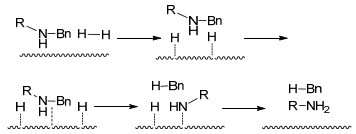
图式 1.
催化氢解脱苄反应机理
Scheme 1.
Reaction mechanism of catalytic hydrogenolysis and debenzylation

氨基的保护和脱除是有机合成中常用的策略, 在药物、天然产物等研究中屡见不鲜.氨基的常见保护基团主要有三类:烷氧羰基保护、酰基保护、烷基保护.苄基作为一种烷基保护基, 可通过适当的卤代物在碱性条件下引入.苄基的脱除也相对容易, 可通过催化氢解、液氨和钠或锂、氧化脱苄、酸解脱苄等方法脱保护.除此之外, 苄基保护的有机胺能在强碱、亲核试剂、有机金属试剂的存在下保持稳定, 因此苄基成为氨基保护最常用的基团之一[1~5].本文将综述氨基的脱苄研究进展.
还原脱苄大都采用催化氢解, 即N-苄基的C—N键在催化剂作用下发生σ-断裂而脱苄.催化氢解具有反应条件温和、产率高等优点, 是目前应用最广泛的脱苄方法. Pd具有优异的吸氢性能, 是氢解最常用的催化剂, 其组成可以是单质、氧化物、盐等的形式存在.为了提高催化性能, Pd通常负载于多孔材料上, 如活性炭、CaCO3、BaSO4和Al2O3等[6, 7].催化氢解根据氢源的不同, Pd催化氢解可分为催化氢解和催化氢转移氢解, 前者是以氢气为氢源, 后者则以甲酸、甲酸铵、环己烯等为氢源.
催化氢解脱苄是较为常用的脱苄方法, 其过程为氢气首先被吸附在催化剂上形成原子氢, 随后苄基保护的有机胺与催化剂络合、C—N键发生断裂、苄基与原子氢形成甲苯, 而胺与原子氢形成脱去苄基的仲胺或伯胺[2, 5, 8, 9](Scheme 1).对于常见的N-苄基, 催化氢解都能较好地脱除, 而且对O-苄基、卤素也有一定的脱除效果.除此之外, 催化氢解以氢气为氢源, 在反应过程中需要一定的压力, 有时为了加快反应速度还需在加热条件下进行.反应温和、产率高是催化氢解脱苄的最大的优势, 但其选择性差、不适用于易被还原的底物是其最主要的缺点. Pd/C和Pd(OH)2/C是典型的催化氢解催化剂, 甲醇、乙醇、乙酸等极性溶剂是适宜的反应溶剂[10~16].此外, 酸性条件可促进有机胺质子化, 防止催化剂失活, 因此催化氢解脱苄也可在酸性条件下进行.
催化氢解在多官能团的氮杂环脱苄研究中优势明显, Yoshida等[17]使用Pd(OH)2/C为催化剂在室温下快速氢解N-苄基胺生成脱苄产物2 (Eq. 1), 而底物中叔丁基二甲基硅烷基、四氢吡喃基和三甲基硅烷基等对酸敏感的基团在该脱苄反应过程中也未被破坏.反应以强极性的甲醇作溶剂, 在101 kPa的氢气压力下氢解, 数小时就能以较高的产率(81%~99%)反应完全.该方法快速、高效、温和, 尤其适宜于含有敏感基团的底物脱苄.
|
|
(1) |
当脱苄产物的稳定性低于反应物时, 简单的催化氢解往往无法得到脱苄产物, 如高能炸药CL-20 (HNIW, 六硝基六氮杂异伍兹烷)的制备. CL-20是目前综合性能最好的单质炸药, 其合成分为六苄基六氮杂异伍兹烷(HBIW, 3)的合成、HBIW的脱苄、脱苄产物的硝化三步[18]. HBIW可通过苄胺和乙二醛缩合制备, 而HBIW中苄基的脱保护是CL-20成功合成的关键. Bellamy[19]系统研究了HBIW的脱苄情况, 发现在乙酸酐中以质量分数为20% Pd(OH)2/C为催化剂氢解HBIW可以得到四乙酰基二苄基异伍兹烷(TADBIW) (Eq. 2), TADBIW是制备HNIW的重要中间体.由于异伍兹烷的稳定性要低于HBIW, 酸酐的加入能够及时捕获异伍兹烷, 生成酰基稳定的TADBIW.因此, 在HBIW脱苄过程中, 使用强酰化试剂的脱苄效果要好于弱酰化试剂和无酰化试剂的脱苄效果.研究还发现, 苄基苯环上无论含有供电子或吸电子基团, 对脱苄反应均是不利的; 而且湿Pearlman催化剂[质量分数为20% Pd(OH)2/C]的催化效果要优于干Pearlman催化剂, 也优于干Degussa催化剂(质量分数为10% Pd/C).
1997年, Wardle等[20]改进了在酸酐中催化氢解的条件, 确定了以Pd(OH)2/C为催化剂, 以二甲基甲酰胺(DMF)和乙酸酐组成的混合溶剂的脱苄体系.在反应中添加溴化物, 可防止催化剂中毒, 促进催化氢解快速进行, 且大大降低催化剂的使用量(Scheme 2).目前, 大量关于HNIW合成的文献[21~24]均采用Pd(OH)2/C氢解的方法, 即首先将HBIW催化氢解得到四乙酰基二苄基异伍兹烷(TADBIW, 4), TADBIW以甲酸为氢供体的条件下继续氢解得到四乙酰基二甲酰基异伍兹烷(5)和其他副产物.
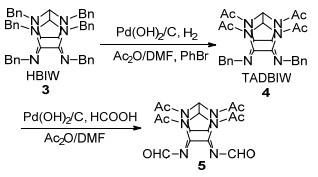
|
|
(2) |
对于催化氢解脱苄, 适当向反应体系中添加酸, 可缩短反应时间、降低贵金属催化剂的用量. 2-氨基吡啶甲基吡咯烷是一类高效选择性神经元型一氧化氮酶抑制剂, 合成这些抑制剂的关键步骤是去除双保护的2-氨基吡啶环衍生物6中的苄基. 2012年Ji等[25]研究发现, 吡啶环相连氮原子上的苄基脱除是困难的, 以Pd/C为催化剂的氢解脱苄和2, 3-二氯-5, 6-二氰基对苯醌(DDQ)为氧化剂氧化脱苄体系均不能脱去N-苄基, 而使用硝酸铈铵(CAN)的氧化脱苄也仅有26%的产率.他们又改用质量分数为20% Pd(OH)2/C催化剂, 将温度升高到60 ℃, 并向体系中加入乙酸, 经过一系列改进使得脱苄产率由最初的26%提高到60% (Eq. 3).乙酸的加入促进吡啶环的质子化, 使氢解反应更易进行.
2014年朱思兰等[26]在合成八氢吡咯并[3, 4-c]吡咯- 2-碳酸叔丁酯(9)的过程中, 使用Pd(OH)2/C催化剂脱去氮原子上的苄基(Eq. 4).该反应条件温和, 以乙醇为溶剂, 常温常压下即可进行, 同时与氮原子相连的酯也未被破坏.
氮杂金刚烷是另一种笼型硝铵类含能化合物的骨架, 罗军等[27]利用Pd(OH)2/C催化氢解方法脱去氮杂金刚烷上的苄基制备了硝基氮杂金刚烷的前体.由于N—H键较活泼, 脱苄产物在酸性条件下不稳定, 该方法也以酸酐为溶剂在Pd(OH)2/C条件下脱苄乙酰化, 得到了2, 4, 9-三乙酰基-2, 4, 9-三氮杂金刚烷- 7-醇(11), 11在硝硫混酸中硝化制得2, 4, 9-三硝基-2, 4, 9-三氮杂金刚烷-7-醇(Eq. 5).
|
|
(3) |
|
|
(4) |
|
|
(5) |
Pd/C催化剂是另一种使用广泛的氢解催化剂, 而溶剂在氢解过程发挥重要的作用, 控制催化氢解的反应溶剂, 得到意想不到的结果. Bailey等[28]在催化氢解合成(-)-raumacline (13)过程中, 使用非醇类溶剂, 通常不反应; 而使用简单醇类溶剂可导致N-烷基化的副反应发生, 得到的是(-)-raumacline和N-烷基化的混合物.他们改用三氟乙醇作溶剂, 能够达到较高的产率并且无N-烷基化的副产物.三氟乙醇作溶剂的催化氢解克服了(-)-raumacline合成中N-烷基化的问题, 并适用于多种底物, 令人惊讶的是该方法在N-甲基存在下还可选择性脱除N-苄基(Eq. 6).
|
|
(6) |
传统的催化氢解脱苄具有反应条件温和、产率高等优点, 但对于低分子量的胺和含多亲水基团有机胺的催化氢解的产率往往很低, 前者是因为低分子量不稳定, 后者是因为其较好的水溶性. Cheng等[29]向催化氢解体系中引入多氯代烷, 较好地克服了小分子胺的不稳定和水溶性的问题(Eq. 7). C—Cl键会在Pd/C催化下发生加氢脱氯协同反应产生HCl和氯代烷, 而有机胺作为HCl的受体能加速氢解反应, 同时改变多氯代烷的用量可显著影响氢解反应的速率.该方法实现了脱苄和成盐两步合二为一的转化, 在不对称合成和天然产物的合成中具有较好的应用.
|
|
(7) |
Quast等[30]在研究2, 4-二氮杂二环[3.1.0]己烷-3-氧(17)的合成和反应活性时, 利用Pd/C在乙醇中催化氢解使环丙基开环的同时还能脱去咪唑啉上的两个苄基(Eq. 8).该催化氢解反应条件温和, 在98 kPa压力、54 ℃的条件下搅拌24 h就能获得产物.
|
|
(8) |
盐酸莫西沙星是一种氟喹诺酮类抗菌药, 临床上用于治疗肺病、肺炎、细菌性鼻窦炎以及皮肤和软组织感染等疾病.国内有很多课题组[31~34]在合成盐酸莫西沙星过程中, 使用Pd/C催化氢解的方法脱去(S, S)-8-苄基- 2-甲基-2, 8-二氮杂双环[4.3.0]壬烷(18)中的N-苄基(Eq. 9).该过程需在2.5~3.0 MPa的压力下进行, 能以超过80%的产率得到(S, S)-2-甲基-2, 8-二氮杂双环[4.3.0]壬烷, 无需纯化即可进行下一步反应.该脱苄方法虽产率高, 但需在高压釜中进行, 对设备提出了一定的要求, 是该脱苄方法的一大缺点.
|
|
(9) |
Pd/C作催化剂的脱苄方法不仅适用于小环化合物, 对于含多苄基的大环化合物同样具有较好的脱苄活性. Désogère等[35]使用Pd/C催化剂成功实现了含三苄基的九元环化合物1, 4, 7-三苄基-1, 4, 7-三氮杂环壬烷(20)的脱苄.该脱苄反应无需加热, 只在室温、乙酸条件下即可完成, 并能以95%高产率得到脱三苄产物1, 4, 7-三氮杂环壬烷(21) (Eq. 10).
|
|
(10) |
催化脱苄除了适用于链状化合物, 单、双杂环化合物和对三环化合物如螺桨烷也有较好的脱苄效果. Torres等[36]在研究螺桨烷的抗甲型流感病毒活性时, 合成了一系列的氮杂螺桨烷.其中, 对苄基保护的氮杂螺桨烷脱苄时, 先将底物22与盐酸反应制成盐酸盐, 而后在甲醇中以Pd/C催化氢解能高效得到脱苄基的螺桨烷盐酸盐(Scheme 3).鉴于螺桨烷盐酸盐的稳定性及氢解效果明显优于螺桨烷本身, 螺桨烷盐酸盐直接脱苄为不稳定化合物的脱苄提供了思路.
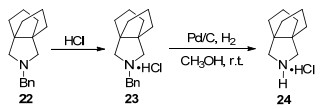
Pd(OH)2/C和Pd/C是催化氢解两种主要的催化剂, 二种催化剂的混合可达到单一催化剂无法实现的效果. Li等[37]则报道了用等量Pd(OH)2/C和Pd/C混合催化剂脱苄基的实例, 该体系对于苄基保护的脂肪胺或芳香胺均具有良好催化效果.混合催化剂的使用可以大幅缩短反应的时间, 对单一催化剂不能催化的反应也具有一定的催化活性.
2018年, Lou等[38]研究了用双金属催化剂代替单一的Pd金属催化剂制备HNIW的反应.该方法不仅可以降低贵金属Pd的用量, 而且对HBIW和TADBIW的脱苄具有选择性.研究发现, 以沉淀还原的方法制备的PdFe双金属催化剂的催化活性要远高于其他双金属如PdNi、PdCu、PdCo等催化剂.研究还表明, 以不同载体所制备的PdFe双金属催化剂对HBIW和TADBIW具有选择性, 以活性炭为载体的PdFe双金属催化剂只对TADBIW有催化活性, 以SiO2和Al2O3为载体的PdFe双金属催化剂HBIW有催化活性, 而以TiO2为载体的PdFe双金属催化剂对HBIW和TADBIW均有脱苄催化活性.
催化氢解是以氢气作氢源的非均相催化反应, 因此为加强氢气在催化剂上的吸附, 在实际使用过程中需在一定的压力下进行.催化氢转移氢解作为催化氢解的一种替代方法, 克服了催化氢解需在压力设备中进行的弊端.某些特定化合物在催化剂作用下发生脱氢作用, 氢原子从供体转移给催化剂, 然后传递给要被氢解的底物, 以这样的氢源进行的脱苄反应称为氢转移氢解[5, 8, 39]. Pd同样是应用最广泛的催化剂活性成分, 甲酸[40, 41]、甲酸铵[42~44]、环己烯[45]和1, 4-环己二烯[46]等可作为氢供体.催化氢转移氢解不需在压力条件下进行, 因此反应较温和, 操作简单, 无需耐压设备, 但氢供体的引入给产物的提纯带来不便, 是该反应的一大缺点.
甲酸作为一种常见有机氢供体, 能与多数有机溶剂混溶, 是优良的氢供体.在氨基酸和多肽合成过程中, 催化氢转移氢解被用来脱除苄基保护基, 由于环己烯和1, 4-环己二烯氢供体与肽及肽衍生物不混溶性, 使得在该条件下的催化氢转移反应不能发生. Elamin等[40]发现利用低浓度的甲酸溶液如体积分数为4.4%的甲酸/甲醇溶液, 在Pd/C催化下10 h就能以较高的产率(90%)脱去苄基赖氨酸25上的N-苄基(Eq. 11).
|
|
(11) |
温和的反应条件对那些温度敏感的底物尤为重要, 1987年Gray等[47]以甲酸为氢供体对N, N-二苄基丙氨酸甲酯(27)进行氢解脱苄(Eq. 12), 该反应在室温下氢解2 h即可完成, 氮原子上的两个苄基以较高的产率同时脱去得到丙氨酸甲酯.
|
|
(12) |
Zheng等[48]在合成β-氨基酸30时, 以催化氢解进行氨基脱保护时始终得到脱苄基、脱卤素的混合物.他们通过改变溶剂、催化剂、氢供体等条件, 最终发现催化剂的种类、溶剂对脱苄产物的影响不大, 氢供体则影响脱苄产物的组成.研究表明, 在Pd/C-HCOOH-CH3OH体系中, 苄基和带有支链保护的苄基均能脱去得到伯胺, 而且苯环上的卤素并未脱除(Eq. 13).因此, 可通过选择适当的氢供体在卤素存在下脱保护.
甲酸铵作氢供体的反应在文献中时有报道, 它本身不具酸性且几乎不与底物发生副反应, 在脱保护之后分离简单.莫西沙星作为第四代喹诺酮类抗菌药物, 因其具有半衰期长、不良反应少及抗菌谱广等优点, 具有很好的应用前景. (S, S)-2, 8-二氮杂双环[4.3.0]壬烷作为莫西沙星的关键中间体, 其快速高效合成是影响莫西沙星生产及应用的重要因素. Chen等[49]在优化合成(S, S)-2, 8-二氮杂双环[4.3.0]壬烷时, 以甲酸铵为氢供体使用催化转移氢解代替传统的高压催化氢解, 在甲醇溶剂中反应4 h即可以90.5%的收率得到(S, S)-2, 8-二氮杂双环[4.3.0]壬烷(32) (Eq. 14).该方法在常压下就可以进行, 大大降低了生产成本, 缩短了反应时间, 很好地促进了莫西沙星在医药领域的应用.
|
|
(13) |
|
|
(14) |
Hedgehog信号通路可在胚胎阶段调节细胞的增殖和分化, 其异常激活与肿瘤、癌症的发展有关, 因此通过Hedgehog信号通路发现新的潜在抗癌药物. Taladegib作为一类已在临床试验的Hedgehog信号通路抑制剂, 其高效合成具有重要的研究价值. Guo等[50]改进、优化Taladegib合成路线, 使用甲酸铵代替氢气, 用苄基保护基代替叔丁氧羰基保护基, 发现了一种新奇、高效合成Taladegib的新方法.该方法以甲酸铵为氢源、Pd/C为催化剂, 50 ℃反应12 h即可高效脱除苄基(Eq. 15).相比传统方法, 该方法反应条件温和、产率高、后处理简单, 适宜于大规模合成.
|
|
(15) |
环己烯和1, 4-环己二烯也可作为催化氢转移的氢供体. Bajwa等[51]使用1, 4-环己二烯实现了含有N-、O-苄基底物的选择性脱N-脱苄, 而O-苄基在反应中能不受影响(Eq. 16).
|
|
(16) |
Babu等[52, 53]还报道了一种新型催化转移氢解方法, 即使用廉价的金属镁、锌与氢供体甲酸铵去除N-Bn、O-Bn和S-Bn衍生物的苄基.在此过程中, 一些取代基如卤素、甲氧基、苯酚、酯、酸、乙烯基和叔丁氧羰基(Boc)均不受影响.该方法使用廉价金属催化很好地克服了贵金属催化的高成本的缺点, 同时避免了自燃, 是一种经济、环保的替代脱苄方案, 具有较好的应用前景.
还原脱苄虽能脱除大多数有机胺的苄基保护基, 但脱除苄基的同时也容易将其他基团脱除, 因此该方法选择性差.氧化脱苄是兼具温和和高选择性的脱苄方法, 具体过程为叔胺被氧化剂氧化形成α-羟基苄胺, 随后α-羟基苄胺进一步分解成仲胺及醛(Scheme 4), 达到脱苄的目的[8, 9, 54].氧化脱苄所用的氧化剂主要有有KMnO4、CrO3、O2、硝酸铈铵(CAN)和2, 3-二氯-5, 6-二氰对苯醌(DDQ)等.
CAN是氧化脱苄中应用最广泛的氧化剂, 其具有反应活性高、条件温和、操作方便、后处理简单、污染小, 具备一定的选择性等特点.由于CAN在多数有机溶剂中溶解度低, 在水中溶解度高, 因此CAN参与氧化脱苄反应大多在水、乙腈的混合溶剂中进行[55].
N, N-二苄基是构成NH2基团保护的重要形式, 已广泛应用到亲核加成或周环反应.然而对于含敏感基团的底物而言, 催化氢解选择性脱保护得到氨基或单N-苄基化合物的途径是困难的.在苄基酯或苄基醚存在下含有两个N-苄基取代基的叔胺37, 很容易用CAN进行选择性脱单苄基[56](Eq. 17).
|
|
(17) |
催化氢解是应用最广的脱苄方法, 但是对于含有不饱和键的底物往往导致双键的还原和脱苄同时发生, 得不到期望的脱苄产物. CAN作为脱苄的氧化剂, 不仅能高效脱除苄基, 而且对不饱和键显示出一定的化学惰性, 能够保留底物中的不饱和建. Torres等[36]使用CAN氧化苄基保护的氮杂[4.3.3]螺桨烷(39), 能够脱去苄基得到裸露的仲胺, 螺桨烷骨架上的双键并未发生变化(Eq. 18).该脱苄方法对于含不饱和键的底物具有良好的选择性, 解决了还原脱苄不具选择性的问题.
|
|
(18) |
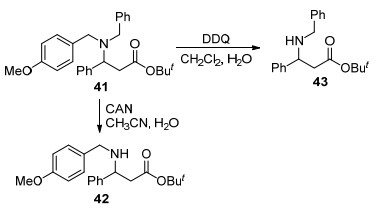
目前, CAN和DDQ已被广泛用作含苄基保护基团脱保护, 如醇[57, 58]和胺[59~61]的脱保护.传统脱苄方法可以容易地实现杂原子苄基脱保护, 但是仅有少数化合物在多官能团存在下能实现化学选择性脱苄. Bull等[62, 63]以不同底物来进行CAN氧化脱苄反应, 详细研究了该脱苄试剂的反应特点.研究发现, CAN和DDQ虽然均能选择性脱除苄基, 但两者的脱苄效果存在一定差距, 前者优先脱除41中未取代的苄基得到42, 后者则更适用于对含给电子基团的苄基脱除(Scheme 5).此外, CAN对叔胺有较好的脱苄效果, 而对仲胺往往是化学惰性的.因此, 在含多苄基的底物中尤其是N, N-二苄基叔胺44, CAN氧化脱苄仅停留在脱单苄的阶段(Scheme 6).
陈树森等[64]选用多种不同氧化剂对HBIW进行了一系列研究, 氧化剂弱, 反应不发生; 氧化剂强, 笼型骨架解体.鉴于HBIW脱苄产物含有裸露的N—H键, 在多数的氧化体系中的稳定性要远低于HBIW, 因此在脱苄过程中必须及时保护脱苄产物.他们采用酸酐为反应介质, 在冰盐浴中用KMnO4氧化HBIW得到了脱苄乙酰化的产物46 (Eq. 19), 而使用CrO3氧化脱苄[65]则往往将苄基氧化成苯甲酰基未得到脱苄的产物.
|
|
(19) |
氧气是另一类重要的脱苄反应氧化剂, 尤其适用于含氮杂环, 在碱性条件下可提高其反应活性. 1983年, Gigg等[66]首次报道了使用t-BuOK和O2用于吡喃葡萄糖苷的快速N-脱苄基化的实例, 近20年后, Haddach和Kong等[67, 68]又用相同氧化试剂实现芳香杂环的N-脱苄基化. Semak等[69]此基础上改进反应条件, 提出了一种环保, 易于处理、经济且高效的脱苄方法, 该方法在碱性介质中使用空气或稀释的氧气作氧化剂, 能快速脱去取代苄基(Eq. 20).除此之外, 所需的脱苄基化产物可直接用于合成而无需进一步纯化, 反应后处理仅需要酸碱中和萃取除去形成的苯甲酸.
|
|
(20) |
目前, 以O2为氧化剂的脱苄方法经过几十年的发展, 形成了O2和t-BuOK的氧化脱苄体系. 2015年, 朱文通等[70]分别以N-取代苄基咔唑和N-取代吲哚为原料, 在碱性条件氧气环境下脱去苄基得到咔唑和吲哚51 (Eq. 21).该方法操作简单、反应温和且安全环保, 在天然产物和药物的合成中有重要应用.
|
|
(21) |
2016年, Page等[71]发现了一种1, 4-二取代三唑通过烷基化脱苄基制备1, 5-二取代三唑的方法, 该方法未使用贵金属催化剂, 简便高效廉价.他们首先用碘代烷对1, 4-二取代三唑进行烷基化, 生成一种反应活性较高的三唑盐53, 然后在t-BuOK的存在下氧化脱去苄基(Scheme 7).研究同时表明, 碘和t-BuOK的存在对成功脱苄有重要作用.
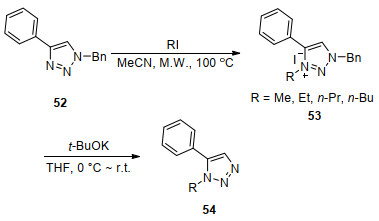
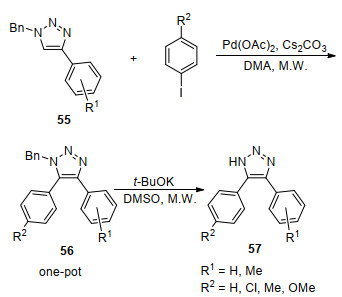
Zhao等[72]在没有配体情况下使用Pd(OAc)2催化剂催化二取代三唑芳基化偶联得到三取代三唑56, 反应混合物不经过分离直接加入强碱t-BuOK实现氧化脱苄(Scheme 8).该反应在微波条件短时间内实现偶联、脱苄一锅转化, 脱苄的底物不需分离提纯, 具有操作简单、产率高(产率60%~90%)、反应条件温和等优点.微波的使用降低了反应的难度, 大大加快了反应的进行, 提高了产率.
Feula等[73]将含有叔和仲苄胺官能团的化合物暴露于碘单质和碳酸铯的氧化条件下, 水解后得到仲胺的脱苄产物, 证实了仲苄基胺通过氧化、水解是脱苄的有效方法.该方法首先用碘单质和碳酸铯作为混合氧化剂在乙腈中氧化得到亚胺, 随后亚胺经水解得到胺59 (Eq. 22).该方案操作简单, 产率高产且无需分离纯化, 能选择性氧化脱去仲苄基胺上的苄基, 而对于叔胺苄基具有较好的化学惰性.该方案不仅能有效地脱去苄基, 还可脱去烷基, 这为苄基、烷基的脱保护策略提供了选择.
|
|
(22) |
偶氮二甲酸二异丙酯(DIAD)也是一种氧化脱苄的试剂, 它不仅能脱去N-苄基, 而且在多种官能团存在下具有选择性. Kroutil等[74, 75]提出了选择性脱N-苄基的新方法, 此类试剂尤其适用于多官能团的杂环化合物.该方法通过苄基保护的底物与DIAD反应可选择性脱除N-苄基(Eq. 23), 而其上的O-苄基并未反应.
|
|
(23) |
Moriyama等[76]开发了一种由碱金属溴化物/过硫酸氢钾复合盐(Oxone)氧化体系脱除N-苄基酰胺和O-苄基醚上苄基的脱苄方法(Eq. 24).该脱苄方法具有很好的底物普适性, 无论是对氮原子上连有吸电子基团如苯磺酰基、硝基苯磺酰基、苯甲酰基, 还是含有甲基、乙基、异丙基、异丁基、环戊基、环己基、环庚基等供电子的烷基, 均有很好的脱苄效果.除此之外, 该体系还能脱去O-苄基醚上苄基得到酮类化合物.该试剂具有对空气稳定、易处理、低毒性且不产生污染环境的产物等特点, 适宜在有机合成中广泛使用.
|
|
(24) |
传统催化氢解和催化氢转移氢解虽能高效脱去氮原子上的苄基, 但对某些底物如N-酰基-N-苄基衍生物或芳香杂环的脱苄往往不成功.可溶性碱金属和有机金属试剂可用于切断N-苄基、O-苄基中C—N、C—O键, 是催化氢解的替代方法.碱金属脱苄的过程是金属的电子转移到反应底物生成负离子自由基, 负离子自由基裂解产生苄基自由基和胺负离子, 苄基自由基获取氢原子生成甲苯, 胺负离子获取质子得到胺[2, 9].近年来, 越来越多的研究表明, 强碱金属和有机金属试剂可用于较难脱苄的底物[77~82], 该方法虽具有高效、快速等特点, 但由于该方法中涉及到活泼金属试剂, 低温、无氧的操作环境, 其苛刻的反应条件大大限制了应用.
有机锂化合物是一类重要的金属有机化合物, 独特的电子分布决定了其强亲核性和强碱性.因此, 有机锂具有对多种官能团进行烷基化、脱除保护基团、合成其它甲基化有机金属试剂、还原其他过渡金属等应用. Suzuki等[83]用甲基锂与苄基取代的吲哚类衍生物64反应, 制得脱苄产物吲哚类似物(Eq. 25).此外, 正丁基锂和二异丙基氨基锂(LDA)等有机锂化合物同样用来作为脱苄基试剂, 且无需在低温下进行, 上述试剂的反应条件较碱金属脱苄温和.因此, 有机锂化合物广泛地用来合成吲哚和吲哚类衍生物.
|
|
(25) |
Rao等[84]在合成atorvastatin过程中, 以催化氢解、催化氢转移、叔丁醇钾氧化体系等不同方法均不能成功脱苄.最终, 他们发现了将金属钠溶解在液态氨中, 并以t-BuOH为质子供体, 在-78 ℃低温下能脱去芳香杂环66上的氮苄基(Eq. 26).该方法虽反应条件苛刻, 并涉及危险试剂和药品, 但对于传统方法较难脱除的N-酰基-N-苄基衍生物和芳香杂环化合物来说不失为一种较好的脱苄方法.
|
|
(26) |
Franceschini等[85, 86]使用强碱金属Li在低温条件下对苄基硫吗啉-3-酮脱保护得到硫吗啉-3-酮69 (Eq. 27), 随之探索了反应时间对脱苄产物收率的影响.结果表明, 随着反应时间的延长, 产物的副产物变多, 致使分离提纯难度加大, 因此合理控制反应时间是控制产物分布的有效方法.该方法虽反应迅速, 产率高, 且对于苄基上含有支链的底物也有较好的脱苄效果, 但其反应条件的苛刻及试剂的危险性严重限制了该方法的实用性.
负载于多孔硅胶(SG)的碱金属是有机胺还原脱烯丙基、苄基、三苯甲基化的有效试剂(Eq. 28)[87], 这类试剂为传统碱金属氨溶液脱保护提供了方便替代品.其中, 乙二胺(EDA)是促进脂族底物反应所必需的添加物, 随后底物可在16~24 h内将其彻底脱保护.该方法具有反应简单, 易于后处理和分离的特点, 反应条件也较碱金属氨溶液脱苄温和.
|
|
(27) |
|
|
(28) |
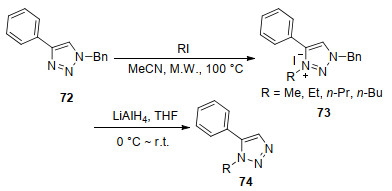
LiAlH4作为强还原剂在有机合成中占据重要的地位, 尤其是用于酯、羧酸和酰胺的还原. Page等[71]发现了一种由1, 4-二取代三唑72通过烷基化而后脱苄基制备1, 5-二取代三唑的方法(Scheme 9), 该方法未使用贵金属催化剂, 简便高效廉价.他们首先用碘代烷对1, 4-二取代三唑进行烷基化, 生成一种反应活性较高的三唑盐.由于三唑盐中含有不饱和的五元杂环, 无法通过使用催化氢解、氢转移氢解进行脱苄.他们尝试使用强还原试剂LiAlH4还原三唑盐, 意外地发现能够脱苄得到1, 5-二取代三唑. LiAlH4进行脱苄虽获得目标物产率较低, 但开拓了一种脱去杂环上苄基的新方法.
截短侧耳素(pleuromutilin)是一类天然存在的抗生素, 具有一定的抗菌活性, 其中截短侧耳素SB-268091已被开发用来治疗鼻窦炎和中耳炎. 2012年, Choudary等[88]报道了抗生素SB-268091的合成, 其中重点介绍了合成奎宁环-4-硫醇配体76新途径.该途径涉及到在液氨中使用金属钠进行还原性脱苄基, 实现了N-苄基和S-苄基同时脱除(Eq. 29).钠与液氨组成的脱苄体系具有较强的脱苄能力, 因此对脱保护不具备选择性, 特别适用于不含敏感基团且较难脱除的苄基化合物的脱保护.
与其他脱苄方法相比, 酸解脱苄反应迅速且操作简单, 因此也是一类重要的脱苄方法.酸解脱苄就是苄基保护的底物在酸的作用下, 脱除苄基得到游离胺或有机胺盐的过程[4].酸解脱苄过程需要在酸性环境中进行, 因此底物需对酸具有较好的耐受力, 常用催化酸解脱苄的酸有三氟乙酸(TFA)、三氟甲磺酸(TfOH)、对甲苯磺酸(p-TsOH)等.酸解脱苄过程操作简单, 反应迅速, 但是仅对某些底物有较好的脱苄效果, 不具备通用性.
|
|
(29) |
吡唑是一类具有广泛药物活性的基团, 近年来大量吡唑衍生物被合成出来.尤其值得注意是含有1-芳基吡唑结构的celecoxib (78), 它是选择性环氧酶-2 (COX-2)抑制剂, 可用于治疗骨关节炎、类风湿性关节炎和急性疼痛等症状. 2012年, Shaw等[89]报道了一种通过Ugi反应/脱苄基化/肼介导的环化的三步反应来合成3-羟基吡唑的方法.该方法脱苄基时, 采用10% TFA(三氟乙酸)/DCE(二氯乙烷)溶液在微波辅助10 min下脱去苄基保护的3-羟基吡唑衍生物的苄基, 产率也超过80% (Eq. 30).相比于其他传统脱苄方式, 该方法快速高效, 且脱苄产物的分离也较简单.
|
|
(30) |
Lee等[90]在制备N-甲基酰胺的过程中, 首先合成出N-甲基-N-苄基酰胺, 随后经酸解脱苄的方法合成了N-甲基酰胺. N-甲基-N-苄基酰胺79以三氟乙酸(TFA)为脱苄试剂得到N-甲基酰胺(Eq. 31), 而酰胺和芳胺上的N-甲基均能保持稳定.该方法对于合成各种含氮酰胺非常有效, 并且应用于多种在酸性下稳定的官能团的脱保护.
|
|
(31) |
三氟乙酸不仅能脱去苄基, 对于取代苄基如对甲氧基苄基也有不错的脱苄效果.吲哚环是许多天然产物和药物的主要骨架, 具有一定替代模式的吲哚的高效构建具有挑战. 2014年, Lam等[91]在研究高效构建吲哚环时, 需对重要中间体5-丁基-2-碘-3-[N-(4-甲氧基苄基)-N-(对甲苯磺酰)氨基]苯基甲磺酸(82)脱保护.他们使用三氟乙酸为脱苄试剂, 在室温环境中搅拌3 h即可脱去对甲氧基苄基(Eq. 32), 产率达94%~99%.该方法既不用使用耐压的设备, 又不需要昂贵的催化剂, 在温和的条件下就可实现脱苄, 极大降低了合成吲哚的成本.
|
|
(32) |
另外, Rombouts等[92]报道了在微波辅助下用三氟甲磺酸对N-苄基酰胺83进行脱苄基的新方法(Eq. 33).该脱苄方法已经显示出相当普遍的适应性, 对于仲、叔, 脂族、芳香族和非环状、环状酰胺均有较好的脱苄活性.
|
|
(33) |
2014年, Lima等[93]报道了kealiiquinone及Leucetta衍生物的合成方法(Eq. 34).在由苄基保护基的咪唑萘醌合成6, 7-二甲氧基-1-甲基-4-苯基-1H-萘并[2, 3-d]-咪唑-2, 5, 8-三酮(86)时, 由于咪唑萘醌化合物中含有羰基、甲氧基和N-甲基, 传统的还原脱苄和氧化脱苄的脱苄方法均不能成功脱保护.随后他们改用TfOH作为酸解脱苄的试剂, 在55 ℃即可脱去苄基得到86, 而骨架上的羰基、甲氧基及N-甲基也并未发生变化.
|
|
(34) |
Chern等[94]发现了用对甲苯磺酸脱去苄基的新方法, 该方法尤其适用于对酸或还原体系敏感的底物. Chern等使用同类底物87对不同的脱苄方法进行比较, 以Pd/C为催化剂的催化氢解和三氟乙酸的酸解脱苄的产率均在22%以下, 而对甲苯磺酸的酸解脱苄却能在90%以上.此外, 对甲苯磺酸还能在N-Fmoc、N-Boc或N-Tr保护基存在下, 选择性脱除苄基, 与传统催化氢解相比, 该方法都具有独特的优势(Eq. 35).
|
|
(35) |
硝解脱苄即底物中的苄基与硝化试剂反应得到硝基或亚硝基取代的衍生物, 该方法实现了含能材料的脱苄和硝化的一步转化, 在含能材料的合成中具有重要应用.硝解脱苄就是硝化试剂首先进攻氮原子, 随后脱去羟胺得到亚胺正离子, 亚胺正离子经水解硝化得到硝基或亚硝基取代的衍生物的过程[8, 9].由于反应常在酸性条件下进行且需使用硝化试剂, 因此底物需具有较好的稳定性, 这大大限制了它的使用.即便如此, 这仍是直接引入硝基的脱苄方法, 在含能领域有着独特的优势.
亚硝解[95, 96]也可用于苄基的脱保护, 即分子中苄基保护基被亚硝基取代.由于亚硝基易被氧化成硝基, 因此亚硝解脱苄在含能领域也有重要应用.于永忠等以四乙酰基二苄基异伍兹烷(4)为原料, 在NOBF4体系中亚硝解得到了亚硝解的脱苄产物.该方法可将催化氢解不易脱除的4, 10位的苄基脱掉, 获得四乙酰基二亚硝基异伍兹烷(Eq. 36), 后者经进一步氧化、硝化可得到HNIW.此后, Bayat等[97~99]又改用N2O4亚硝化试剂将4, 10位不易脱除的苄基脱除, 得到四乙酰基二亚硝基异伍兹烷.
|
|
(36) |
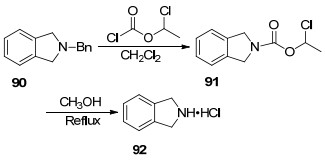
取代脱苄就是一种以易脱除的基团取代底物中的苄基得到取代衍生物, 而后将易取代的基团脱去得到裸露的N—H键的方法.取代脱苄的取代基一般优先选择氯甲酸酯, 用来进行脱苄的氯甲酸酯有氯甲酸甲酯、氯甲酸乙酯和氯甲酸苯酯, 其中苯酯的脱苄效率要高于甲酯和乙酯[100].苄基保护的叔胺与氯甲酸酯生成氨基甲酸酯, 随后氨基甲酸酯在一定条件下反应得到仲胺.
1993年Yang等[101]利用氯甲酸氯乙酯取代叔胺90上的苄基, 而后经CH3OH回流得到仲胺的盐酸盐(Scheme 10).在反应中, 氯甲酸氯乙酯与叔胺形成季铵盐, 随后分子内脱去一分子氯苄得到化合物91, 化合物91与甲醇加成脱羧后制得仲胺盐酸盐92.氯甲酸酯脱苄也是一类重要的脱保护方法, 它适用于多种含氮五元及六元杂环叔胺, 氯甲酸酯的毒性限制了该方法的使用.
1994年, See等[102]报道了将HBIW与氯甲酸三甲基硅基乙酯在四氢呋喃或乙醚中进行取代脱苄反应, 可制得六取代的六氮杂异伍兹烷93(Eq. 37).该方法所需温度低, 时间长, 并且涉及到有毒的光气, 因此实用价值较小.
|
|
(37) |
Wright等[103]也利用氯甲酸乙酯作脱苄试剂, 在以苯为溶剂回流条件下对氮苄基氨基甲酸酯化合物94进行脱苄得到氨基甲酸酯衍生物(Eq. 38).该方法尤其适用于制备氨基甲酸酯衍生物, 同时发现苯环上含有吸电子基团或是供电子基团对于脱苄均反应是不利的.
|
|
(38) |
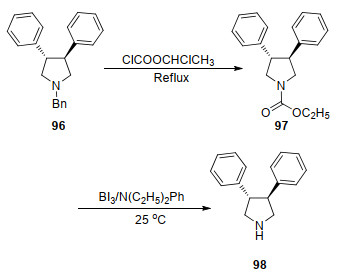
Olofson等[104]发现氯甲酸氯乙酯与叔胺生成季铵盐, 该季铵盐可在甲醇的回流条件下脱去烷基得到胺的盐酸盐[103].因此, 可利用该方法高效脱除氮原子上的烷基, 并生成季铵盐.对于脱保护之后不稳定的化合物, 季铵盐的生成可可大大提高产物的稳定性, 在不稳定化合物的合成中具有独特的优势.鉴于烷基保护基的脱除难度大于苄基的脱除, 因此该方法也可用来脱除苄基. Kanth等[105]使用氯甲酸氯乙酯取代了三取代吡咯衍生物96上的苄基生成氨基甲酸酯衍生物, 在I3B/N(C2H5)Ph存在下以80%总产率生成二取代吡咯衍生物98 (Scheme 11).
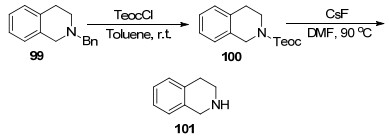
TeocCl [Teoc: C(C=O)O(CH2)2TMS]也可作为脱苄试剂, 它也能高效取代苄基生成N-Teoc衍生物, 随后N-Teoc衍生物通过CsF在90 ℃下即可脱去Teoc保护基, 得到四氢异喹啉101. Igarashi等[106]合成奎宁类生物碱前体时, 使用Teoc为保护基取代苄基并用CsF脱除实现了高效构建奎宁环(Scheme 12).
天然产物(NPs)是生物活性小分子的重要来源, 其为发现新药物提供了可能.在过去30年批准的新药中, 大约三分之一的小分子药物是基于天然产物及其衍生物. NPs除了具有复杂的结构和化学多样性外, 还因其惊人的药物分子特性而获得大量关注. Annamalai等[107]合成了六类具有一定生物活性化合物, 试图由此建立一个具有生物活性的核心结构的化合物库.他们在合成十氢-1, 6-二氮杂萘类化合物103过程中, 由于氮杂萘上含有酯, 使用传统的催化氢解不能使苄基保护的氮杂萘脱保护.经过大量的实验, 他们使用1-氯甲酸氯乙酯成功脱去N-苄基(Eq. 39), 脱苄产率最高可超过90%.
|
|
(39) |
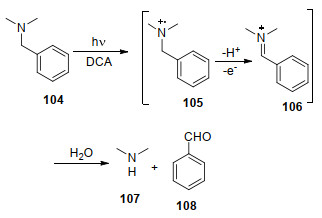
Pandey和Rani等[108]发现了单电子转移脱苄的新方法, 即以9, 10-二氰基蒽作为电子受体, 底物在光照条件下由基态络合物发生单电子转移, 同时自身被氧气及碳亲核试剂进攻后得到亚胺正离子, 亚胺正离子再进行水解得到脱苄的产物107和苯甲醛(Scheme 13), 反应的产率在75%~90%之间.该方法适应多种底物如环叔胺和脂肪叔胺, 同时羟基及酯基能在该反应中保持稳定.
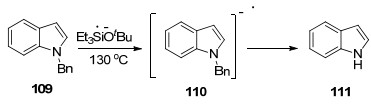
叔丁醇钾-三乙基硅烷体系不仅能够实现芳烃和杂环芳烃的硅烷基化, 而且还能还原性断裂C—N, C—O键和芳香硫醚的C—S键. Smith等[109]系统研究了叔丁醇钾-三乙基硅烷体系脱保护的反应机理, 包含了单电子转移和氢化物传递的过程. Et3SiH与t-BuOK首先形成良好的电子供体, 电子经过转移形成中间体110, 最后通过氢化物的传递得到脱苄产物111 (Scheme 14)以及副产物甲苯.该体系由于具有较强的供电子能力, 还可作为电子供体还原不饱和键双键和叁键, 更能脱去甲基、烯丙基、苄基、萘基等保护基.
格氏试剂具有强亲核性, 在有机反应中具有重要应用, 是增加碳链长度和进行衍生化的重要试剂. Golantsov等[110]在使用2-氰基吲哚(112)合成含有手性取代基的吲哚衍生物过程中, 发现该反应得到的并非是单一的吲哚酮, 而是含有手性取代基的吲哚酮和脱去苄基的吲哚酮的混合物(Eq. 40), 从而得到了意外的脱苄结果.进一步研究表明, 改变反应条件可控制两者的生成比例, 在乙醚中往往得到两者的混合物, 而在甲苯中仅得到脱苄基吲哚酮.此外过渡金属试剂(CuCN)的加入使产物更多地向含有手性取代基的吲哚酮转化.该方法虽底物的适应性较低, 但其完成某些含氮化合物衍生化和脱苄一步转化, 减少了反应步骤.
|
|
(40) |
近年来, 因生物催化脱保护不需高温、高压和有毒的试剂, 对环境影响小, 同时具有高选择性脱保护的特点, 该方法已成为脱保护的一大热点.其中, 水解或氧化还原酶已经被证明可用于多种官能团选择性修饰, 酰基转移酶、酯酶、脂肪酶和蛋白酶被认为是烷基酯脱保护的有效酶, 氧化还原酶也已显示出N-脱保护反应的潜力. Montero等[111]报道了利用漆酶(Laccase)有效脱除N-苄基反应的研究, 漆酶可在水溶液与不同的化学氧化剂兼容, 这扩大了它们在许多工艺中的适用性.以漆酶和2, 2, 6, 6-四甲基哌啶-氮-氧化物(TEMPO)组成的混合氧化体系可以实现脂肪胺、环状和芳香胺高选择性温和脱保护(Eq. 41).该反应对N-苄基化的仲胺表现出较好的反应活性, 但它不会改变O-苄基化的醇或N-苄基化的叔胺, 这是其他化学脱保护方法较难实现的, 适合广泛用于合成化学, 如氨基酸或核苷化学.
|
|
(41) |
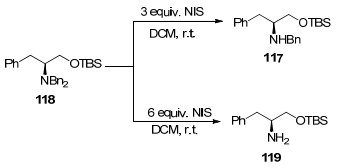
N-碘代琥珀酰胺(NIS)是一种高效、温和、方便且可控的脱苄试剂, 其能进行N-二苄胺的选择性脱苄基. Grayson等[112]在进行N-卤化生成亚胺的研究过程中, 发现连续卤化、消除和水解可能为N-脱苄基化提供途径.他们进一步研究证实, N-氯代丁二酰亚胺脱苄效果不佳, 但N-碘代丁二酰亚胺(NIS)能较好实现二苄胺的脱苄基化(Scheme 15).当底物118与NIS的物质的量比为1:3时得到的是脱单苄产物, 而物质的量比增加至1:6时则得到较多的脱二苄产物.该试剂在多种底物中具有实用性和选择性, 尤其是在合成含多官能团苄氨基醇类化合物, 可以通过调节NIS的用量来控制反应产物.
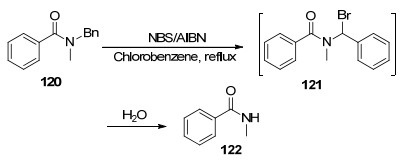
Baker等[113]报道了一种使用N-溴代丁二酰亚胺(NBS)/偶氮二异丁腈(AIBN)试剂脱去酰胺苄基的新方法, 该方法苄基保护的底物与NBS发生溴代反应生成中间体, 而后中间体经水解后发生C—N键的断裂, 生成脱苄产物122 (Scheme 16).脱保护的过程中形成的苄卤中间体水解是脱苄反应的关键, 因此该体系仅对氮原子上的苄基有较好的反应活性, 对于N-甲基和N-苯基均不能反应.
Yanada等[114]在合成羟吲哚类化合物124时, 分别尝试了以BBr3、t-BuOK/O2、CAN等氧化脱苄方法, 遗憾的是以上方法均不能脱去氮原子上的苄基. NBS/AIBN组成的体系能以95%的产率脱去苄基得到羟吲哚(Eq. 42), 该体系为多种天然产物的全合成尤其是含氮杂环脱保护提供了一种重要的策略.
综上所述, 脱苄的方法有很多, 各有特点.催化氢解和催化氢转移氢解脱苄是使用最广泛的方法, 其优点是反应温和, 产率高, 但也存在选择性差、成本高, 往往不适用于含不饱和键底物的缺点.催化氢解一般需要在加压环境中进行, 对反应设备要求高.催化氢转移氢解过程中使用的氢供体, 不利于产物的分离提纯.对于在反应条件下不稳定的化合物, 可通过预先成盐或是用其他试剂及时捕捉脱苄产物的方法实现脱保护.碱金属脱苄反应活性高, 产率好, 可用于底物结构稳定、苄基不易脱除的转化; 但其反应条件苛刻, 涉及有毒危险试剂, 严重制约了其使用范围.取代脱苄将苄基转换为易脱除的基团, 大大降低了脱苄过程的难度, 但不足之处是增加了转化的步骤.氧化脱苄虽然操作方便, 后处理简单, 且具备一定的化学选择性, 但在氧化过程中往往会发生一些副反应, 给脱苄产物的分离、提纯带来了一定的困难, 同时也对底物的稳定性提出了特殊要求.硝解脱苄得到特定的硝基或亚硝基化合物, 可直接用来制备含能材料, 因此在含能材料领域应用较多.其他脱苄方法虽然效率高, 但仅针对特定的反应底物, 适应性小.酶催化脱苄是目前兼具高效、选择性、环保等特点且前景较好的脱苄方法, 值得进行深入研究.但在前期研究中需进行酶活性筛选, 筛选难度大, 强度较高, 严重制约了其应用.因此, N-苄基脱除方法要根据具体反应底物的结构进行合理选择, 选用不同的脱苄策略.
|
|
(42) |
荣国斌, 有机合成中的保护基, 华东理工大学出版社, 上海, 2004, pp.494~654.Rong, G.-B. Protective Groups in Organic Synthesis, East China University of Science and Technology Press, Shanghai, 2004, pp.494~654(in Chinese).
黄培强, 靳立人, 陈安齐, 有机合成, 高等教育出版社, 北京, 2006, pp.385~389.Huang, P.-Q.; Jin, L.-R.; Chen, A.-Q. Organic Synthesis, Higher Education Press, Beijing, 2006, pp.385~389(in Chinese).
吴毓林, 姚祝军, 胡泰山, 现代有机合成化学, 科学出版社, 北京, 2006, pp.27~73.Wu, Y.-L.; Yao, Z.-J.; Hu, T.-S. Modern Organic Synthetic Chemistry, Science Press, Beijing, 2006, pp.27~73(in Chinese).
武钦佩, 李善茂, 保护基化学, 化学工业出版社, 北京, 2007, pp.239~277.Wu, Q.-P.; Li, S.-M. Protective Chemistry, Chemical Industry Press, Beijing, 2007, pp.239~277(in Chinese).
Mackie R. K., Smith D. M., Aitken R. A., Meng G.有机合成指南, 化学工业出版社, 北京, 2009, pp.210~223.Mackie, R.K.; Smith, D.M.; Aitken, R.A.; Meng, G. Guidebook to Organic Synthesis, Chemical Industry Press, Beijing, 2009, pp.210~223(in Chinese).
戴云生, 董守安, 潘再富, 陈家林, 工业催化, 2011, 19, 4. doi: 10.3969/j.issn.1008-1143.2011.04.002Dong, Y.-S.; Dai, S.-A.; Pan, Z.-F.; Chen, J.-L. Ind.Catal. 2011, 19, 4(in Chinese). doi: 10.3969/j.issn.1008-1143.2011.04.002
Bayat, Y.; Ebrahimi, H.; Fotouhi, F.F. Org. Process Res. Dev.2012, 16, 1733. doi: 10.1021/op300162d
贾会平, 欧育湘, 陈博仁, 徐永江, 陈江涛, 含能材料, 1998, 6(4), 145. http://www.cnki.com.cn/Article/CJFDTotal-HNCL804.000.htmJia, H.-P.; Ou, Y.-X.; Chen, B.-R.; Xu, Y.-J.; Chen, J.-T. Chin. J. Energ. Mater.1998, 6(4), 145(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-HNCL804.000.htm
邱文革, 于永忠, 合成化学, 1998, 6, 34.Qiu, W.-G.; Yu, Y.-Z. Chin.J.Synth.Chem. 1998, 6, 34(in Chinese).
Davies, S.G.; Ichihara, O. Tetrahedron Lett.1999, 40, 9313. doi: 10.1016/S0040-4039(99)01954-1
Couturier, M.; Andresen, B.M.; Jorgensen, J.B.; Tucker, J.L.; Busch, F.R.; Brenek, S.J.; Dube, P.; Ende, D.J.; Negri, J.T.P. Org. Process Res. Dev.2002, 6, 42. doi: 10.1021/op0102161
Amat, M.; Llor, N.; Hidalgo, J.; Escolano, C.; Bosch, J. J. Org. Chem.2003, 68, 1919. doi: 10.1021/jo0266083
Davies, S.G.; Nicholson, R.L.; Price, P.D.; Roberts, P.M.; Rueesell, E.D.; Savory, E.D.; Smith, A.D.; Thomson, J.E. Synlett 2004, 901.
Srimurugan, S.; Murugan, K.; Chen, C. Chem. Pharm. Bull.2009, 57, 1421. doi: 10.1248/cpb.57.1421
Davies, S.G.; Fletcher, A.M.; Hughes, D.G.; Lee, J.A.; Price, P.D.; Roberts, P.M.; Russell, A.J.; Smith, A.D.; Thomson, J.E.; Williams, O.M.H. Tetrahedron 2011, 67, 9975. doi: 10.1016/j.tet.2011.09.038
Leitch, D.C.; Greene, T.F.; O'Keeffe, R.; Lovelace, T.C.; Powers, J.D.; Searle, A.D. Org. Process Res. Dev.2017, 21, 1806. doi: 10.1021/acs.oprd.7b00261
Yoshida, K.; Nakajima, S.; Wakamatsu, T.; Ban, Y. Heterocycles 1988, 27, 1167. doi: 10.3987/COM-88-4525
公绪滨, 孙成辉, 庞思平, 张静, 李玉川, 赵信岐, 有机化学2012, 32, 486. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341112.shtmlGong, X.-B.; Sun, C.-H.; Pang, S.-P.; Zhang, J.; Li, Y.-C.; Zhao, X.-Q. Chin. J. Org. Chem.2012, 32, 486(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341112.shtml
Bellamy A.J. Tetrahedron 1995, 51, 4711.
Wardle, R.B.; Edwards, W.W. WO 97/20785, 1997.
Koskin, A.P.; Simakova, I.L.; Parmon, V.N. Russ. Chem. Bull.2007, 56, 2370. doi: 10.1007/s11172-007-0377-5
Kalashnikov, A.I.; Sysolyatin, S.V.; Sakovich, G.V.; Surmacheva, I.A.; Surmachev, V.N.; Lapinab, Y.T. Russ. Chem. Bull.2009, 58, 2164. doi: 10.1007/s11172-009-0295-9
Fotouhi-Far, F.; Bashiri, H.; Hamadanian, M. Propellants Explos., Pyrotech.2017, 42, 213. doi: 10.1002/prep.201600035
Fotouhi-Far, F.; Bashiri, H.; Hamadanian, M.; Keshavarz, M.H. Inorg. Nano-Met. Chem.2017, 47, 1489. doi: 10.1080/24701556.2017.1357593
Ji, H.-T.; Jing, Q.; Huang, J.-W.; Silverman, R.B. Tetrahedron 2012, 68, 1359. doi: 10.1016/j.tet.2011.12.013
朱思兰, 朱晶; 张伟萍, 陈程, 史海健, 合成化学, 2014, 22, 664. doi: 10.3969/j.issn.1005-1511.2014.05.022Zhu, S.-L.; Zhu, J.; Zhang, W.-P.; Cheng, C.; Shi, H.-J. Chin.J.Synth.Chem. 2014, 22, 664(in Chinese). doi: 10.3969/j.issn.1005-1511.2014.05.022
Hou, T.-J.; Zhang, J.; Wang, C.-J.; Luo, J. Org. Chem. Front.2017, 4, 1819. doi: 10.1039/C7QO00357A
Bailey, P.D.; Beard, M.A.; Dang, H.P.T.; Phillips, T.R.; Price, R.A.; Whittaker, J.H. Tetrahedron Lett.2008, 49, 2150. doi: 10.1016/j.tetlet.2008.01.104
Cheng, C.-J.; Sun, J.-W.; Xing, L.-X.; Xu, J.-M.; Wang, X.-Y.; Hu, Y.-F. J. Org. Chem.2009, 74, 5671. doi: 10.1021/jo9007282
Quast, H.; Walter, S.J.; Mueller, B. Heterocycles 2012, 85, 2449. doi: 10.3987/COM-12-12543
高金华, 硕士论文, 浙江工业大学, 杭州, 2012.Gao, J.-H. M.S.Thesis, Zhejiang University of Technology, Hangzhou, 2012(in Chinese).
张明光, 马堰启, 夏正君, 陈再新, 中国医药工业杂志, 2014, 45, 404. http://www.cnki.com.cn/Article/CJFDTotal-ZHOU201405003.htmZhang, M.-G.; Ma, Y.-Q.; Xia, Z.-J.; Chen, Z.-X. Chin. J. Pharm.2014, 45, 404(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-ZHOU201405003.htm
卢定强, 王维胞, 凌岫泉, 解杰, 沈东, 现代化工, 2014, 34, 33. doi: 10.3969/j.issn.0253-4320.2014.02.008Lu, D.-Q.; Wang, W.-B.; Ling, X.-Q.; Xie, J.; Shen, D. Mod. Chem. Ind.2014, 34, 33(in Chinese). doi: 10.3969/j.issn.0253-4320.2014.02.008
喻理德, 徐其雄, 王星, 江西师范大学学报(自然科学版), 2017, 45, 507. http://www.cnki.com.cn/Article/CJFDTOTAL-CAPE201705011.htmYu, L.-D.; Xu, Q.-X.; Wang, X. J. Jiangxi Norm. Univ.(Nat. Sci. Ed.)2017, 45, 507(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-CAPE201705011.htm
Désogère, P.; Rousselin, Y.; Poty, S.; Bernhard, C.; Goze, C.; Boschetti, F.; Denat, F. Eur. J. Org. Chem.2014, 35, 7831.
Torres, E.; Leiva, R.; Gazzarrini, S.; Carrizo, M.R.; Vivas, M.F.; Moroni, A.; Naesens, L.; Vázquez, S. ACS Med. Chem. Lett.2014, 5, 831. doi: 10.1021/ml500108s
Li, Y.; Manickam, G.; Ghoshal, A.; Subramaniam, P. Synth. Commun.2006, 36, 925. doi: 10.1080/00397910500466199
Lou, D.-Y.; Wang, H.-S.; Liu, S.; Li, L.; Zhao, W.; Chen, X.-H.; Wang, J.-L.; Li, X.-Q.; Wu, P.-F.; Yang, J. Catal. Commun.2018, 109, 29.
舒畅, 郑纯智, 王日杰, 张继炎, 化学通报, 2004, 67. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxtb200401073Shu, C.; Zheng, C.-Z.; Wang, R.-J.; Zhang, J.-Y. Chemistry 2004, 67(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxtb200401073
Elamin, B.; Anantharamaiah, G.M.; Royer, G.P.; Means, G.E. J. Org. Chem.1979, 44, 3442. doi: 10.1021/jo01333a048
Ram, S.; Spicer, L.D. Tetrahedron Lett.1987, 28, 515. doi: 10.1016/S0040-4039(00)95769-1
Anwer, M.K.; Spatola, A.F. Tetrahedron Lett.1981, 22, 4369. doi: 10.1016/S0040-4039(01)82959-2
Anwer, M.K.; Spatola, A.F.; Bossinger, C.D.; Flanigan, E.; Liu, R.-C.; Olsen, D.B.; Stevenson, D. J. Org. Chem.1983, 48, 3503. doi: 10.1021/jo00168a025
Adger, B.M.; O'Farrell, C.; Lewis, N.J.; Mitchell, M.B. Synthesis 1987, 53. doi: 10.1002/chin.198722132
Khan, S.A.; Sivanandaiah, K.M. Synthesis 1978, 750. doi: 10.1055/s-1978-24877
Felix, A.M.; Heimer, E.P.; Lambros, T.J.; Tzougraki, C.; Meienhofer, J. J. Org. Chem.1978, 43, 21, 4194.
Gray, B.D.; Jeffs, P.W. J. Chem. Soc., Chem. Commun.1987, 18, 1329. doi: 10.1039/C39870001329
Zheng, Y.; Wu, G.; Zhu, X.R.; Li, Y.-J.; Ma, Y.-H.; Zhao, X.; Lu, T.; Zhu, Y.-Q. Chem. Res. Chin.Univ. 2011, 27, 224.
Chen, S.-P.; Liu, D.-Q.; Si, L.-L.; Chen, L.-G.; Yan, X.-L. Synth. Commun.2017, 47, 3, 238.
Guo, M.-L.; Hong, K.-H.; Lv, Y.-F.; Ding, Y.; Li, C.-C.; Xu, H.; Qi, W.-X.; Chen, J.-Q.; Ji, M.; Cai, J. J. Chem. Res.2017, 2, 112.
Bajwa, J.S.; Slade, J.; Repic, O. Tetrahedron Lett.2000, 41, 6025. doi: 10.1016/S0040-4039(00)01013-3
Babu, S.N.N.; Srinivasa, G.R.; Santhosh, D.C.; Gowda, D.C. J. Chem. Res.2004, 66. doi: 10.3184/030823404323000846
Babu, S.N.N.; Srinivasa, G.R.; Santhosh, D.C.; Gowda, D.C. Synth. Commun.2004, 34, 1831. doi: 10.1081/SCC-120034165
Kim, S.S.; Lin, G.; Yang, J.W. Bull. Korean Chem. Soc.2004, 25, 249. doi: 10.5012/bkcs.2004.25.2.249
Davies, S.G.; Mortimer, D.A.B.; Mulvaney, A.W.; Russell, A.J.; Skarphedinsson, H.; Smith, A.D.; Vickers, R.J. Org. Biomol. Chem.2008, 6, 1625. doi: 10.1039/b802204f
Hungerhoff, B.; Samanta, S.S.; Roels, J.; Metz, P. Synlett 2000, 77. doi: 10.1002/chin.200017064
Oikawa, Y.; Yoshioka, T.; Yonemitsu, O. Tetrahedron Lett.1982, 23, 885. doi: 10.1016/S0040-4039(00)86974-9
Oikawa, Y.; Yoshioka, T.; Yonemitsu, O. Tetrahedron Lett.1982, 23, 889. doi: 10.1016/S0040-4039(00)86975-0
Singh, S.B. Tetrahedron Lett.1995, 36, 2009. doi: 10.1016/0040-4039(95)00214-W
Yamaura, M.; Suzuki, T.; Hashimoto, H.; Yoshimura, J.; Okamoto, T.; Shin, C.M. Bull. Chem. Soc. Jpn.1985, 58, 1413. doi: 10.1246/bcsj.58.1413
Davies, S.G.; Ichihara, O. Tetrahedron Lett.1998, 39, 6045. doi: 10.1016/S0040-4039(98)01193-9
Bull, S.D.; Davies, S.G.; Fenton, G.; Mulvaney, A.W.; Prasad, R.S.; Smith, A.D. Chem. Commun.2000, 337. doi: 10.1002/chin.200113073
Bull, S.D.; Davies, S.G.; Fenton, G; Mulvaney, A.W.; Prasad, R.S.; Smith, A.D. J. Chem. Soc., Perkin Trans. 1 2000, 3765. doi: 10.1002/chin.200113073
陈树森, 邱文革, 于永忠, 火炸药学报, 2000, 2, 4.Chen, S.-S.; Qiu, W.-G.; Yu, Y.-Z. Chin.J.Explos.Propellants 2000, 2, 4(in Chinese).
Qiu, W.-G.; Chen, S.-S; Yu, Y.-Z. Chin. J. Chem.1999, 17, 554.
Gigg, R.; Conant, R. J. Chem. Soc. Chem. Commun.1983, 465.
Haddach, A.A.; Kelleman, A.; Deaton-Rewolinski, M.V. Tetrahedron Lett.2002, 43, 399. doi: 10.1016/S0040-4039(01)02192-X
Kong, W.; Fu, C.; Ma, S. Chem.-Eur. J.2011, 17, 13134. doi: 10.1002/chem.201102251
Semak, V.; Escolano, C.; Arroniz, C. Tetrahedron:Asymmetry 2010, 21, 2542. doi: 10.1016/j.tetasy.2010.09.014
朱文通, 甘海峰, 陈正帮, 郭凯, 合成化学, 2015, 23, 977. doi: 10.15952/j.cnki.cjsc.1005-1511.2015.10.0977Zhu, W.-T.; Gan, H.-F.; Chen, Z.-B.; Guo, K. Synth.Chem.2015, 23, 977(in Chinese). doi: 10.15952/j.cnki.cjsc.1005-1511.2015.10.0977
Page, P.C.B.; Stephenson, G.R.; Harvey, J.; Slawin, A.M.Z. Synlett 2016, 27, 2500. doi: 10.1055/s-0035-1562603
Zhao, F.; Tian, W.-H.; Luo, F.; Cheng, H.-L.; Jiang, Y.-B.; Chen, Z. Synth. Commun.2016, 46, 20, 1678.
Feula, A.; Fossey, J.S. RSC Adv.2013, 3, 16, 5370. doi: 10.1039/C2RA22279E
Kroutil, J.; Trnka, T.; Cerny, M. Org. Lett.2000, 2, 1681. doi: 10.1021/ol005746g
Kroutil, J.; Trnka, T.; Cerny, M. Synthesis 2004, 446. doi: 10.1002/chin.199844207
Moriyama, K.; Nakamura, Y.; Togo, H. Org. Lett.2014, 16, 3812. doi: 10.1021/ol501703y
Ghosh, S.; Shashidhar, J. Tetrahedron Lett.2009, 50, 1177. doi: 10.1016/j.tetlet.2008.11.115
Amat, M.; Pérez, M.; Minaglia, A.T.; Casamitjana, N.; Bosch, J. Org. Lett.2005, 7, 3653. doi: 10.1021/ol051242c
Amat, M.; Escolano, C.; Gómez-Esqué, A.; Lozano, O.; Llor, N.; Griera, R.; Bosch, J. Tetrahedron:Asymmetry 2006, 17, 1581. doi: 10.1016/j.tetasy.2006.05.018
Torregrosa, R.; Pastor, I.M.; Yus, M. Tetrahedron 2007, 63, 947. doi: 10.1016/j.tet.2006.11.032
Morales, C.L.; Pagenkopf, B.L. Org. Lett.2008, 10, 157. doi: 10.1021/ol702376j
Garst, M.E.; Dolby, L.J.; Esfandiari, S.; Fedoruk, N.A.; Chamberlain, N.C.; Avey, A.A. J. Org. Chem.2000, 65, 7098. doi: 10.1021/jo0008136
Suzuki, H.; Tsukuda, A.; Kondo, M.; Aizawa, M.; Senoo, Y.; Nakajima, M.; Watanabe, T.; Yokoyama, Y.; Murakami, Y. Tetrahedron Lett.1995, 36, 1671. doi: 10.1016/0040-4039(95)00126-W
Rao, T.S.; Pandey, P.S. Synth. Commun.2004, 34, 3121. doi: 10.1081/SCC-200028570
Franceschini, N.; Da N.S.; Miel, H.; Sonnet, P. Heterocycl. Commun.2005, 11, 509.
Franceschini, N.; Da N.S.; Miel, H.; Sonnet, P. Tetrahedron:Asymmetry 2003, 14, 3401. doi: 10.1016/j.tetasy.2003.09.010
Nandi, P.; Dye, J.L.; Jackson, J.E. Tetrahedron Lett.2009, 50, 3864. doi: 10.1016/j.tetlet.2009.04.058
Choudary, B.; Giles, R.G.; Jovic, F.; Lewis, N.; Moore, S.; Urquhart, M. Org. Process Res. Dev.2012, 16, 1927. doi: 10.1021/op300263w
Shaw, A.Y.; Mclaren, J.A.; Nichol, G.S.; Hulme, C. Tetrahedron Lett.2012, 53, 2592. doi: 10.1016/j.tetlet.2012.03.033
Lee, S.H.; Mu, Y.; Kim, G.W.; Kim, J.S.; Park, S.H.; Jin, T.; Lee, K.Y.; Ham, W.H. Heterocycles 2013, 87, 1749. doi: 10.3987/COM-13-12747
Lam, T.Y.; Wang, Y.-P.; Danheiser, R.L. J. Org. Chem.2014, 45, 9396.
Rombouts, F.; Franken, D.; Martinez-Lamenca, C.; Braeken, M.; Zavattaro, C.; Chen, J.S.; Trabanco, A.A. Tetrahedron Lett.2010, 51, 4815. doi: 10.1016/j.tetlet.2010.07.022
Lima, H.M.; Sivappa, R.; Yousufuddin, M.; Lovely, C.J. J. Org. Chem.2014, 79, 2481. doi: 10.1021/jo4027337
Chern, C.Y.; Huang, Y.P.; Kan, W.M. Tetrahedron Lett.2003, 44, 1039. doi: 10.1016/S0040-4039(02)02738-7
于永忠, 管晓培, 含能材料, 1999, 7, 1. doi: 10.3969/j.issn.1006-9941.1999.01.001Yu, Y.-Z.; Guan, X.-P. Chin. J. Energ. Mater.1999, 7, 1(in Chinese). doi: 10.3969/j.issn.1006-9941.1999.01.001
吕连营, 欧育湘, 王建龙, 陈博仁, 精细化工, 2003, 20, 577. doi: 10.3321/j.issn:1003-5214.2003.10.001Lv, L.-Y.; Ou, Y.-X.; Wang, J.-L.; Chen, B.-R. Fine Chem.2003, 20, 577(in Chinese). doi: 10.3321/j.issn:1003-5214.2003.10.001
Bayat, Y.; Hajimirsadeghi, S.S.; Pourmortazavi, S.M. Org. Process Res. Dev.2011, 15, 810. doi: 10.1021/op200056j
Bayat, Y.; Ebrahimi, H.; Fotouhi-Far, F. Org. Process Res. Dev.2012, 16, 1733. doi: 10.1021/op300162d
Bayat, Y.; Hajighasemali, F. Propellants Explos. Pyrotech.2016, 41, 893. doi: 10.1002/prep.201500347
欧育湘, 刘进全, 高能量密度化合物, 国防工业出版社, 北京, 2005, pp.96~110.Ou, Y.-X.; Liu, J.-Q. High Energy Density Compound, National Defence Industrial Press, Beijing, 2005, pp.96~110(in Chinese).
Yang, B.W.; O'Rourke D, J.L.I. Synlett 1993, 195.
See, Y.B. JP 321962, 1994.
Wright, W.B.; Brabander, H.J. J. Org. Chem.1961, 26, 10 doi: 10.1021/jo01060a003
Olofson, R.A.; Martz, J.T.Senet, J.P.; Piteau, M.; Malfroot, T. J. Org. Chem.1984, 49, 2081. doi: 10.1021/jo00185a072
Kanth, J.V.B,; Reddy, C.K.; Periasamy, M. Synth. Commun.1994, 24, 313. doi: 10.1080/00397919408011190
Igarashi, J.; Kobayashi, Y. Tetrahedron Lett.2005, 46, 6381. doi: 10.1016/j.tetlet.2005.06.171
Annamalai, M.; Hristeva, S.; Bielska, M.; Ortega, R.; Kumar, K. Molecules 2017, 22, 827. doi: 10.3390/molecules22050827
Pandey, G.; .Rani, K.S. Tetrahedron Lett.1988, 29, 4157. doi: 10.1016/S0040-4039(00)80443-8
Smith, A.J.; Young, A.; Rohrbach, S.; O'Connor, E.F.; Allison, M.; Wang, H.-S.; Poole, D.L.; Tuttle, T.; Murphy, J.A. Angew. Chem., Int. Ed.2017, 56, 13747. doi: 10.1002/anie.201707914
Golantsov, N.E.; Karchava, A.V.; Yurovskaya, M.A. Chem. Heterocycl. Compd. 2006, 42, 1021. doi: 10.1007/s10593-006-0198-8
Montero, M.L.; Rodríguez, D.A.; Gotor, F.V.; Fernández, V.G.; Lavandera, I. Green Chem.2015, 46, 2794.
Grayson, E, J.; Davis, B.G. Org. Lett.2005, 7, 2361. doi: 10.1021/ol050624f
Baker, S.R.; Parsons A.F.; Wilson M. Tetrahedron Lett.1998, 39, 331. doi: 10.1016/S0040-4039(97)10480-4
Yanada, R.; Obika, S.; Kobayashi Y.; Inokuma, T.; Oyama, M.; Yanada, K.; Takemoto Y. Adv. Synth. Catal.2005, 347, 1632. doi: 10.1002/adsc.200505147

图式 1 催化氢解脱苄反应机理
Scheme 1 Reaction mechanism of catalytic hydrogenolysis and debenzylation

图式 2 六苄基六氮杂异伍兹烷催化氢解脱苄
Scheme 2 Hexabenzylhexaazaisowurtzitane catalytic hydrogenolysis debenzylation

图式 3 [3.3.3]螺桨烷催化氢解脱苄
Scheme 3 [3.3.3]propellane catalytic hydrogenolysis debenzylation

图式 5 两种氧化剂脱苄反应比较活性
Scheme 5 Comparison of the activity of two oxidants for debenzylation

图式 7 氧化脱苄合成1, 5-二取代三唑衍生物
Scheme 7 Oxidative debenzylation synthesis of 1, 5-disubsti- tuted triazole derivatives

图式 9 LiAlH4为脱苄试剂的强碱脱苄
Scheme 9 Strong alkali debenzylation of LiAlH4 as debenzylation reagent

图式 10 氯甲酸氯乙酯为脱苄试剂脱苄
Scheme 10 Debenzylation of chloroethyl chloroformate as debenzylation reagent

图式 11 氯甲酸氯乙酯为脱苄试剂合成二苯基吡咯
Scheme 11 Synthesis of diphenylpyrrole by chloroethyl chloroformate as debenzylation reagent

图式 14 叔丁醇钾和三乙基硅烷为脱苄试剂的脱苄
Scheme 14 Debenzylation of potassium tert-butoxide and triethylsilane as debenzylation reagent

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:











































 下载:
下载:







 下载:
下载: