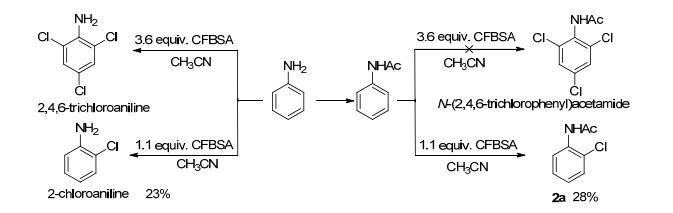
Scheme 1.
Chlorination of aniline and acetanilide

A range of functional molecules, from natural products to synthetic drugs, can be afforded through transition metal catalyzed transformations.[1] Palladium-catalyzed C—H activation is particularly a valuable tool to construct C—C and C—X bonds in synthetic chemistry.[2] During the process of palladium-catalyzed cycle, oxidant plays a significant role, such as aryl iodide, [3] air, [4] K2S2O8, [5] oxone, [6] TBHP, [7] electrochemical anodic oxidation, [8] etc. In addition, several fluorine-containing reagents also exhibit oxidant characteristics. Yu and co-workers[9] exploited para- selective C—H arylation of monosubstituted arenes using a F+ reagent, 1-fluoro-2, 4, 6-trimethylpyridinium triflate (NFTMPT), as a bystanding oxidant. N-chloro-N-fluorobenzenesulfonylamide (NFSI) is widely acted as both oxidant and nitrogen source to achieve palladium- catalyzed C(sp3)—H sulfonamidation.[10] Liu et al.[11] applies SelectFluor as a strong oxidant to Pd-catalyzed functionalization of alkenes. Our group also reported Pd-catalyzed tandem carboxylation and direct intramolecular coupling with SelectFluor as an oxidant.[12] In this paper, we applied N- chloro-N-fluorobenzenesulfonylamide (CFBSA)[13] as both oxidant and chlorine source into Pd catalyzed C(sp2)—H chlorination of anilides.
Over the past decade, significant achievements have been achieved on aromatic chlorination. Many chlorinating reagents have been developed and applied to chlorination of C(sp2)—H on the benzene ring. Chlorinated acetanilide is generally afforded by N-chlorosuccinimide (NCS), [14] CuCl2, [15] t-BuOCl[16] and a secondary ammonium salt organocatalyst.[17] Koenig et al.[18] reported an efficient and practical system for C—H chlorination utilizing a photocatalytic oxidative process based on the organic dye riboflavin tetraacetate that mimicks the enzymatic process. Moreover, Bedford and co-workers[19] reported a solvent-free aromatic C—H chlorination reaction. Very recently, a visible light-mediated regioselective halogenation of anilides was discovered by Ghosh et al.[20] Despite of various methods available for chlorination, the speed and completeness under mild condition continue to be challenging.
Due to the strong electronegativity of the fluorine atom, the chlorine atom of CFBSA showed an electropositive feature. Therefore, CFBSA is equipped with the property of oxidant. Several studies show that it is also an efficient chlorinating reagent, such as aminochlorination of alke- nes, [21, 23] chlorination of carbonyl and nitrogen- containing aromatic heterocyclic compounds.[13] Particularly, aniline could be chlorinated to afford 2, 4, 6- trichloroaniline with CFBSA as chlorinating reagent.[13] Although chlorination with CFBSA yielded monochlorinated product, the reaction system is complex and the result is unsatisfactory with monochlorinated product 2a in 28% yield (Scheme 1). Inspired by previous research, this study attempted to directedly chlorinate the C(sp2)—H of arenes using CFBSA as both oxidant and chlorine source. Therefore, acetanilide/Pd(OAc)2/CFBSA was selected to realize the ortho-selective chlorination.
One equivalent of CFBSA reacted with acetanilide in CH3CN and 1, 2-dichloroethane (DCE) (Table 1, Entries 1~2). The yield was low because the substrate was not consumed completely. Surprisingly, when using 20 mol% Pd(OAc)2 as catalyst, most of the substrate was consumed, but the yield still remained low (Table 1, Entry 3). And the reaction system became more simple, when monitored by TLC. Different additives were added to the reaction to promote the transformation of substrates, including CH3COOH, CF3SO3H, HCl, NaOCl, K2CO3, FeCl3, CuCl2, and p-toluenesulfonic acid (PTSA) (Table 1, Entries 4~11), and the results showed that PTSA was the best choice for the reaction. Different solvents were also tested, including ether, ester, nitrile, and toluene (Table 1, Entries 11~18). DCE was found to be the most suitable solvent, which yielded 64% of the desired product at room temperature. Furthermore, the yield of ortho-chlorinated acetanilide was lowered to 31% when the temperature was increased to 80 ℃ (Table 1, Entries 19~20). The product 2a could also be obtained at 62% yield in ice water bath, however, the time of the reaction was prolonged beyond 24 h (Table 1, Entry 21). After further screening, the loading of Pd(OAc)2 was reduced to 6 mol% with 72% isolated yield, which largely affected the reaction (Table 1, Entries 22~23). Notably, the reaction could also proceed with 74% desired product yield while lowering the amount of PTSA to 0.4 equiv. (Table 1, Entries 24~26). As a result, the optimal reaction conditions were determined as follows: acetanilide (1.0 equiv.), CFBSA (1.0 equiv.), Pd(OAc)2 (6 mol%), PTSA (0.4 equiv.), DCE as solvent, and room temperature (Table 1, Entry 25).
 下载:
导出CSV
下载:
导出CSV
 |
||||||
| Entrya | Catalyst/mol% | Additive/equiv. | Solvent | T/℃ | Time/h | Yieldb/% |
| 1 | — | — | CH3CN | 25 | 24 | 28 |
| 2 | — | — | DCE | 25 | 24 | 32 |
| 3 | 20 | — | DCE | 25 | 12 | 47 |
| 4 | 20 | CH3COOH (1.0) | DCE | 25 | 8 | 15 |
| 5 | 20 | CF3SO3H (1.0) | DCE | 25 | 8 | 36 |
| 6 | 20 | HCl (1.0) | DCE | 25 | 12 | 28 |
| 7 | 20 | NaOCl (1.0) | DCE | 25 | 12 | 23 |
| 8 | 20 | FeCl3 (1.0) | DCE | 25 | 8 | 21 |
| 9 | 20 | CuCl2 (1.0) | DCE | 25 | 8 | 16 |
| 10 | 20 | K2CO3 (1.0) | DCE | 25 | 8 | n.d. |
| 11 | 20 | PTSA (1.0) | DCE | 25 | 3 | 64 |
| 12 | 20 | PTSA (1.0) | Toluene | 25 | 8 | 46 |
| 13 | 20 | PTSA (1.0) | Ethyl acetate | 25 | 12 | 52 |
| 14 | 20 | PTSA (1.0) | Methanol | 25 | 12 | 27 |
| 15 | 20 | PTSA (1.0) | 2-Methoxyethanol | 25 | 12 | 35 |
| 16 | 20 | PTSA (1.0) | 1, 4-Dioxane | 25 | 12 | 25 |
| 17 | 20 | PTSA (1.0) | CH3CN | 25 | 12 | 21 |
| 18 | 20 | PTSA (1.0) | CCl4 | 25 | 10 | 32 |
| 19 | 20 | PTSA (1.0) | DCE | 40 | 2 | 33 |
| 20 | 20 | PTSA (1.0) | DCE | 80 | 2 | 31 |
| 21 | 20 | PTSA (1.0) | DCE | 0 | 24 | 62 |
| 22 | 6 | PTSA (1.0) | DCE | 25 | 6 | 72 |
| 23 | 4 | PTSA (1.0) | DCE | 25 | 10 | 55 |
| 24 | 6 | PTSA (0.2) | DCE | 25 | 12 | 57 |
| 25 | 6 | PTSA (0.4) | DCE | 25 | 6 | 74 |
| 26 | 6 | PTSA (1.4) | DCE | 25 | 6 | 54 |
| a Reaction conditions: acetanilide (0.3 mmol), CFBSA (1 equiv.), additive, catalyst, solvent (4 mL), and temperature. b Isolated yield. | ||||||
With the optimized conditions in hand (Table 1, Entry 25), virous substituted-anilides were surveyed and the results were listed in Table 2. Substrates with both electron-donating and electron-withdrawing groups on the aromatic ring were attempted. As shown, the results indicate that the electronic characteristics of substituents on the aromatic ring did not considerably influence the yields of desired products. A series of acetanilides bearing halogen atoms at para position of aromatic ring provided desired chlorinated products at 62%~73% yields (2c, 2d, and 2k). meta-Chlorinated substrate can also provide an ortho-chlorinated product with 62% yield (2j); however, the ortho-chlorinated acetanilide can not even react under this condition. Furthermore, substrates with alkyl groups, such as methyl (2b), isopropyl (2m), and t-butyl (2n) groups, could achieve desired acetanilides with moderate yields. The steric hindrance effect on the para position of arene had no effect on this reaction owning to the compariable yields of 2n and 2b. Acetanilide with a methyl group at the meta position yielded 2r at 46%. Acetanilides with methoxy group on the benzene ring yielded corresponding products at relatively low yields (2e, 2f, and2g). The methoxy group in 2e has an electron- withdrawing effect to the ortho position hydrogen atom, while the methoxy group in 2f shows electron-donating conjugation effect and improves the reactivity of the ortho-position hydrogen. The low yield of 2g may due to the steric hindrance effect. The chlorination of strong electron-withdrawing acetanilides achieved different results. para-Trifluoromethyl (2i) and trifluoromethoxy (2u) substituted acetanilide produced the corresponding desired products at 67% and 65% yields, respectively. However, para-cyano acetanilide (2v) could not react with CFBSA at 70 ℃ overnight. Under optimized reaction conditions, the para-phenyl substrate 1o was successfully chlorinated at the ortho position and yielded 2o in 60%. para-Carbonyl substituted acetanilide yielded 2h at 82%, while meta-carbonyl substituted substrate yielded the corresponding product 2s in 46%. However, the yield of substrate with both methoxy and carbonyl groups provided 2t in 46%. When the para position of the aromatic ring is occupied by groups that can be conjugated with a benzene ring, the reaction showed great consequences. The conjugation effect could enrich the density of the electron cloud on the benzene ring, which increased the activity of C(sp2)—H and promoted the completion of the reaction. Actually, trace amount of para-chlorinated products can be obtained when para position is unoccupied (2f, 2j and 2r). In addition to the acetyl group, the benzoyl group (2p), pivalyl group (2q), and phenylpropanyl group (2l) could be utilized as directing groups. These substrates yielded chlorinating acetanilides in moderate yields. However, the reactions of ortho-substituted substrates could not occur even by heating the reaction to 70 ℃, when the ortho position has been occupied (2w, 2x, and 2y).
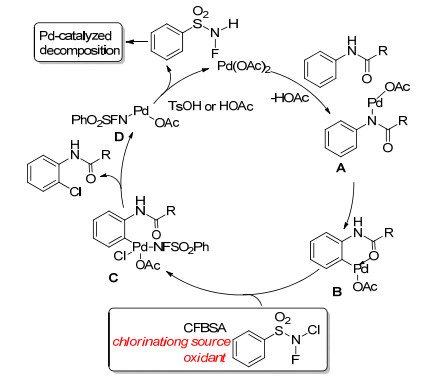
On the basis of the obtained results, a plausible mechanism for ortho-chlorination of acetanilide was proposed. A Pd(Ⅱ)/Pd(Ⅳ) catalytic process was supposed in Scheme 2. Intermediate B was considered to originate from C—H insertion of intermediate A. Intermediate C is formed from oxidative addition of intermediate B using CFBSA. PTSA provides protons to promote both the catalytic process and ligand exchange. A desired ortho-chlorinated product is afforded from intermediate C, undergoing a reductive elimination process. Intermediate D was transformed to Pd(OAc)2 and provided N-fluoroben- zenesulfonamide. However, this was not observed when monitoring the reaction with TLC. Mechanism experi- ments showed that N-fluorobenzenesulfonamide was decomposed under Pd(OAc)2.
Under the optimized conditions, several chlorinating reagents were chosen for comparison of reactivity with CFBSA, and the results are shown in Table 3. For N- chloro-N-methoxybenzenesulfonamide (CMOBSA)[22] and NCS (Table 3, Entries 2~3), 1a can not consumed completely after stirring overnight and formed 2a at moderate yields. Differently, in Bedford's method, the chlorination of acetanilide with NCS in toluene gives 80% isolated yield.[19] With t-BuOCl as chlorinating reagent, 60% yield of 2a was obtained (Table 3, Entry 4). Moreover, the use of trichloroisocyanuric acid yielded 2a with 53% yield (Table 3, Entry 5).
下载:
导出CSV
 |
||
| Entry | Chlorinating reagent | Isolated yieldb/% |
| 1 | CFBSA | 74 |
| 2 | CMOBSA | 41 |
| 3 | NCS | 58 |
| 4 | t-BuOCl | 60 |
| 5 | Trichloroisocyanuric acid | 53 |
| a The reaction conditions are as follows: acetanilide (1.0 equiv., 0.03mmol), Pd(OAc)2 (6 mol%), chlorinating reagent (1.0 equiv.), PTSA (0.4 equiv.), DCE, room temperature. b Isolated yields. | ||
In summary, a mild and simple palladium catalyzed C—H activation and chlorination of anilides using CFBSA was developed in our laboratory. CFBSA shows unique property comparing with other common chlorinating reagents. (1) CFBSA contains both chlorine and fluorine atoms, and (2) CFBSA shows chlorination activity rather than fluorination activity in this reaction. Especially, ortho- monochlorinated products can be afforded when para- position was occupied. CFBSA shows ortho/para sele- ctivity when para-position was not occupied; (3) the byproduct N-fluorobenzenesulfonylamine can be decom- posed in the presence of Pd(OAc)2, which promotes accomplishment of the reaction. This report will expand the possibility of CFBSA as a high-efficient chlorinating reagent and oxidant in catalysis process. Further detailed mechanistic investigation is currently proceeding.
All the starting chemicals were commercially available and used without further purification. Substrates were purchased from Energy chemical Co. Ltd. Damas-beta Co. Ltd. Fluorinating reagent (Selectfluor) and N-fluorobenze- nesulfonamide were purchased from Shanghai Science Bio-pharmaceutical Co. Ltd. DCE was freshly distilled over P2O5. Flash column chromatography was performed using silica gel (300~400 mesh).
To a solution of acetanilide 1 (0.3 mmol, 1.0 equiv.), Pd(OAc)2 (0.018 mmol, 6 mol%) and PTSA (0.12 mmol, 0.4 equiv.) in DCE (4 mL) were added CFBSA (0.3 mmol, 1.0 equiv.). The reaction mixture was stirred until the starting material was consumed. The product 2 was obtained by flash column chromatography on silica gel [eluent: V(petroleum ether):V(ethyl acetate)=20:1].
N-(2-Chlorophenyl)acetamide (2a): White solid (74% yield), m.p. 90.7~91.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.35 (d, J=8.3 Hz, 1H), 7.62 (bs, 1H), 7.36 (dd, J=8.0, 1.5 Hz, 1H), 7.30~7.23 (m, 1H), 7.05~7.01 (m, 1H), 2.24 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.4, 134.7, 129.1, 127.9, 124.7, 122.6, 121.7, 25.0; IR (KBr) ν: 3238, 1661, 1552, 1526, 1439, 1354, 1346 1061 cm-1; HRMS-EI calcd for C8H8ClNO 169.0294, found 169.0293.
N-(2-Chloro-4-methylphenyl)acetamide(2b): White solid (54% yield), m.p. 115.7~117.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.18 (d, J=8.4Hz, 1H), 7.52 (bs, 1H), 7.17 (s, 1H), 7.06 (dd, J=8.5, 1.9 Hz, 1H), 2.29 (s, 3H), 2.22 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.3, 134.9, 132.1, 129.4, 128.4, 122.6, 121.7, 24.9, 20.8; IR (KBr) ν: 3277, 2934, 1659, 1534, 1358, 1294, 1058 cm-1; HRMS-EI calcd for C9H10ClNO 183.0451, found 183.0452.
N-(2, 4-Dichlorophenyl)acetamide (2c): White solid (73% yield), m.p. 144.7~146.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.34 (d, J=8 Hz 1H), 7.55 (bs, 1H), 7.38 (d, J=4 Hz, 1H), 7.25 (dd, J=8, 4 Hz, 1H), 2.24 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 168.4, 133.5, 129.2, 128.8, 128.0, 123.2, 122.5, 24.9; IR (KBr) ν: 3275, 1668, 1607. 1517, 1370, 1305, 1078, 1059 cm-1. HRMS-EI calcd for C8H7Cl2NO 202.9905, found 202.9903.
N-(4-Bromo-2-chlorophenyl)acetamide (2d): White solid (62% yield), m.p. 146.7~148.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.26 (d, J=8.9 Hz, 1H), 7.58 (bs, 1H), 7.50 (d, J=2.2 Hz, 1H), 7.36 (dd, J=8.9, 2.2 Hz, 1H), 2.23 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.4, 133.9, 131.5, 130.9, 123.3, 122.7, 116.3, 25.0; IR (KBr) ν: 3281, 3028, 2768, 2334, 1666, 1584, 1371, 1294, 1080 cm-1; HRMS-EI calcd for C8H7BrClNO 246.9400, found 246.9397.
N-(4-Methoxyphenyl)acetamide (2e): White solid (41% yield), m.p. 114.6~116.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.12 (d, J=9.1 Hz, 1H), 7.42 (bs, 1H), 6.91 (d, J=2.8 Hz, 1H), 6.81 (dd, J=9.1, 2.9 Hz 1H), 3.77 (s, 3H), 2.21 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 168.3, 156.4, 128.0, 124.3, 123.5, 114.6, 113.3, 55.8, 24.7; IR (KBr) ν: 3276, 2915, 1657, 1533, 1373, 1280, 1049 cm-1; HRMS-EI calcd for C9H10ClNO2 199.0400, found 199.0398.
N-(2-Chloro-5-methoxyphenyl)acetamide (2f): White solid (52% yield), m.p. 100.5~102.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.05 (d, J=3.0 Hz, 1H), 7.62 (bs, 1H), 7.22 (d, J=8.9 Hz, 1H), 6.59 (dd, J=8.9, 2.9 Hz, 1H), 3.79 (s, 3H), 2.23 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 168.5, 159.0, 135.3, 129.2, 113.8, 111.1, 106.7, 55.7, 25.0; IR (KBr) ν: 2931, 1675, 1591, 1526, 1466, 1313, 1275, 1225, 1035 cm-1; HRMS-EI calcd for C9H10ClNO2 199.0400, found 199.0401.
N-(2-Chloro-5-methoxy-4-methylphenyl)acetamide (2g): White solid (28% yield), m.p. 119.3~121.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.00 (s, 1H), 7.54 (bs, 1H), 7.09 (s, 1H), 3.83 (s, 3H), 2.23 (s, 3H), 2.14 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.4, 156.8, 133.1, 129.9, 123.5, 112.8, 103.9, 55.8, 25.1, 15.8; IR (KBr) ν: 3270, 1667, 1591, 1522, 1392, 1167 cm-1; HRMS-EI calcd for C10H12ClNO2 213.0557, found 213.0556.
Methyl 4-acetamido-3-chlorobenzoate (2h): White solid (82% yield), m.p. 107.8~109.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.49 (d, J=8.7 Hz, 1H), 8.03 (d, J=1.9 Hz, 1H), 7.91 (dd, J=8.7, 2.0 Hz, 1H), 7.81 (bs, 1H), 3.91 (s, 3H), 2.28 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.5, 165.6, 138.6, 130.4, 129.4, 126.1, 122.0, 120.4, 52.4, 25.1; IR (KBr) ν: 3290, 2957, 1732, 1666, 1526, 1435, 1275, 1225, 1118 cm-1; HRMS-EI calcd for (C10H10ClNO3) 227.0349, found 227.0347.
N-(2-Chloro-4-(trifluoromethyl)phenyl)acetamide (2i): White solid (67% yield), m.p. 131.5~133.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.55 (d, J=8.7 Hz, 1H), 7.76 (bs, 1H), 7.62 (d, J=2.0 Hz, 1H), 7.51 (dd, J=8.7, 2.0 Hz, 1H), 2.27 (s, 3H); 13C NMR (151 MHz, CDCl3) δ:168.6, 137.7, 126.3 (q, J=4.1 Hz), 125.1 (q, J=4.0 Hz), 124.3, 122.5, 122.3, 121.1, 25.1; 19F NMR (376 MHz, CDCl3) δ: -62.30 (s, 3F); IR (KBr) ν: 3279, 3017, 1672, 1531, 1495, 1404, 1325, 1307, 1279, 1170, 1119, 1080 cm-1; HRMS-EI calcd for (C9H7ClF3NO) 237.0168, found 237.0169.
N-(2, 5-Dichlorophenyl)acetamide (2j): White solid (37.8 mg, 62% yield), m.p. 129.5~131.2 ℃; 1H NMR (400 MHz, CDCl3)δ: 8.45 (s, 1H), 7.61 (bs, 1H), 7.31~7.24 (m, 1H), 7.01 (ddd, J=8.6 Hz, 2.5 Hz, 1.3 Hz, 1H), 2.18 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 168.4, 135.5, 133.7, 129.7, 124.6, 121.4, 120.5, 25.0; IR (KBr) ν: 3281, 2359, 1666, 1579, 1520, 1462, 1404, 1369, 1277, 1092, 1053 cm-1; HRMS-EI calcd for (C8H7Cl2NO) 202.9905, found 202.9908.
N-(2-Chloro-4-fluorophenyl)acetamide (2k): White solid (67% yield), m.p. 105.6~107.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.28 (dd, J=9.2 Hz, 5.6 Hz, 1H) 7.48 (bs, 1H), 7.12 (dd, J=8.1 Hz, 2.9 Hz, 1H), 6.99 (ddd, J=9.2 Hz, 7.9 Hz, 2.9 Hz, 1H), 2.23 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.4, 159.3, 157.7, 131.1, 123.2 (d, J=8.6 Hz), 116.3 (d, J=26.1 Hz), 114.7 (d, J=22Hz), 24.8; 19F NMR (376 MHz, CDCl3) δ: -116.34 (s, 1F); IR (KBr) ν: 3279, 2920, 2849, 1666, 1535, 1487. 1393, 1304, 1248, 1190, 1043 cm-1; HRMS-EI calcd for C8H7ClFNO 187.0200, found 187.0201.
N-(2-Chlorophenyl)-3-phenylpropanamide (2l): White solid (53% yield), m.p. 118.9~120.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.36 (d, J=8.0 Hz, 1H) 7.56 (bs, 1H), 7.37-7.18 (m, 7H), 7.02 (td, J=7.8 Hz, 1.6 Hz, 1H), 3.08 (t, J=7.7 Hz, 2H), 2.75 (t, J=7.7 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ: 170.4, 140.4, 134.6, 129.0, 128.7, 128.4, 127.8, 126.5, 124.7, 122.7, 121.8, 39.6, 31.5; IR (KBr) ν: 3274, 1655, 1526, 1440, 1378, 1354, 1300, 1062 cm-1; HRMS-EI calcd for C15H14ClNO 259.0764, found 259.0765.
N-(2-Chloro-4-isopropylphenyl)acetamide (2m):White solid (50% yield), m.p. 111.2~113.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.19 (d, J=8.4 Hz, 1H), 7.56 (bs, 1H), 7.21 (d, J=1.7 Hz, 1H), 7.11 (dd, J=8.5 Hz, 2.1 Hz, 1H), 2.89-2.79 (m, 1H), 2.21 (s, 3H), 1.22 (s, 3H) 1.20 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 168.3, 146.0, 132.3, 126.9, 125.9, 122.8, 122.0, 33.6, 24.8, 23.9; IR (KBr) ν: 3262, 3036, 2959, 1659, 1587, 1537, 1404, 1371, 1307, 1059, 1016 cm-1; HRMS-EI calcd for C11H14ClNO 211.0764, found 211.0764.
N-(4-(tert-Butyl)-2-chlorophenyl)acetamide (2n): White solid (49% yield), m.p. 147.9~150.1 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.22 (d, J=8.6 Hz, 1H), 7.53 (bs, 1H), 7.35 (d, J=2.1 Hz, 1H), 7.28 (dd, J=8.7 Hz, 2.2 Hz, 1H), 2.23 (s, 3H), 1.29 (s, 9H); 13C NMR (101 MHz, CDCl3) δ: 168.3, 148.4, 132.0, 126.0, 124.9, 122.6, 121.6, 34.6, 31.3, 24.9; IR (KBr) ν: 3264, 3225, 2963, 1659, 1585, 1537, 1387, 1275, 1057 cm-1; HRMS-EI calcd for (C12H16ClNO) 225.0920, found 225.0921.
N-(3-Chloro-[1, 1'-biphenyl]-4-yl)acetamide (2o): White solid (44.1 mg, 60% yield); m.p. 140.3~142.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.42 (d, J=8.5 Hz, 1H), 7.67 (bs, 1H), 7.60 (d, J=2.0 Hz, 1H) 7.56-7.52 (m, 2H) 7.50 (dd, J=8.6 Hz, 2.1 Hz, 1H), 7.43 (t, J=7.5 Hz, 2H), 7.35 (t, J=7.3 Hz, 1H), 2.26 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.4, 139.3, 137.9, 133.7, 129.0, 127.8, 127.4, 126.9, 126.4, 123.0, 121.9, 25.0; IR (KBr) ν: 3285, 2918, 2849, 1663, 1526, 1499, 1385, 1298, 1059 cm-1; HRMS-EI calcd for (C14H12ClNO) 245.0607, found 245.0605.
N-(2-Chlorophenyl)benzamide (2p): White solid (73% yield); m.p. 104.8~106.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.56 (dd, J=8.3 Hz, 1.7 Hz, 1H), 8.46 (s, 1H), 7.92 (dd, J=7.1 Hz, 1.9 Hz, 2H), 7.57 (dd, J=8.3 Hz, 6.1 Hz, 1H), 7.51 (td, J=7.0 Hz, 6.1 Hz, 1.7 Hz, 2H), 7.41 (dd, J=8.0 Hz, 1.7 Hz, 1H), 7.32 (t, J=7.9 Hz, 1H), 7.08 (td, J=7.8 Hz, 3.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 165.3, 134.8, 134.7, 132.3, 129.1, 129.0, 127.9, 127.2, 124.8, 123.2, 121.7; IR (KBr) ν: 3283, 1658, 1531, 1440, 1308, 1048 cm-1; HRMS-EI calcd for (C13H10ClNO) 231.0451, found 231.0450.
N-(2-Chlorophenyl)pivalamide (2q): White solid (74% yield); m.p. 75.2~77.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.40 (dd, J=8.3 Hz, 1.5 Hz, 1H), 8.01 (bs, 1H), 7.35 (dd, J=8.0 Hz, 1.5 Hz, 1H), 7.28~7.24 (m, 1H), 7.05~7.98 (m, 1H), 1.34 (s, 9H); 13C NMR (101 MHz, CDCl3) δ: 176.8, 134.9, 129.0, 127.9, 124.5, 123.0, 121.5, 40.3, 27.7; IR (KBr) ν: 3428, 1670, 1591, 1578, 1510, 1439, 1305, 1291, 1163, 1047 cm-1; HRMS-EI calcd for C11H14ClNO 211.0764, found 211.0765.
N-(2-Chloro-5-methylphenyl)acetamide (2r):White solid (46% yield); m.p. 92.7~94.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.18 (s, 1H), 7.57 (bs, 1H), 7.22 (d, J=8.2 Hz, 1H), 6.84 (dd, J=8.4 Hz, 2.1 Hz, 1H), 2.33 (s, 3H), 2.23 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.4, 138.0, 134.2, 128.6, 125.6, 122.3, 119.7, 25.0, 21.4; IR (KBr) ν: 3267, 3043, 2922, 1663, 1585, 1533, 1541, 1472, 1415, 1373, 1140, 1055, 1026 cm-1; HRMS-EI calcd for (C9H10ClNO) 183.0451, found 183.0450.
Methyl 3-acetamido-4-chlorobenzoate (2s):White solid (35% yield); m.p. 120.6~123.1 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.96 (s, 1H), 7.72 (dd, J=8.4 Hz, 2.1 Hz, 1H), 7.64 (bs, 1H), 7.43 (d, J=8.3 Hz, 1H), 3.90 (s, 3H), 2.26 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.4, 166.2, 134.7, 129.9, 129.2, 127.5, 125.8, 122.8, 52.5, 24.9; IR (KBr) ν: 3262, 2959, 1724, 1670, 1533, 1431, 1369, 1250, 1103, 1024 cm-1; HRMS-EI calcd for C10H10ClNO3 227.0349, found 227.0348.
Methyl 4-acetamido-5-chloro-2-methoxybenzoate (2t): White solid (46% yield); m.p. 152.7~155.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.30 (s, 1H), 7.87 (s, 1H), 7.77 (bs, 1H), 3.92 (s, 3H), 3.86 (s, 3H), 2.27 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.8, 165.0, 159.4, 139.1, 132.1, 115.1, 112.5, 104.3, 56.5, 52.2, 25.3; IR (KBr) ν: 3420, 1718, 1705, 1576, 1404, 1228, 1103, 1053 cm-1. HRMS-EI calcd for C11H12ClNO4 257.0455, found 257.0453.
N-(2-Chloro-4-(trifluoromethoxy)phenyl)acetamide (2u): White solid (65% yield), m.p. 104.4~106.1 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.42 (d, J=9.1 Hz, 1H), 7.58 (br, 1H), 7.29~7.27 (m, 1H), 7.19~7.13 (m, 1H), 2.25 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 168.4, 144.7, 133.6, 123.1, 122.2 (d, J=35.2 Hz), 121.3, 120.7, 119.6, 24.9; 19F NMR (376 MHz, CDCl3) δ: -58.2 (s, 3F); IR (KBr) ν: 3294, 1664, 1535, 1485, 1395, 1285, 1207, 1149, 1051 cm-1; HRMS-EI calcd for C9H7ClF3NO2 253.0117, found 253.0119.
Supporting Information Optimization of reaction conditions. 1H NMR, 13C NMR and 19F NMR spectra of compounds. TLC pictures of each reaction. Mechanism experiments. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Kinsinger, T.; Kazmaier, U. Org. Lett. 2018, 20, 7726.
(b) Nasrollahzadeh, M.; Issaabadi, Z.; Tohidi, M. M.; Sajadi, S. M. Chem. Rev. 2018, 18, 165.
(c) Ding, H.; Li, J.; Guo, Q.; Xiao, Y. Chin. J. Org. Chem. 2017, 37, 3112.
(d) Wang, J.; Li, F.; Yu, X.; Liu, L.; Ding, J.; Xie, P.; Wang, J. Chin. J. Org. Chem. 2018, 38, 1638.
(e) Davie, E. A. C.; Mennen, S. M.; Xu, Y.; Miller, S. J. Chem. Rev. 2007, 107, 5759.
(a) Seifert, S.; Schmidt, D.; Shoyama, K.; Wuerthner, F. Angew. Chem., Int. Ed. 2017, 56, 7595.
(b) Li, H.; Shi, Z.-J. Prog. Chem. 2010, 22, 1414.
(c) Liang, J.-Y.; Shen, S.-J.; Xu, X.-H.; Fu, Y.-L. Org. Lett. 2018, 20, 6627.
(d) Manikandan, T. S.; Ramesh, R.; Semeril, D. Organometallics 2019, 38, 319.
(e) Wang, Y.; Zeng, J.; Cui, X. Chin. J. Org. Chem. 2010, 30, 181.
(a) Sehnal, P.; Taylor, R. J. K.; Fairlamb, I. J. S. Chem. Rev. 2010, 110, 824.
(b) Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147.
(a) Dilauro, G.; Quivelli, A. F.; Vitale, P.; Capriati, V.; Perna, F. M. Angew. Chem., Int. Ed. 2019, 58, 1799.
(b) Youn, S. W.; Kim, Y. H.; Jo, Y. H. Adv. Synth. Catal. 2019, 361, 462.
(a) Mei, C.; Lu, W. J. Org. Chem. 2018, 83, 4812.
(b) Wang, G.-W.; Yuan, T.-T.; Wu, X.-L. J. Org. Chem. 2008, 73, 4717.
(c) Karthikeyan, J.; Cheng, C.-H. Angew. Chem., Int. Ed. 2011, 50, 9880.
(a) Gao, X.-A.; Yan, R.-L.; Wang, X.-X.; Yan, H.; Li, J.; Guo, H.; Huang, G.-S. J. Org. Chem. 2012, 77, 7700.
(b) Xing, X.; O'Connor, N. R.; Stoltz, B. M. Angew. Chem., Int. Ed. 2015, 54, 11186.
(c) Saito, F.; Aiso, H.; Kochi, T.; Kakiuchi, F. Organometallics 2014, 33, 6704.
(a) Lu, O.; Huang, J.; Li, J.; Qi, C.; Wu, W.; Jiang, H. Chem. Commun. 2017, 53, 10422.
(b) Khatun, N.; Modi, A.; Ali, W.; Patel, B. K. J. Org. Chem. 2015, 80, 9662.
Ma, C.; Zhao, C.-Q.; Li, Y.-Q.; Zhang, L.-P.; Xu, X.-T.; Zhang, K.; Mei, T.-S. Chem. Commun. 2017, 53, 12189. doi: 10.1039/C7CC07429H
Wang, X.; Leow, D.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 13864. doi: 10.1021/ja206572w
(a) Dong, Y.; Liu, G. J. Org. Chem. 2017, 82, 3864.
(b) Jin, L.; Zeng, X.; Li, S.; Hong, X.; Qiu, G.; Liu, P. Chem. Commun. 2017, 53, 3986.
(a) Chen, C.-H.; Luo, Y.-X.; Fu, L.; Chen, P.-H.; Lan, Y.; Liu, G.-S. J. Am. Chem. Soc. 2018, 140, 1207.
(b) Chen, C.-H.; Chen, P.-H.; Liu, G.-S. J. Am. Chem. Soc. 2015, 137, 15648.
Liu, R.; Lu, Z.-H.; Hu, X.-H.; Li, J.-L.; Yang, X.-J. Org. Lett. 2015, 17, 1489. doi: 10.1021/acs.orglett.5b00376
Lu, Z.-H.; Li, Q.-W.; Tang, M.-H.; Jiang, P.-P.; Zheng, H.; Yang, X.-J. Chem. Commun. 2015, 51, 14852. doi: 10.1039/C5CC05052A
(a) Kim, K.; Jung, Y.; Lee, S.; Kim, M., Shin, D.; Byun, H.; Cho, S. J.; Song, H.; Kim, H. Angew. Chem., Int. Ed. 2017, 56, 6952.
(b) Bedford, R. B.; Haddow, M. F.; Mitchell, C. J.; Webster, R. L. Angew. Chem., Int. Ed. 2011, 50, 5524.
Wan, X.-B.; Ma, Z.-X.; Li, B.-J.; Zhang, K.-Y.; Cao, S.-K.; Zhang, S.-W.; Shi, Z.-J. J. Am. Chem. Soc. 2006, 128, 7416. doi: 10.1021/ja060232j
Lengyel, I.; Cesare, V.; Stephani, R. Synth. Commun. 1998, 28, 1891. doi: 10.1080/00397919808007021
Xiong, X. D.; Yeung, Y. Y. Angew. Chem., Int. Ed. 2016, 55, 16101. doi: 10.1002/anie.201607388
Hering, T.; Muehldorf, B.; Wolf, R.; Koenig, B. Angew. Chem., Int. Ed. 2016, 55, 5342. doi: 10.1002/anie.201600783
Bedford, R. B.; Engelhart, J. U.; Haddow, M. F.; Mitchell, C. J.; Webster, R. L. Dalton Trans. 2010, 39, 10464. doi: 10.1039/c0dt00385a
Singh, H.; Sen, C.; Sahoo, T.; Ghosh, S. C. Eur. J. Org. Chem. 2018, 4748.
Pu, X.-Q.; Zhao, H.-Y.; Lu, Z.-H.; He, X.-P.; Yang, X.-J. Eur. J. Org. Chem. 2016, 4526.
Pu, X.-Q.; Li, Q.-W.; Lu, Z.-H.; Yang, X.-J. Eur. J. Org. Chem., 2016, 5937.
Zhao, H.-Y.; Pu, X.-Q.; Yang, X.-J. Chin. J. Chem. 2017, 35, 1417. doi: 10.1002/cjoc.v35.9
Table 1. Optimization of reaction conditions for C—H halogenation of acetanilide
|
|
||||||
| Entrya | Catalyst/mol% | Additive/equiv. | Solvent | T/℃ | Time/h | Yieldb/% |
| 1 | — | — | CH3CN | 25 | 24 | 28 |
| 2 | — | — | DCE | 25 | 24 | 32 |
| 3 | 20 | — | DCE | 25 | 12 | 47 |
| 4 | 20 | CH3COOH (1.0) | DCE | 25 | 8 | 15 |
| 5 | 20 | CF3SO3H (1.0) | DCE | 25 | 8 | 36 |
| 6 | 20 | HCl (1.0) | DCE | 25 | 12 | 28 |
| 7 | 20 | NaOCl (1.0) | DCE | 25 | 12 | 23 |
| 8 | 20 | FeCl3 (1.0) | DCE | 25 | 8 | 21 |
| 9 | 20 | CuCl2 (1.0) | DCE | 25 | 8 | 16 |
| 10 | 20 | K2CO3 (1.0) | DCE | 25 | 8 | n.d. |
| 11 | 20 | PTSA (1.0) | DCE | 25 | 3 | 64 |
| 12 | 20 | PTSA (1.0) | Toluene | 25 | 8 | 46 |
| 13 | 20 | PTSA (1.0) | Ethyl acetate | 25 | 12 | 52 |
| 14 | 20 | PTSA (1.0) | Methanol | 25 | 12 | 27 |
| 15 | 20 | PTSA (1.0) | 2-Methoxyethanol | 25 | 12 | 35 |
| 16 | 20 | PTSA (1.0) | 1, 4-Dioxane | 25 | 12 | 25 |
| 17 | 20 | PTSA (1.0) | CH3CN | 25 | 12 | 21 |
| 18 | 20 | PTSA (1.0) | CCl4 | 25 | 10 | 32 |
| 19 | 20 | PTSA (1.0) | DCE | 40 | 2 | 33 |
| 20 | 20 | PTSA (1.0) | DCE | 80 | 2 | 31 |
| 21 | 20 | PTSA (1.0) | DCE | 0 | 24 | 62 |
| 22 | 6 | PTSA (1.0) | DCE | 25 | 6 | 72 |
| 23 | 4 | PTSA (1.0) | DCE | 25 | 10 | 55 |
| 24 | 6 | PTSA (0.2) | DCE | 25 | 12 | 57 |
| 25 | 6 | PTSA (0.4) | DCE | 25 | 6 | 74 |
| 26 | 6 | PTSA (1.4) | DCE | 25 | 6 | 54 |
| a Reaction conditions: acetanilide (0.3 mmol), CFBSA (1 equiv.), additive, catalyst, solvent (4 mL), and temperature. b Isolated yield. | ||||||
 下载: 导出CSV
下载: 导出CSV
Table 3. Test of reactivity of various chlorinating reagents on acetanilidea
|
|
||
| Entry | Chlorinating reagent | Isolated yieldb/% |
| 1 | CFBSA | 74 |
| 2 | CMOBSA | 41 |
| 3 | NCS | 58 |
| 4 | t-BuOCl | 60 |
| 5 | Trichloroisocyanuric acid | 53 |
| a The reaction conditions are as follows: acetanilide (1.0 equiv., 0.03mmol), Pd(OAc)2 (6 mol%), chlorinating reagent (1.0 equiv.), PTSA (0.4 equiv.), DCE, room temperature. b Isolated yields. | ||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: