环利用、开发与氧化剂相容的氮源等仍是需面临的问题.
目前以醇为原料, 通过“氧化氮化”法直接合成腈类化合物已有很多报道[27], 该法主要包含三个过程: (1)醇在氧化剂作用下氧化生成相应的醛; (2)醛与氮源发生氮化反应; (3)重排生成相应的腈.氧化剂主要有I2/ DHI[28, 29]、DDQ[30]、TEMPO/PhI(OAc)2[31, 32]以及金属氧化物MnO2/Al2O3[33, 34]等, 氮源有氨气[35]、铵盐[36]等(Scheme 1 (1)).因此, 氧化剂与氮源的适应性成为反应条件优化的关键.
有机电合成已成为有机合成中的一个重要组成部分, 通过C—H键电化学活化构建C—X (X=C、N、O、S)键合成具有高附加值的化学品受到了广泛研究[37], 因此利用电子作为氧化还原试剂合成腈类化合物极具吸引力. Yuan等[38]报道了以NH4I作为电催化剂和氮源, 实现了从醛到腈的间接电化学合成, 该方法简单高效, 但碘化铵用量较大对环境不友好. Li等[39]报道了以2, 2, 6, 6-四甲基哌啶氮氧基自由基(TEMPO)作为电催化剂, 醋酸铵(NH4OAc)作为氮源, 实现了从芳醇到腈的间接电氧化合成(Scheme 1(2)).目前, 通过醇直接电氧化氰化反应合成腈类化合物未见文献报道.
在醇/醛电化学氰化工作的启发下, 本文提出了由芳醇直接电化学氧化生成醛, 再进一步氰化转化为腈的电化学合成法(Scheme 1(3)).以羟胺为反应氮源, 四丁基高氯酸铵(Bu4NClO4)为支持电解质, 采用恒电流电解技术, 研究了从芳香伯醇到芳腈的直接电氧化氰化合成过程, 这种在阴阳两极同时发生电化学转化的成对电氧化还原合成路线极大简化了反应步骤.
在以0.05 mol•L-1 Bu4NClO4作为支持电解质的情况下, 考察了水的体积含量对p-MeOC6H4CH2OH电氧化行为的影响, 结果如图 1所示.在没有额外加入水的情况下, 1.60 V处出现微弱的氧化峰.当水体积含量为10%时, 氧化峰电流明显增加, 氧化峰电位正移到1.74 V左右.而随着水含量继续增加, p-MeOC6H4CH2OH的氧化电位发生负移.当水含量增加到30%时, p-MeO-C6H4CH2OH的氧化峰电位负移到1.63 V左右, 由于这种比例的水与乙腈的混合溶剂与反应底物之间达到了一种互溶状态, 有利于反应底物的扩散, 加快了电极与活性物种之间电荷传递速率, 峰电流增强, 峰电位下降.当水含量超过40%后, p-MeOC6H4CH2OH的氧化峰电流有所下降, 因为过多的水导致有机反应底物在混合溶剂中溶解度下降, 从而使电氧化性能下降.

图 2是不同硫酸浓度对p-MeOC6H4CH2OH电氧化行为的影响.发现体系中的酸性强弱对p-MeOC6H4-CH2OH电氧化过程的峰电流和峰电位均有较大的影响.无硫酸加入时, 在1.83 V左右出现氧化峰.在硫酸浓度≤0.35 mol•L-1时, 随着硫酸浓度的增加, p-MeO-C6H4CH2OH的氧化电位发生负移, 氧化峰电流逐步增强, 这是由于溶液的离子强度增强所致, 同时体系中H+浓度增大, 有利于阴极上析氢反应的进行.当硫酸浓度为0.35 mol•L-1时, p-MeOC6H4CH2OH的氧化电位为1.57 V左右.随着硫酸浓度进一步增加, p-MeOC6H4-CH2OH的氧化峰电流出现微弱下降, 表明适量的H+浓度有利于p-MeOC6H4CH2OH阳极氧化反应的进行.
在0.15 mol•L-1 H2SO4和30% H2O的情况下考察不同温度对p-MeOC6H4CH2OH的电氧化行为的影响, 见图 3.研究发现, 随着温度的升高, 氧化峰电流逐渐增大, 氧化峰电位有所负移.说明提高温度在一定程度上能加快反应底物向电极表面的扩散, 有利于p-MeO-C6H4CH2OH的电氧化过程.考虑到羟胺在高温下容易分解以及腈类化合物的水解反应, 没有继续研究更高温度下的影响, 因此在后续恒电流电解反应中最高电解温度控制在60 ℃.

为了进一步探究p-MeOC6H4CH2OH电氧化氰化反应的最佳反应条件, 在CV的研究基础上, 进行了恒电流电解实验, 考察了支持电解质浓度、电流密度、电极材料、反应温度、酸浓度、溶剂等对p-MeOC6H4CH2OH转化率和目标产物对甲氧基苯腈(p-MeOBN)收率的影响, 结果列于表 1中.
 下载:
导出CSV
下载:
导出CSV
 |
|||||||
| Entry | Bu4NClO4 equiv. | Anode/Cathode | j/(mA•cm-2) | Solvent | T/℃ | Conv. b/% | Yieldc/% |
| 1 | 0.3 | Pt/Pb | 10.0 | DMF/H2O | 60 | 85 | 40 |
| 2 | 0.6 | Pt/Pb | 10.0 | DMF/H2O | 60 | 98 | 72 |
| 3 | 1.0 | Pt/Pb | 10.0 | DMF/H2O | 60 | 97 | 83 |
| 4 | 1.2 | Pt/Pb | 10.0 | DMF/H2O | 60 | 99 | 81 |
| 5 | 1.0 | Pt/Pb | 12.5 | DMF/H2O | 60 | 97 | 78 |
| 6 | 1.0 | Pt/Pb | 15.0 | DMF/H2O | 60 | 96 | 70 |
| 7 | 1.0 | Pt/Pb | 10.0 | DMF/H2O | 30 | 33 | 16 |
| 8 | 1.0 | Pt/Pb | 10.0 | DMF/H2O | 40 | 96 | 55 |
| 9 | 1.0 | Pt/Pb | 10.0 | DMF/H2O | 50 | 98 | 74 |
| 10 | 1.0 | Pt/C | 10.0 | DMF/H2O | 60 | 99 | 42 |
| 11 | 1.0 | Pt/Pt | 10.0 | DMF/H2O | 60 | 99 | 78 |
| 12 | 1.0 | Pt/Pb | 10.0 | CH3OH/H2O | 60 | 90 | 13 |
| 13 | 1.0 | Pt/Pb | 10.0 | C2H5OH/H2O | 60 | 95 | 48 |
| 14 | 1.0 | Pt/Pb | 10.0 | CH3CN/H2O | 60 | 99 | 60 |
| 15 | 1.0 | Pt/Pb | 10.0 | DMSO/H2O | 60 | 98 | 90 |
| 16d | 1.0 | Pt/Pb | 10.0 | DMSO/H2O | 60 | 99 | 36 |
| 17d | 1.0 | Pt/Pb | 10.0 | DMSO/H2O | 60 | 99 | 22 |
| a p-MeOC6H4CH2OH (1.5 mmol), NH2OH (3.0 equiv), H2SO4 (0.15 mol•L-1), solvent/H2O (30%, 30 mL) was electrolyzed at a constant current in an undivided cell, t=8 h, Pt/Pb (2 cm×2 cm); b Conversions were determined by GC; c GC yields of nitriles; d H2SO4 (0.25, 0.35 mol•L-1). | |||||||
在有机电合成过程中, 需要加入支持电解质提高体系的导电性确保反应的顺利进行.在不同Bu4NClO4浓度下, p-MeOC6H4CH2OH电氧化氰化反应的结果见表 1 Entries 1~4.结果表明, 在加入不同当量的支持电解质下, p-MeOC6H4CH2OH均具有良好的转化率(85%~99%), 但对p-MeOBN的选择性影响较大.在加入0.3 equiv.的Bu4NClO4时, p-MeOBN的收率仅为40%.随着支持电解质当量的增加, 收率从40%提高到83%, 但考虑到过量的Bu4NClO4使得体系的黏度增加, 阻碍反应物种的扩散, 同时目标产物的收率也有所下降, 因此选择1.0 equiv.的Bu4NClO4为最佳用量.
随着电流密度的增大, p-MeOC6H4CH2OH可以有效转化(转化率≥96%), 但p-MeOBN的收率却有所下降(表 1, Entries 3, 5~6).通过GC-MS检测, 发现随着电流密度的增加, 产物中醛的进一步氧化产物对甲氧基苯甲酸的量也增加[40, 41].
温度是影响反应底物在体系中溶解度及扩散速率的重要因素, 其对p-MeOC6H4CH2OH电氧化氰化反应的影响见表 1 (Entries 3, 7~9).结果发现, 随着温度的升高, p-MeOC6H4CH2OH的转化率和p-MeOBN的收率均明显提高.
电极材料是电化学反应的场所, 不同阴极材料对p-MeOC6H4CH2OH电氧化氰化行为的影响见表 1 (Entries 3, 10, 11).以Pt作为阳极, 在三种阴极材料上, p-MeOC6H4CH2OH的转化率很高(≥97%), 但p-MeOBN的收率却有较大差别.当石墨作为阴极时, p-MeOBN的收率仅为42%.以铂作为阴极时, 收率提高到78%.以铅作为阴极时, 收率明显提高到83%.显然, 与铂或石墨相比, 铅是一种适合电氧化氰化反应的阴极材料, 同时也表明p-MeOC6H4CH2OH电氧化氰化过程中, 在阴极上除了析氢反应, 还涉及一步反应中间产物的转化过程, 这将在后续的机理探讨中进行佐证.
溶剂既影响反应底物在体系中的溶解性能, 也影响电化学反应的后续步骤, 因此电合成反应的选择性受溶剂的影响较大.对比p-MeOC6H4CH2OH在不同溶剂中电氧化氰化反应结果(表 1, Entries 3, 12~15), 发现在不同溶剂中, p-MeOC6H4CH2OH的转化率都很高(≥90%), 但p-MeOBN的收率差别较大.在甲醇溶剂中副反应明显, 在GC-MS检测中发现有56%的甲氧基化产物.在乙醇和乙腈溶剂中, p-MeOBN的收率明显提高, 但在产物中检测到一定量的相应酸和肟.当DMSO作溶剂时, p-MeOBN的收率最高为90%.
在酸浓度对p-MeOC6H4CH2OH电氧化影响的CV曲线中(图 2), 发现随着酸浓度的增加, 氧化峰电流逐渐增大.但通过恒电流电解发现, 当酸浓度大于0.15 mol•L-1时, p-MeOC6H4CH2OH的转化率高达99%, 而p-MeOBN的收率却明显下降(表 1, Entries 3, 16, 17), 在产物中检测到约50%的醛及相应酸, 这说明在强酸性条件下不仅使醛容易进一步氧化, 也使阴极的析氢反应加剧, 从而影响醛与羟胺缩合后的中间体进一步转化为相应腈的步骤, 致使反应停留在醛的这一步.这也进一步说明了阴极电极材料对p-MeOC6H4CH2OH电氧化氰化反应产生影响的原因.这些结果表明, p-MeOC6H4CH2-OH电氧化氰化合成相应腈的反应很可能是一个同时利用阳极和阴极的成对电化学合成反应.

基于以上结果, 对不同取代基取代的芳香伯醇进行了普适性研究, 在实验范围内, 将不同反应底物分别在溶剂DMSO和CH3CN中取得的较好电解结果列于表 2中.不论是供电子基团(OMe, Me, t-Bu)还是弱吸电子基团(F, Cl, Br), 取代位置为邻、对位时, 芳香伯醇在电氧化氰化反应中均具有很好的转化率(≥96%), 其相应目标产物腈的收率在79%~92%.当取代基为强吸电子基团CF3时, 转化率有所下降(93%), 相应目标产物对三氟甲基苯甲腈的收率为61% (表 2, Entry 12).而间位取代的芳香醇在转化率和产物收率上比邻、对位取代的效果差(表 2, Entries 3, 9), 这可能是由于p-Π共轭效应的影响, 使得邻、对位取代的芳香醇在直接电氧化过程中生成的鎓阳正离子更稳定, 有利于反应的顺利进行.
下载:
导出CSV
 |
|||||
| Entry | Product | j/(mA•cm-2) | Time/h | Conv. b/% | Yieldc/% |
| 1 |  |
10.0 | 10 | 99 | 90 |
| 2 |  |
17.5 | 9 | 99 | 85 |
| 3 |  |
17.5 | 13 | 59 | 15 |
| 4 |  |
17.5 | 13 | 99 | 92 |
| 5 |  |
12.5 | 9 | 99 | 89 |
| 6 |  |
12.5 | 11 | 96 | 85 |
| 7 |  |
17.5 | 10 | 97 | 89 |
| 8 |  |
20.0 | 11 | 98 | 90 |
| 9 |  |
20.0 | 11 | 89 | 59 |
| 10 |  |
20.0 | 11 | 99 | 86 |
| 11 |  |
20.0 | 10 | 99 | 79 |
| 12 |  |
17.5 | 13 | 93 | 61 |
直接以不同对位取代的芳香醛为反应底物进行电氧化氰化反应, 结果列于表 3中.供电子基(OMe, Me)和吸电子基(F)取代的醛, 该电氧化氰化反应的转化率都很高(≥95%), 主要产物为相应的芳腈, 但收率比以相同取代基的醇为反应底物时要低, 其中供电子基取代的醛在电氧化氰化过程中易进一步生成相应的酸, 而吸电子基取代的醛在产物中还有其他物质生成.这可能是由于在酸性条件下, 电极表面高浓度的醛更容易进一步失去电子氧化为相应的酸, 以及由于醛与羟胺很容易生成肟, 从而增加了高浓度的肟与乙腈等发生其他副反应的机会[42].因此在本实验中通过原位生成醛, 进一步发生氰基化反应是一种更有效的氰化方法.
下载:
导出CSV
 |
||||||
| Entry | Substrte | Time/h | Conv. b/% | Yieldc/% | ||
| a | b | c | ||||
| 1 |  |
8 | 98 | 85 | 9 | n.d. |
| 2 |  |
9 | 95 | 78 | 6 | 11 |
| 3 |  |
9 | 98 | 75 | n.d. | 23 |
| a Aromatic aldehydes (1.5 mmol), NH2OH-H2SO4 (3.0 equiv.), Bu4NClO4 (1.0 equiv.), Pt (2 cm×2 cm), Pb (2 cm×2 cm), j (10 mA/cm2), solvent/H2O (30%, 30 mL), 60 ℃, Undivided cell; b Conversions were determined by GC; c GC yields of nitriles; n.d.=no detection. | ||||||
为了佐证电流对p-MeOC6H4CH2OH电氧化性能的影响, 在一室型电解槽中不通电流情况下, 发现p-MeOC6H4CH2OH几乎未发生转化, 说明电流是芳香醇发生电氧化反应的必要条件.根据不同的阴极材料及体系中酸性强弱对电解产物的影响结果, 推测在阴极除了析氢反应, 还有中间体的还原反应, 进而生成目标产物p-MeOBN.为了佐证这一假设, 在其他条件一样的情况下, 采用H型隔膜电解槽分别在阴极室和阳极室进行p-MeOC6H4CH2OH的恒电流电解实验, 发现在阴极室反应底物几乎未发生转化.而在阳极室中, 主要检测到86%的对甲氧基苯甲醛, 这说明p-MeOC6H4CH2OH在阳极发生电化学氧化反应生成p-MeOBA.当直接以p-MeOBA为反应底物, 采用H型隔膜电解槽在阴极室进行恒电流电解时, 反应停留在生成对甲氧基苯甲醛肟(p-MeOBAO, 收率为88%)这一步, 而没有进一步转化为相应的腈.
根据以上实验结果, 我们认为p-MeOC6H4CH2OH在阳极发生氧化反应生成相应醛后, 与羟胺生成p-MeOBAO, 随之在阳极和阴极的共同作用下进而转化为p-MeOBN, 可能的反应机理见Scheme 2. p-MeOC6H4CH2OH在阳极先失去两个电子得到鎓阳离子2, 在水溶液中生成氧鎓离子3, 继而脱去H+生成缩醛4.由于缩醛极不稳定, 在酸性条件下重排脱掉一分子水生成活性中间体醛5[43, 44], 醛与溶液中的羟胺发生亲核加成反应生成醛肟6.然后, 醛肟在阳极作用下进一步失去两个电子和两个质子生成腈氧化物中间体7[45], 再经过阴极得到两个电子还原生成目标产物腈8[46~48].

提出了一种在无隔膜电解槽中以羟胺(HAM)为反应氮源, 芳香醇为反应底物, 通过原位生成醛, 继而在阴阳两极上进一步氰化转化的成对电化学合成芳腈化合物的方法.以Bu4NClO4为电解质, 在0.15 mol•L-1 H2SO4及30%水的有机溶剂体系中, 不同取代芳香醇具有一定的普适性, 相应腈的收率在61~92%之间.该成对电氧化还原合成路线极大地缩短了从醇出发合成腈类化合物的合成步骤.
循环伏安测试采用上海辰华电化学工作站(CHI660D), 恒电流电解采用固纬直流电源(GWINSTEK GPD-4303S), 产物分析采用气相质谱联用仪GC-MS (Thermo Trace ISQ instrument with TG 5MS capillary column), 气相色谱分析仪GC (Agilent 7890 A with Hp-5 capillary column and a FID), 物质的熔点采用SGWX-4A显微熔点仪测定, 核磁共振谱由Bruker-500 MHz型核磁共振仪测定, CDCl3为溶剂, 分别在500、125 MHz条件下得到1H NMR、13C NMR谱图.反应中所用到的反应原料、溶剂均为市售无水分析纯试剂, 电极材料购自于天津艾达恒晟科技有限公司.
循环伏安(CV)测试, 在无隔膜的一室型单电解池(10 mL)中采用三电极体系进行.工作电极(WE)为铂电极(φ=3 mm), 辅助电极(CE)为铂片电极(2 cm×2 cm), 参比电极(RE)为Ag/AgCl电极, 于室温下进行CV测试.扫描速率为50 mV•s-1, 扫描范围为0.5~2.0 V.溶液为含有0.05 mol•L-1四丁基高氯酸铵的DMF水溶液, 底物浓度为0.05 mol•L-1.
恒电流电解(CCE)实验在配备磁子搅拌的一室型恒温电解槽(50 mL)中采用双电极体系进行.电极材料分别为铂片电极(2 cm×2 cm), 铅片电极(2 cm×2 cm), 石墨棒电极(φ=3 mm).电解液为含有体积分数为30%的水和0.15 mol•L-1 H2SO4的DMF或DMSO溶液(30 mL), 反应底物为对甲氧基苯甲醇(0.05 mol•L-1), 反应氮源为羟胺(0.15 mol•L-1), 支持电解质为四丁基高氯酸铵(0.05 mol•L-1), 反应温度为60℃, 恒电流电解8 h.电解产物经过无水乙醚萃取(20 mL×3), 有机相经过水洗涤(20 mL×3), 最后用无水硫酸钠干燥.产物的定性分析采用气质联用仪GC-MS进行, 定量分析采用气相色谱分析仪GC进行, 分析方法为面积归一化法.目标产物经柱层析提纯[展开剂: V(石油醚):V(乙酸乙酯)=15:1].
所有化合物表征数据均与参考文献一致[1~3, 12b, 19, 39]
4-甲氧基苯腈(2a)[1]:白色固体, m.p. 58~59 ℃; 1H NMR (500 MHz, CDCl3) δ: 3.86 (s, 3 H), 6.96 (d, J=8.5 Hz, 2H), 7.59 (d, J=8.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 55.52, 104.00, 114.76, 119.17, 133.95, 162.86; GC-MS (EI, 70 eV) m/z: 133.05 [M]+.
4-甲基苯腈(2b)[12b]:白色固体, m.p. 34~35 ℃; 1H NMR (500 MHz, CDCl3) δ: 2.42 (s, 3H), 7.27 (d, J=8.0 Hz, 2H), 7.53 (d, J=8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 21.79, 109.37, 119.10, 129.02 132.03, 143.66; GC-MS (EI, 70 eV) m/z: 117.16 [M]+.
3-甲基苯腈(2c)[19]:无色液体. 1H NMR (500 MHz, CDCl3) δ: 2.39 (s, 3H), 7.35 (t, J=8.0 Hz, 1H), 7.36 (d, J=8.0 Hz, 1H), 7.41 (d, J=8.0 Hz, 1H), 7.44(s, 1H); 13C NMR (125 MHz, CDCl3) δ: 21.01, 112.16, 118.89, 128.90, 129.15, 132.35, 133.55, 139.14; GC-MS (EI, 70 eV) m/z: 117.01 [M]+.
2-甲基苯腈(2d)[12b]:无色液体. 1H NMR (500 MHz, CDCl3) δ: 2.54 (s, 3H), 7.27 (t, J=8.0 Hz, 1H), 7.32 (d, J=8.0 Hz, 2H), 7.48 (t, J=7.5 Hz, 1H), 7.59 (d, J=7.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 20.40, 112.76, 126.18, 130.20, 132.46, 132.59, 141.59; GC-MS (EI, 70 eV) m/z: 117.13 [M]+.
苯腈(2e)[1]:无色液体. 1H NMR (500 MHz, CDCl3) δ: 7.46~7.49 (m, 2H), 7.59~7.63 (m, 1H), 7.64~7.66 (m, 2H); 13C NMR (125 MHz, CDCl3) δ: 112.39, 118.76, 129.06, 132.07, 132.71; GC-MS (EI, 70 eV) m/z: 103.11 [M]+.
4-叔丁基苯腈(2f)[1]:浅黄色液体. 1H NMR (500 MHz, CDCl3) δ: 1.33 (s, 9H), 7.47~7.50 (m, 2H), 7.57~7.59 (m, 2H); 13C NMR (125 MHz, CDCl3) δ: 30.86, 35.18, 109.27, 119.04, 126.10, 131.88, 156.58; GC-MS (EI, 70 eV) m/z: 159.18 [M]+.
4-氟苯腈(2g)[12b]:浅黄色固体, m.p. 29~30 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.16~7.21(m, 2H), 7.67~7.71 (m, 2H); 13C NMR (125 MHz, CDCl3) δ: 108.57, 116.72, 117.96, 134.61, 163.89; GC-MS (EI, 70 eV) m/z: 121.02 [M]+.
4-氯苯腈(2h)[12b]:白色固体, m.p. 90~91 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.47 (m, 2H), 7.60 (m, 2H); 13C NMR (125 MHz, CDCl3) δ: 110.80, 117.92, 129.68, 133.36, 139.54; GC-MS (EI, 70 eV) m/z: 137.01 [M]+.
3-氯苯腈(2i)[39]:白色固体, m.p. 38~42 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.44 (t, J=8.0 Hz, 1H), 7.55~7.57 (m, 1H), 7.58~7.61 (m, 1H), 7.64 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: 114.00, 117.38, 130.27, 130.47, 131.91, 133.20, 135.25; GC-MS (EI, 70 eV) m/z: 136.96 [M]+.
2-氯苯腈(2j)[12b]:浅黄色固体, m.p. 43~44 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.38 (ddd, J=8.25, 7.25, 1.5 Hz, 1H), 7.51~7.57 (m, 2H), 7.68 (dd, J=7.5, 1.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ: 113.52, 115.93, 127.14, 130.08, 133.83, 134.04, 136.95; GC-MS (EI, 70 eV) m/z: 136.93 [M]+.
4-溴苯腈(2k)[2]:白色固体, m.p. 111~112 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.53 (d, J=8.5 Hz, 2H), 7.64 (d, J=8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 111.26, 118.03, 128.01, 132.64, 133.40; GC-MS (EI, 70 eV) m/z: 181.03 [M]+.
4-三氟甲基苯腈(2l)[12b]:白色固体, m.p. 41~42 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.76 (d, J=8.5 Hz, 2H), 7.81 (d, J=8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 116.14, 117.42, 121.99, 124.16, 126.22, 132.70, 134.49, 134.75; GC-MS (EI, 70 eV) m/z: 171.17 [M]+.
辅助材料(Supporting Information) 目标产物腈2a~2l的气相色谱图、质谱、氢谱、碳谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Tsuchiya, D.; Kawagoe, Y.; Moriyama, K.; Togo, H. Org. Lett. 2013, 15, 4194. doi: 10.1021/ol401906q
Yadav, D. K. T.; Bhanage, B. M. Eur. J. Org. Chem. 2013, 23, 5106.
Kawagoe Y.; Moriyama, K.; Togo, H. Eur. J. Org. Chem. 2014, 19, 4115.
Wang, M.-X. Acc. Chem. Res. 2015, 48, 602. doi: 10.1021/ar500406s
Nobuhito, K.; Takeshi, O. ACS Catal. 2016, 6, 989. doi: 10.1021/acscatal.5b02184
Hodgson, H. H. Chem. Rev. 1947, 40, 251. doi: 10.1021/cr60126a003
Koelsch, C. F.; Whitney, A. G. J. Org. Chem. 1941, 06, 795. doi: 10.1021/jo01206a002
Takagi, K.; Okamoto, T.; Sakakibara, Y. Chem. Lett. 1973, 2, 471. doi: 10.1246/cl.1973.471
Ellis, G. P.; Romney-Alexander, T. M. Chem. Rev. 1987, 87, 779. doi: 10.1021/cr00080a006
Zanon, J.; Klapars, A.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 2890. doi: 10.1021/ja0299708
Arvela, R. K.; Leadbeater, N. E. J. Org. Chem. 2003, 68, 9122. doi: 10.1021/jo0350561
(a) Zhou, W.; Xu, J.-J.; Zhang, L.-R.; Jiao, N. Org. Lett. 2010, 12, 2888.
(b) Yu, Z.-W.; Li, L.-Y.; Shen, Z.-M. Chin. J. Org. Chem. 2017, 37, 1273.
Rogic, M. M.; Van Peppen, J. F.; Klein, K. P.; Demmin, T. R. J. Org. Chem. 1974, 39, 3425.
Yamaguchi, K.; Fujiwara, H.; Ogasawara, Y.; Kotani, M.; Mizuno, N. Angew. Chem. Int. Ed. 2007, 46, 3922. doi: 10.1002/anie.200605004
Rokade, B. V.; Prabhu, K. R. J. Org. Chem. 2012, 77, 5364. doi: 10.1021/jo3008258
Denton, W. I.; Bishop, R. B.; Caldwell, H. P.; Chapman, H. D. Ind. Eng. Chem. 1950, 42, 796. doi: 10.1021/ie50485a019
Cavani, F.; Parrinello, F.; Trifiro, F. J. Molecuhr. Catal. 1987, 43, 117. doi: 10.1016/0304-5102(87)87025-6
Kumar, C. P.; Reddy, K. R.; Rao, V. V.; Chary, K. V. R. Green Chem. 2002, 4, 513. doi: 10.1039/B206581A
Zhang, Y.; Wang, J.-B. Angew. Chem. Int. Ed. 2013, 52, 10573. doi: 10.1002/anie.201305731
Murahashi, S.; Komiya, N.; Terai, H. J. Am. Chem. Soc. 2003, 125, 15312. doi: 10.1021/ja0390303
Murahashi, S. I.; Nakae, T.; Terai, H.; Komiya, N. J. Am. Chem. Soc. 2008, 130, 11005. doi: 10.1021/ja8017362
Liu, W-P.; Ackermann, L. Chem. Commun. 2014, 50, 1878. doi: 10.1039/c3cc49502g
Do, H. Q.; Daugulis, O. Org. Lett. 2010, 12, 2517. doi: 10.1021/ol100772u
Shu, Z.; Ji, W.; Wang, J. Angew. Chem. Int. Ed. 2014, 53, 2186. doi: 10.1002/anie.201309791
Li, J.; Ackermann, L. Angew. Chem. Int. Ed. 2015, 54, 3635. doi: 10.1002/anie.201409247
Gong, T.-J.; Xiao, B.; Fu, Y. J. Am. Chem. Soc. 2013, 135, 10630. doi: 10.1021/ja405742y
Jiao, N. Nitrogenation Strategy for the Synthesis of N-containing Compounds, Springer, Singapore, 2017, p. 70.
Chaitanya, M.; Yadagiri, D.; Anbarasan, P. Org. Lett. 2013, 15, 4960. doi: 10.1021/ol402201c
Iida, S.; Togo, H. Tetrahedron 2007, 63, 8274. doi: 10.1016/j.tet.2007.05.106
Rokade, B. V.; Malekar, S. K.; Prabhu, K. R. Chem. Commun. 2012, 48, 5506. doi: 10.1039/c2cc31256e
Zhu, C.; Sun, C.; Wei, Y. Synthesis 2010, 4235.
Vatèle J. M. Synlett 2014, 25, 1275. doi: 10.1055/s-0033-1341124
Ishida, T.; Watanabe, H.; Takei, T.; Hamasaki, A.; Tokunaga, M.; Harut, M. Appl. Catal. A. 2012, 425, 85.
Oishi, T.; Yamaguchi, K.; Mizuno, N. Angew. Chem. Int. Ed. 2009, 48, 6286. doi: 10.1002/anie.200900418
Preger, Y.; Root, T. W.; Stahl, S. S. ACS Omega 2018, 3, 6091. doi: 10.1021/acsomega.8b00911
Yadav, D. K. T.; Bhanage, B. M. Eur. J. Org. Chem. 2013, 23, 5106.
Wu, Y.-X.; Xi, Y.-C.; Zhao, M.; Wang, S.-Y. Chin. J. Org. Chem. 2018, 38, 2590. doi: 10.6023/cjoc201804001
Qu, Q.-H.; Gao, X.-F.; Gao, J.; Yuan, G.-Q. Sci. Chin. Chem. 2015, 58, 747. doi: 10.1007/s11426-015-5331-z
Fan, Z.-Q.; Yang, X.-J.; Shen, Z.-L.; Li, M.-C. J. Electrochem. Soc. 2017, 164, 54.
Zhu, Y.-H.; Zhu, Y.; Zeng, H.-Y.; Chen, Z.-Y.; R. D. Little.; Ma, C.-A. J. Electroanal. Chem. 2015, 751, 105.
Zhu, Y.-H.; Chen, Z.-Y.; Zhang, J.-Q.; Wu, Q.-Q.; Ma, C.-A.; Little, R. D. Electrochim. Acta 2016, 207, 308. doi: 10.1016/j.electacta.2016.04.126
Chiou, S.; Hoque, A. K. M. M.; Shine H. J. J. Org. Chem. 1990, 55, 3227. doi: 10.1021/jo00297a045
Zhang, L.; Chen, H.; Zha, Z.-G.; Wang, Z.-Y. Chem. Commun. 2012, 48, 6574. doi: 10.1039/c2cc32800c
Meng, L.; Su, J.-H.; Zha, Z.-G.; Zhang, L.; Zhang, Z.-L.; Wang, Z.-Y. Chem. Eur. J. 2013, 19, 5542. doi: 10.1002/chem.201204207
Zhao, H.-Bo.; Xu, P.; Song, J.-S.; Xu, H.-C. Angew. Chem. Int. Ed. 2018, 57, 15153.
Petrosyan, V. A.; Niyazymbetov, M. E.; Ul'yanova, E. V. Russ. Chem. Bull. 1989, 38, 1548. doi: 10.1007/BF00978458
Petrosyan, V. A.; Niyazymbetov, M. E.; Ul'yanova, E. V. Russ. Chem. Bull. 1990, 39, 546. doi: 10.1007/BF00959580
Hartmera, M. F.; Waldvogel, S. R. Chem. Commun. 2015, 51, 16346. doi: 10.1039/C5CC06437F

图 1 不同水体积含量下p-MeOC6H4CH2OH (0.05 mol•L-1)电氧化循环伏安曲线(v=50 mV•s-1)
Figure 1 Cyclic voltammograms of p-MeOC6H4CH2OH at different volume proportion of H2O (v=50 mV•s-1)

图 3 不同温度下p-MeOC6H4CH2OH (0.05 mol•L-1)电氧化循环伏安曲线(v=50 mV•s-1)
Figure 3 Cyclic voltammograms of p-MeOC6H4CH2OH at different temperature (v=50 mV•s-1)

图 2 不同硫酸浓度下p-MeOC6H4CH2OH (0.05 mol•L-1)电氧化循环伏安曲线(v=50 mV•s-1)
Figure 2 Cyclic voltammograms of p-MeOC6H4CH2OH at different concentration of H2SO4 (v=50 mV•s-1)

图式 2 可能的电氧化氰化反应机理
Scheme 2 Plausible reaction mechanism of the electro-oxida-tion cyanidation
表 1 p-MeOC6H4CH2OH电合成p-MeOBN反应条件的优化a
Table 1. Optimization of the electro-synthesis of p-MeOBN from p-MeOC6H4CH2OH
|
|||||||
| Entry | Bu4NClO4 equiv. | Anode/Cathode | j/(mA•cm-2) | Solvent | T/℃ | Conv. b/% | Yieldc/% |
| 1 | 0.3 | Pt/Pb | 10.0 | DMF/H2O | 60 | 85 | 40 |
| 2 | 0.6 | Pt/Pb | 10.0 | DMF/H2O | 60 | 98 | 72 |
| 3 | 1.0 | Pt/Pb | 10.0 | DMF/H2O | 60 | 97 | 83 |
| 4 | 1.2 | Pt/Pb | 10.0 | DMF/H2O | 60 | 99 | 81 |
| 5 | 1.0 | Pt/Pb | 12.5 | DMF/H2O | 60 | 97 | 78 |
| 6 | 1.0 | Pt/Pb | 15.0 | DMF/H2O | 60 | 96 | 70 |
| 7 | 1.0 | Pt/Pb | 10.0 | DMF/H2O | 30 | 33 | 16 |
| 8 | 1.0 | Pt/Pb | 10.0 | DMF/H2O | 40 | 96 | 55 |
| 9 | 1.0 | Pt/Pb | 10.0 | DMF/H2O | 50 | 98 | 74 |
| 10 | 1.0 | Pt/C | 10.0 | DMF/H2O | 60 | 99 | 42 |
| 11 | 1.0 | Pt/Pt | 10.0 | DMF/H2O | 60 | 99 | 78 |
| 12 | 1.0 | Pt/Pb | 10.0 | CH3OH/H2O | 60 | 90 | 13 |
| 13 | 1.0 | Pt/Pb | 10.0 | C2H5OH/H2O | 60 | 95 | 48 |
| 14 | 1.0 | Pt/Pb | 10.0 | CH3CN/H2O | 60 | 99 | 60 |
| 15 | 1.0 | Pt/Pb | 10.0 | DMSO/H2O | 60 | 98 | 90 |
| 16d | 1.0 | Pt/Pb | 10.0 | DMSO/H2O | 60 | 99 | 36 |
| 17d | 1.0 | Pt/Pb | 10.0 | DMSO/H2O | 60 | 99 | 22 |
| a p-MeOC6H4CH2OH (1.5 mmol), NH2OH (3.0 equiv), H2SO4 (0.15 mol•L-1), solvent/H2O (30%, 30 mL) was electrolyzed at a constant current in an undivided cell, t=8 h, Pt/Pb (2 cm×2 cm); b Conversions were determined by GC; c GC yields of nitriles; d H2SO4 (0.25, 0.35 mol•L-1). | |||||||
 下载: 导出CSV
下载: 导出CSV
表 2 不同取代芳香伯醇电氧化氰化合成腈的反应a
Table 2. Electro-oxidation cyanation of aromatic primary alcohols to corresponding nitriles
|
|
|||||
| Entry | Product | j/(mA•cm-2) | Time/h | Conv. b/% | Yieldc/% |
| 1 | |
10.0 | 10 | 99 | 90 |
| 2 | |
17.5 | 9 | 99 | 85 |
| 3 | |
17.5 | 13 | 59 | 15 |
| 4 | |
17.5 | 13 | 99 | 92 |
| 5 | |
12.5 | 9 | 99 | 89 |
| 6 | |
12.5 | 11 | 96 | 85 |
| 7 | |
17.5 | 10 | 97 | 89 |
| 8 | |
20.0 | 11 | 98 | 90 |
| 9 | |
20.0 | 11 | 89 | 59 |
| 10 | |
20.0 | 11 | 99 | 86 |
| 11 | |
20.0 | 10 | 99 | 79 |
| 12 | |
17.5 | 13 | 93 | 61 |
下载: 导出CSV
表 3 不取代芳香醛电氧化氰化合成腈a
Table 3. Electro-oxidation cyanation of aromatic aldehydes to corresponding nitriles
|
||||||
| Entry | Substrte | Time/h | Conv. b/% | Yieldc/% | ||
| a | b | c | ||||
| 1 | |
8 | 98 | 85 | 9 | n.d. |
| 2 | |
9 | 95 | 78 | 6 | 11 |
| 3 | |
9 | 98 | 75 | n.d. | 23 |
| a Aromatic aldehydes (1.5 mmol), NH2OH-H2SO4 (3.0 equiv.), Bu4NClO4 (1.0 equiv.), Pt (2 cm×2 cm), Pb (2 cm×2 cm), j (10 mA/cm2), solvent/H2O (30%, 30 mL), 60 ℃, Undivided cell; b Conversions were determined by GC; c GC yields of nitriles; n.d.=no detection. | ||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: