图 1.
有代表性的手性配体支架
Figure 1.
Representative chiral ligand scaffolds

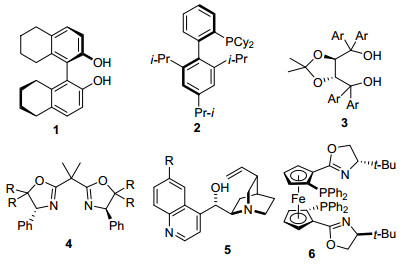
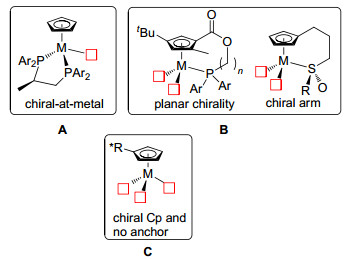
自从1951年发现二茂铁以来, 环戊二烯基(Cp)配体与金属所形成的配合物极大地促进了金属有机化学的发展[1]. η5-环戊二烯基配体与过渡金属的配位十分稳定, 键强度高达118 kcal/mol[2, 3], 能够与所有的过渡金属和大部分f区金属形成配合物[4].目前人们已经制备出了大量的环戊二烯基或者它的常用衍生物五甲基环戊二烯基(Cp*)与过渡金属配位形成的三明治型和半三明治型配合物[5].含有手性配体的金属有机配合物在不对称催化反应中发挥着重要的作用[6], 如催化不对称烯丙基化、不对称氢(胺)化、不对称环加成反应.有代表性的几类常用手性配体[7~9]包括BINAP, BINOL及其氢化衍生物1, XPhos 2, TADDOLS 3, 双噁唑啉4, 金鸡纳生物碱衍生物5, 面手性二茂铁膦6等(图 1).
由于环戊二烯基配体的稳定性和结构可修饰性, 环戊二烯基配体与过渡金属合成的配合物在烯烃聚合[10, 11]、功能材料[12]和燃速催化[13]等方面不断取得新的进展, 然而如何在环戊二烯基配体上引入手性环境是金属有机化学家的挑战性课题之一, 尤其是设计合成具有高对映选择性的手性环戊二烯基配体.手性环戊二烯基配体的发展滞后于其它几类有机配体, 如传统的单膦和双膦配体、亚胺类配体、卡宾以及多齿类氮配体[5], 主要原因是手性环戊二烯基配体的设计与合成具有一定的困难和挑战性.本文主要对手性环戊二烯过渡金属配合物的设计、合成及在不对称催化反应中的应用做一介绍.
过渡金属配合物的手性中心可以来源于中心金属和配体, 由于过渡金属配合物可发生解离、交换等反应, 合成中心金属具有手性的配合物相对困难, 使用起来也不方便, 所以, 大多数成功的手性过渡金属配合物的手性来源于配体.环戊二烯基配合物大体上有三种方法来获得手性[4]: (1)一个非手性的环戊二烯基和一个外来的手性双齿配体A类配合物(chiral-at-metal)[14]; (2)环戊二烯基与膦或亚砜配体形成侧臂的B类配合物[15, 16]; (3)手性环戊二烯基配体C类配合物(图 2). CpM配合物催化的反应大多需要金属上具有最大数目配位点, 可以看出C类配合物金属上具有最多配位点.
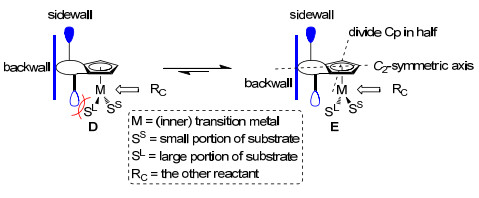
Kagan等[17]首次将C2对称的手性双膦配体Diop应用于α-乙酰胺丙烯酸的不对称还原反应中, 并取得了70%~80%的光学产率.早期的研究发现与对称性较差的催化剂相比, 含C2对称配体的催化剂总体上取得的对映选择性更高, 这是源于使用C2对称的催化剂可以有效地减少竞争的非对映异构体过渡态的数量, 改善手性识别过程[18]. 2012年Cramer等[5]提出合成C类配合物的关键在于手性环戊二烯基配体的设计, 他们认为设计高对映选择性的手性环戊二烯基配体应考虑的条件有: (1)使用C2对称的1, 2-二取代环戊二烯基衍生物Cpx, 来避免金属从Cp的两面配位产生非对映异构体; (2)与Cp平面垂直的具有大空间位阻的基团形成后墙(backwall), 堵住了反应物RC进攻的一个面, 使得RC只能从另一面进攻; (3)大位阻基团上的取代基向中心金属方向延伸形成边墙(sidewall), 从而限制了底物的旋转, 使得底物中较大的基团SL在空间位阻较小的一侧, 如图中E的构象成为优势构象(图 3).
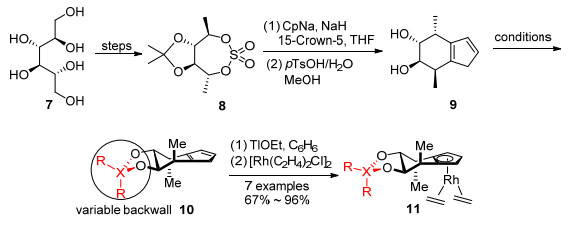
将配体的刚性、空间和电子效应、对称性因素等条件考虑在内, Cramer小组[5]在2012年报道了手性环戊二烯基配体10.从商品化原料(2S, 5S)-2, 5-己二醇或D-甘露醇出发合成环硫酸酯[19~21], 缩酮在酸性条件下分解生成二醇9, 再把羟基活化为易离去的磺酸酯后能够引入不同的基团, 如硅烷基、氧杂蒽基、苯基等, 以达到调控配体后墙的目的(Scheme 1).
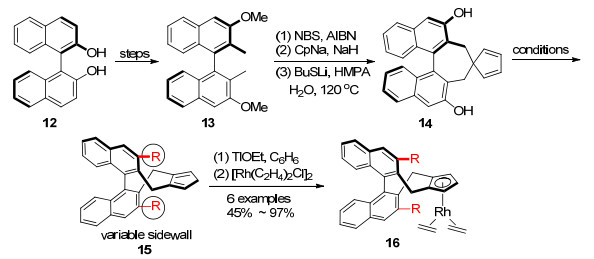
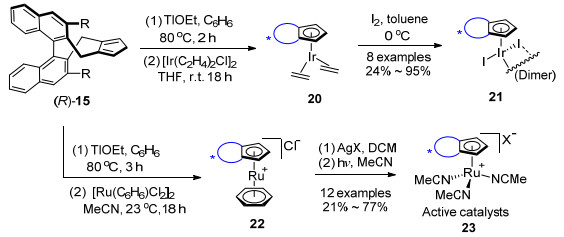
2013年该课题组[22]以(R)-1, 1'-联萘酚为原料制备了第二代手性环戊二烯配体15, 配体15具有固定的后墙二萘基部分和可调控的边墙3, 3'位R取代基. R取代基是较大体积的烷氧基、硅氧基或不同的芳基[23, 24] (Scheme 2).手性环戊二烯配体10和15均在乙醇铊的作用下去质子化, 与[Rh(C2H4)2Cl]2反应得到配合物11和16.
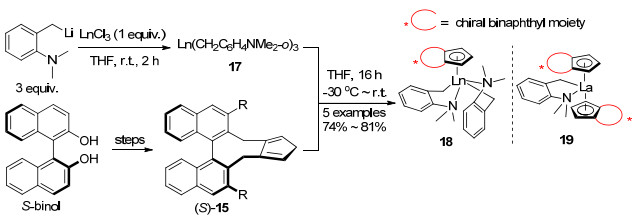
二萘基型手性环戊二烯配体15不仅可以与金属Rh配位, 还可以成功地与Ru(Ⅱ), Ir(Ⅲ), Sc(Ⅲ)等过渡金属以及Gd(Ⅲ)等镧系金属配位. 2014年侯召民等[25, 26]报道了Ln(CH2C6H4NMe2-o)3的四氢呋喃(THF)溶液加入到由(S)-联萘酚制备的(S)-15的THF溶液中, 通过酸碱反应一锅法合成了半三明治型稀土金属配合物18.若(S)-15与17的物质的量比为2:1, 则能够得到夹心型手性镧配合物19[27] (Scheme 3).
2015年Cramer等[28]首先使用手性环戊二烯基配体(R)-15与Ir(Ⅲ)起始原料如IrCl3配位, 没有得到CpxIrCl2配合物.改用与合成Rh(Ⅰ)配合物16类似的方法, 即先制备出Ir(Ⅰ)配合物, 再将其氧化为Ir(Ⅲ)配合物.与Rh(Ⅰ)配合物需要添加氧化剂过氧化二苯甲酰(BzO)2不同, Ir(Ⅰ)配合物更容易被碘分子氧化生成二聚体[(CpxIrI2)]221.同年该课题组[29]利用15的Tl衍生物, 与[Ru(C6H6)2Cl]2反应得到稳定的手性配合物22, 22中的Cl-能够与弱配位的PF6-或SbF6-进行抗衡离子的置换, 再经过光照条件下与乙腈配体交换得到阳离子Ru(Ⅱ)配合物23 (Scheme 4).
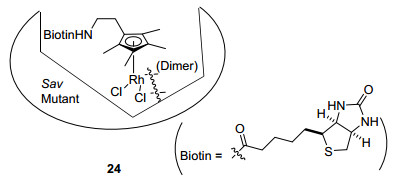
Ward和Rovis等[30]在2012年利用生物素-亲和素(Biotin-Avidin)作用策略创建手性环境.不带糖基的链霉亲和素能够与四分子生物素特异性结合, 通过在中心金属Rh附近不同位置引入谷氨酸残基或天冬氨酸残基, 来合成工程链霉亲和素蛋白(engineered streptavidin).常温下在3-(N-吗啉)丙磺酸缓冲液(MOPS)中, 向0.66 mol%工程链霉亲和素蛋白(Sav Mutant)的去离子水溶液中加入环戊二烯基生物素衍生物[RhCp*biotinCl2]2的DMSO溶液搅拌混合, 即可现场生成人工金属酶24(图 4).与其它的手性环戊二烯基配合物相比, 链霉亲和素蛋白的几何形状可容纳金属有机配合物, 也为底物的结合与激活留下了足够的空间位置, 不足之处是酶的催化活性受到温度、溶剂、介质含水量等实验变量的严格限制.
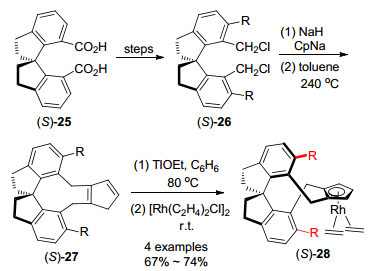
2016年, 游书力等[31]报道了1, 1'-氢化螺二茚(spirobiindane)为支架的手性环戊二烯配体27 (SCps), 氢化螺二茚配体比二萘基配体具有更加延伸的边墙.合成此手性配体是由周其林等[32]合成的氢化螺二茚二羧酸(S)-25出发, 经过羧基的邻位碘化[33], 还原和氯代反应得到(S)-26, 用CpNa亲核进攻并在高温下重排得到(S)-27, 再与[Rh(C2H4)2Cl]2反应生成Rh(Ⅰ)配合物(S)-28 (Scheme 5).
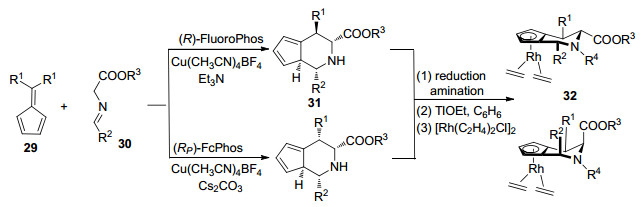
2017年, Antonchick和Waldmann等[34]介绍了哌啶基手性环戊二烯配体31 (JasCp), 此类配体结合了Cramer与Ward等[5, 30]所设计配体的优点, 既具有广泛应用性, 又有快速可调的空间结构, 且合成步骤不复杂.在不同手性膦催化剂存在下, 亚甲基环戊二烯29与亚胺基酯30经过1, 3-偶极环加成反应生成不同构型的配体31[35], 与Rh(Ⅰ)配位得到32 (Scheme 6).
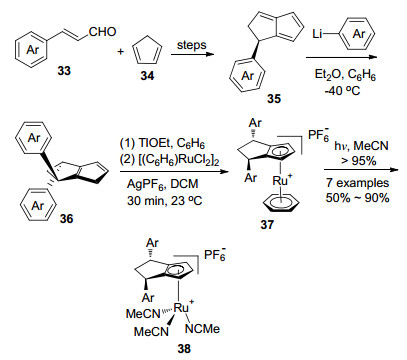
在Hayashi等[36]合成35工作的基础上, 2018年Cramer小组[37]合成了一类新型的手性环戊二烯配体36, 并制备出该配体与Ru配位形成的配合物38.这类配体合成简单(Scheme 7), 分子量小, C2对称, 结构可修饰.
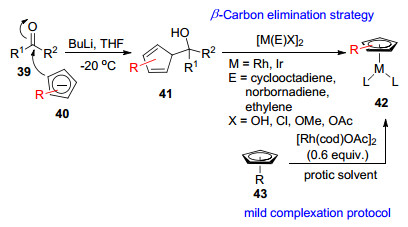
2017年, Cramer小组[38]用环戊二烯基醇41使Rh(Ⅰ)或Ir(Ⅰ)金属源二聚体的桥键断裂, 二者发生分子间缩合脱去一个小分子如H2O、MeOH, 再进行β-C消除得到42.该课题组[39]在2019年利用不同的手性环戊二烯基配体与金属源在质子溶剂存在下, 原位(in situ)生成催化剂前体42.这两种合成方法均避免了铊盐的使用, 并且可以原位生成催化剂前体, β-C消除策略不足的是通常需要41为多取代的位阻较大的环戊二烯醇(Scheme 8).
2014年侯召民课题组[25]以半三明治型稀土金属配合物18a为催化剂, 在简便易得的硼试剂[Ph3C]- [B(C6F5)4]存在下催化α-吡啶与不同取代的1-烯烃加成得到烷基化吡啶46 (Eq. 1).在不影响产率情况下, 对映选择性高达96% ee. 2016年该课题组[26]报道了首例环丙烯与不同取代仲胺48的分子间不对称氢胺化反应, 以100%的原子利用率, 高达97%的产率和99% ee生成胺基取代的环丙烷产物(Eq. 2).该反应无需加入硼试剂.
|
|
(1) |
|
|
(2) |
|
|
(3) |
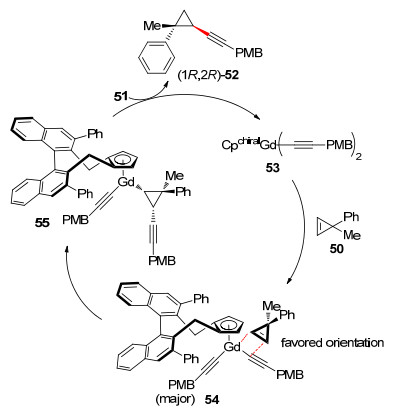
2018年该课题组[40]使用手性Gd催化剂18c催化端炔与环丙烯的不对称加成反应(Eq. 3).他们发现稀土金属阳离子和Cpx配体对于该反应的活性和对映选择性都有重要影响.金属离子Gd3+ (0.938 )的离子半径介于Y3+和Sm3+之间, 在相同配体条件下, Gd3+所形成的配合物有更好的催化结果. 18c为催化剂时, 产物52的产率达到96%, ee值高达99%.他们提出了该反应可能的催化循环:配合物18c与2分子炔烃配位形成53后进入催化循环.环丙烯双键配位插入在中心金属Gd与炔基碳之间生成55, 空间位阻效应使环丙烯配位方向具有专一选择性, 55再与1分子炔烃配位释放出产物52完成催化循环(Scheme 9).
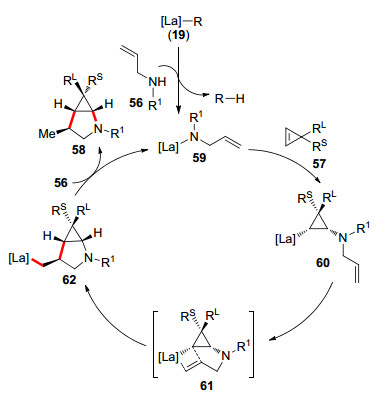
2017年侯召民等[27]使用手性环戊二烯La配合物19催化胺基烯烃56与环丙烯57的反应合成氢胺化产物/双环化物(Eq. 4).他们发现配合物具有更大体积的金属离子时, 有更好的催化结果.当使用19作为催化剂时, 可以专一性地得到双环化产物, 而没有停留在氢胺化产物阶段60.作者提出19催化的胺基烯烃与环丙烯反应的催化循环:配合物中心金属La与胺基烯烃上的氮原子配位, 脱去1分子RH形成59后进入催化循环.环丙烯双键配位插入到金属La与氮原子之间, 胺基烯烃的烯丙基双键再一次配位形成中间态61, 经过环化、质子化完成催化循环(Scheme 10).该机理表明更大体积的金属La可以使分子内烯烃双键快速与配合物中心金属发生配位插入.
Cramer[5]和Rovis[30]两个课题组在2012年分别用环己基型Rh(Ⅰ)配合物11c和金属酶24催化苯甲酰胺邻位C-H官能团化, 对映选择性合成二氢异喹啉65 (Eq. 5).值得注意的是, 这两种催化方法的底物烯烃64范围可以互补.当烯烃为富电子的苯乙烯时, 用配合物11c作催化剂, 取得91%的产率和高达94% ee.当烯烃是缺电子的丙烯酸酯时, 使用人工金属酶24作催化剂, 以95%产率和82% ee得到相应的产物65.与二取代的配合物11c相比, 24中的Cp*单元具有更强的空间要求和丰富的电子性质.
|
|
(4) |
|
|
(5) |
二萘基型配体通过与不同的过渡金属进行配位, 可形成CpRh(Ⅰ)、CpIr(Ⅲ)、CpRu(Ⅱ)等配合物. 2013年Cramer课题组[22]用CpxRh(Ⅰ)配合物催化了芳基C(sp2)—H烯丙基化反应(Eq. 6). N-甲氧基苯甲酰胺66与联烯67为底物, 用(BzO)2将16c氧化为可与底物配位的Rh(Ⅲ)催化剂.他们发现用手性Cpx配合物11c催化该反应时, 产物具有较低的对映选择性, 但在相同条件下用16c作催化剂, 不仅增加了产率, 而且使光学选择性大幅度提高, 并且底物普适性较好.
|
|
(6) |
|
|
(7) |
|
|
(8) |
|
|
(9) |
2014年Cramer等[41]在相当温和的条件下用二萘基配合物CpxRh(Ⅰ)催化N-新戊酰氧基苯甲酰胺69与两类重氮酯70构建五元氮杂环的反应(Eq. 7).与烯烃两个碳原子的配位插入不同, 该反应仅插入了重氮基团离去所生成卡宾中间体的一个碳原子.值得注意的是, 此反应提高ee值的关键是, 重氮酯碳-氮双键两边的基团具有不同的空间位阻, 从而使底物重氮酯与催化剂的配位方向有选择性.
游书力等[42]在2015年报道了首例β-萘酚和炔烃的不对称环化去芳构化反应(Eq. 8), 以16a作为手性催化剂, Cu(OAc)2和氧气为氧化剂, 碳酸钾为碱, 成功合成了螺茚衍生物74, 在不影响产率条件下, ee值高达84%.同年Lam等[43]利用不对称[3+2]螺环化反应, 以高区域选择性合成了螺茚衍生物77 (Eq. 9), 且不需要加入(BzO)2作为氧化剂.与游书力课题组所用底物72不同, 该反应底物75由于烯醇互变产生两个不同位置的羟基作为导向基(directing group), 其中酮羰基更倾向于形成烯醇, 与中心金属Rh配位脱去1分子AcOH.此反应螺茚衍生物产率达到94%, ee值高达97%.
Cramer课题组[44]在2017年介绍了用二萘基配合物CpxIr(Ⅲ) 21a催化二芳基氧化膦78和叠氮化物79反应生成手性膦产物(Eq. 10).他们发现添加剂手性羧酸与手性Cpx配体二者的协同作用可以促使向底物氧化膦传递手性信息, 且手性羧酸的构型与手性Cpx配合物的构型一致可以得到高ee值, 为合成手性膦化合物提供了一种有别于传统光学拆分消旋底物的新途径.同年该课题组[45]使用二萘基配合物23a催化1-炔基三氮烯与双环烯烃的[2+2]环加成反应(Eq. 11).以23a为催化剂, Bu4NCl为抗衡离子有效地获得产物烯基三氮烯84, 84作为一类独特的烯基阳离子替代物, 在酸性条件下能够与多种亲核试剂反应从而引入不同的官能团.
|
|
(10) |
|
|
(11) |
2016年, 游书力等[31]以1, 1'-氢化螺二茚手性环戊二烯配体所形成的配合物(S)-28a为催化剂, (BzO)2为氧化剂, 研究了1-萘基苯并异喹啉与烯烃的脱氢Heck氧化偶联反应(Eq. 12).用(R)-16a催化时获得轴手性联芳基产物87的ee值最高只能达到80%[46], 而用(S)-28a作为催化剂时在不影响产率的情况下ee值可达到90%, 这也证明了螺二茚配体比二萘基配体有更加延伸的边墙.
|
|
(12) |
2017年Antonchick和Waldmann等[34]用哌啶基手性环戊二烯配体所形成的Rh配合物32c催化C—H芳基化合成轴手性联芳基产物(Eq. 13).底物苯甲酰胺88和重氮萘酮89具有高反应活性, 且底物89在产物芳基-芳基键相邻位置提供两个邻位取代基羟基和羰基, 从而有助于轴向手性稳定化.以32c作为催化剂, 该反应的产率可达到93%, 在不影响产率条件下, ee值高达89%.
|
|
(13) |
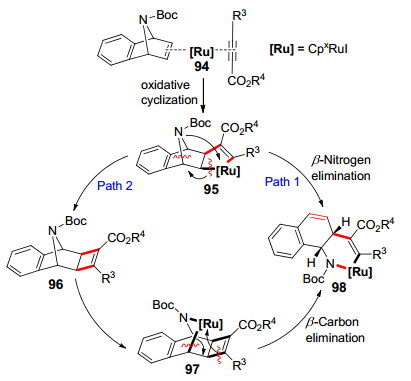
2018年Cramer等[37]用新型二芳基环戊基CpxRu(Ⅱ)配合物38a催化环烯烃91与炔基酯92反应构建二氢苯并吲哚(Eq. 14).作者提出该反应的机理有两种可能的途径:配合物[Ru]([Ru]=CpxRuI)与烯烃双键和炔基三键配位、氧化环化形成95后, 可能直接β-N消除得到98, 也可能是先进行还原消除生成[2+2]环加成产物96, 再有1分子配合物[Ru]氧化插入形成97, 经过β-C消除同样可以得到98.最后98还原消除释放出配合物[Ru]和产物93(Scheme 11).
|
|
(14) |
本文综述了六类手性环戊二烯配体所形成配合物的合成及在不对称催化反应中的主要应用, 从应用中可以看出二萘基类手性环戊二烯配体, 在不对称催化领域显示出更广泛的适用性, 可与前过渡金属和后过渡金属配位.最近几年的研究中, 催化剂结构的优化是通过不同基团修饰环戊二烯配体以获得手性, 以期在催化反应时得到更高对映选择性的产物.由于这些手性环戊二烯配体尚未商业化, 其冗长的制备过程阻碍了它们更广泛的应用.要使手性环戊二烯配体得到更广泛的应用, 可以设想另外的一些发展方向, 比如在配体的不同位置进行修饰, 或者是与其它过渡金属配位.未来, 如何快速找到手性环戊二烯配合物与不对称反应之间的构效关系?如何设计出具有高对映选择性、高效的、稳定的新型环戊二烯基配体骨架, 并找出从稳定前体出发的最容易的合成路线仍然是一个巨大的挑战.进一步探讨手性环戊二烯过渡金属配合物在催化过程中的反应机理.
Togni, A.; Halterman, R. L. In Metallocenes: Synthesis, Reactivity, applications, Vol. 2, Eds.: Jutzi, P.; Edelmann, F. T., Wiley-VCH, New York, 1998, pp. 1~102.
Halterman, R. L.; Vollhardt, K. P. C. Organometallics 1988, 7, 883. doi: 10.1021/om00094a015
Halterman, R. L. Chem. Rev. 1992, 92, 965. doi: 10.1021/cr00013a011
Newton, C. G.; Kossler, D.; Cramer, N. J. Am. Chem. Soc. 2016, 138, 3935. doi: 10.1021/jacs.5b12964
Ye, B.-H.; Cramer, N. Science 2012, 338, 504. doi: 10.1126/science.1226938
(a) Jang, H.-J.; Romiti, F.; Torker, S.; Hoveyda, A. H. Nat. Chem. 2017, 9, 1269.
(b) Zhu, S.-F.; Zhou, Q.-L. Acc. Chem. Res. 2017, 50, 988.
(c) Vanable, E. P.; Kennemur, J. L.; Joyce, L. A.; Ruck, R. T.; Schultz, D. M.; Hull, K. L. J. Am. Chem. Soc. 2019, 141, 739.
(d) Ambler, B. R.; Turnbull, B. W. H.; Suravarapu, S. R.; Uteuliyev, M. M.; Huynh, N. O.; Krische, M. J. J. Am. Chem. Soc. 2018, 140, 9091.
Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H. In Comprehensive Asymmetric Catalysis, Vol. Ⅰ~Ⅲ, Eds.: Brown, J. M.; Buchwald, S. L., Springer-Verlag, New York, 1999, pp. 1~10.
Wu, P.-L.; Jia, M.-Q.; Lin, W.-L.; Ma, S.-M. Org. Lett. 2018, 20, 554. doi: 10.1021/acs.orglett.7b03637
(a) Zhu, G.-Q.; Wang, P. Chin. J. Synth. Chem. 2018, 26, 66 (in Chinese).
(朱广乾, 王鹏, 合成化学, 2018, 26, 66.)
(b) Inoue, F.; Myuto, K.; Yadav, M. R.; Nakao, Y. Angew. Chem., Int. Ed. 2017, 56, 13307.
(c) Lin, Z.-Q.; Wang, W.-Z.; Yan, S.-B.; Duan, W.-L. Angew. Chem., Int. Ed. 2015, 54, 6265.
(d) Wang, Z.-B.; Yin, H.-L.; Fu, G. C. Nature 2018, 563, 379.
(e) Zheng, S.-C.; Wang, Q.; Zhu, J.-P. Angew. Chem., Int. Ed. 2019, 58, 1494.
(f) Yao, K.; Yuan, Q.-J.; Qu, X.-X.; Liu, Y.-G.; Liu, D.-L.; Zhang, W.-B. Chem. Sci. 2019, 10, 1767.
Nishiura, M.; Guo, F.; Hou, Z.-M. Acc. Chem. Res. 2015, 48, 2209.
Li, S.-H.; Liu, D.-T.; Wang, Z.-C.; Cui, D.-M. ACS Catal. 2018, 8, 6086. doi: 10.1021/acscatal.8b00885
Zhang, H.; Zhou, B.; Li, H.; Qu, D.-H.; Tian, H. J. Org. Chem. 2013, 78, 2091. doi: 10.1021/jo302107a
Liu, X.-L.; Li, J.-Z.; Bi, F.-Q.; Zhang, W.-Q.; Gao, Z.-W.; Zhang, G.-F. Eur. J. Inorg. Chem. 2015, 9, 1496.
Bauer, E. B. Chem. Soc. Rev. 2012, 41, 3153. doi: 10.1039/c2cs15234g
Matsushima, Y.; Onitsuka, K.; Kondo, T.; Mitsudo, T.; Takahashi, S. J. Am. Chem. Soc. 2001, 123, 10405. doi: 10.1021/ja016334l
Trost, B. M.; Rao, M.; Dieskau, A. P. J. Am. Chem. Soc. 2013, 135, 18697. doi: 10.1021/ja411310w
Kagan, H. B.; Dang, T. P. J. Am. Chem. Soc. 1972, 94, 6429. doi: 10.1021/ja00773a028
Whitesell, J. K. Chem. Rev. 1989, 89, 1581. doi: 10.1021/cr00097a012
Ye, B.-H.; Cramer, N. Acc. Chem. Res. 2015, 48, 1308. doi: 10.1021/acs.accounts.5b00092
Zhang, A.-B.; RajanBabu, T. V. Org. Lett. 2004, 6, 1515. doi: 10.1021/ol0495063
Li, W.-G.; Zhang, Z.-G.; Xiao, D.-M.; Zhang, X.-M. J. Org. Chem. 2000, 65, 3489. doi: 10.1021/jo000066c
Ye, B.-H.; Cramer, N. J. Am. Chem. Soc. 2013, 135, 636. doi: 10.1021/ja311956k
Ooi, T.; Kameda, M.; Maruoka, K. J. Am. Chem. Soc. 2003, 125, 5139. doi: 10.1021/ja021244h
Ahmed, I.; Clark, D. A. Org. Lett. 2014, 16, 4332. doi: 10.1021/ol502126r
Song, G.-Y.; Wylie, W. N. O.; Hou, Z.-M. J. Am. Chem. Soc. 2014, 136, 12209. doi: 10.1021/ja504995f
Teng, H.-L.; Luo, Y.; Wang, B.-L.; Zhang, L.; Nishiura, M.; Hou, Z.-M. Angew. Chem., Int. Ed. 2016, 55, 15406. doi: 10.1002/anie.v55.49
Teng, H.-L; Luo, Y.; Nishiura, M.; Hou, Z.-M. J. Am. Chem. Soc. 2017, 139, 16506. https://www.researchgate.net/publication/317609479_Asymmetric_Yttrium-Catalyzed_Csp-H_Addition_of_2-Methyl_Azaarenes_to_Cyclopropenes
Dieckmann, M.; Jang, Y.-S.; Cramer, N. Angew. Chem., Int. Ed. 2015, 54, 12149. doi: 10.1002/anie.201506483
Kossler, D.; Cramer, N. J. Am. Chem. Soc. 2015, 137, 12478. doi: 10.1021/jacs.5b08232
Hyster, T. K.; Knorr, L.; Ward, T. R.; Rovis, T. Science 2012, 338, 500. doi: 10.1126/science.1226132
Zheng, J.; Cui, W.-J.; Zheng, C.; You, S.-L. J. Am. Chem. Soc. 2016, 138, 5242. doi: 10.1021/jacs.6b02302
(a) Zhu, S.-F.; Yang, Y.; Wang, L.-X.; Liu, B.; Zhou, Q.-L. Org. Lett. 2005, 7, 2333.
(b) Xie, J.-H.; Wang, L.-X.; Fu, Y.; Zhu, S.-F.; Fan, B.-M.; Duan, H.-F.; Zhou, Q.-L. J. Am. Chem. Soc. 2003, 125, 4404.
(c) Li, S.-Y.; Zhang, J.-W.; Li, X.-L.; Cheng, D.-J.; Tan, B. J. Am. Chem. Soc. 2016, 138, 16561.
Mei, T.-S.; Giri, R.; Maugel, N.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 5215. doi: 10.1002/anie.v47:28
Jia, Z.-J.; Merten, C.; Gontla, R.; Daniliuc, C. G.; Antonchick, A. P.; Waldmann, H. Angew. Chem., Int. Ed. 2017, 56, 2429. doi: 10.1002/anie.201611981
(a) Potowski, M.; Bauer, J. O.; Strohmann, C.; Antonchick, A. P.; Waldmann, H. Angew. Chem., Int. Ed. 2012, 51, 9512.
(b) Potowski, M.; Antonchick, A. P.; Waldmann, H. Chem. Commun. 2013, 49, 7800.
(c) He, Z.-L.; Teng, H.-L.; Wang, C.-J. Angew. Chem., Int. Ed. 2013, 52, 2934.
(a) Gotoh, H.; Masui, R.; Ogino, H.; Shoji, M.; Hayashi, Y. Angew. Chem., Int. Ed. 2006, 45, 6853.
(b) Gotoh, H.; Ogino, H.; Ishikawa, H.; Hayashi, Y. Tetrahedron 2010, 66, 4894.
Wang, S.-G.; Park, S. H.; Cramer, N. Angew. Chem. 2018, 130, 5557. doi: 10.1002/ange.v130.19
Smits, G.; Audic, B.; Wodrich, M. D.; Corminboeuf, C.; Cramer, N. Chem. Sci. 2017, 8, 7174. doi: 10.1039/C7SC02986A
Audic, B.; Wodrich, M. D.; Cramer, N. Chem. Sci. 2019, 10, 781. doi: 10.1039/C8SC04385J
Teng, H.-L.; Ma, Y.-H.; Zhan, G.; Nishiura, M.; Hou, Z.-M. ACS Catal. 2018, 8, 4705. doi: 10.1021/acscatal.8b01189
Ye, B.-H.; Cramer, N. Angew. Chem. 2014, 126, 8030. doi: 10.1002/ange.201404895
Zheng, J.; Wang, S.-B.; Zheng, C.; You, S.-L. J. Am. Chem. Soc. 2015, 137, 4880. doi: 10.1021/jacs.5b01707
Chidipudi, S. R.; Burns, D. J.; Khan, I.; Lam, H. W. Angew. Chem., Int. Ed. 2015, 54, 13975. doi: 10.1002/anie.201507029
Jang, Y.-S.; Dieckmann, M.; Cramer, N. Angew. Chem., Int. Ed. 2017, 56, 15088. doi: 10.1002/anie.201708440
Kossler, D.; Perrin, F. G.; Suleymanov, A. A.; Kiefer, G.; Scopelliti, R.; Severin, K.; Cramer, N. Angew. Chem., Int. Ed. 2017, 56, 11490. doi: 10.1002/anie.v56.38
Zheng, J.; You, S.-L. Angew. Chem., Int. Ed. 2014, 53, 13244. doi: 10.1002/anie.201408805

图式 3 二萘基型Cpx稀土配合物的合成
Scheme 3 Synthesis of binaphthyl-derived Cpx rare-earth complexes

图式 10 19催化的胺基烯烃与环丙烯的催化循环
Scheme 10 A catalytic cycle of cyclopropenes with aminoalkenes by 19

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:















 下载:
下载:












 下载:
下载: