8-异戊烯基黄酮类化合物是天然黄酮类化合物的重要成员, 它们存在于许多天然药用植物中, 具有抗肿瘤、抗炎抗菌、抗骨质疏松、保护心血管舒张和提高机体免疫力等生理活性[1 2 .异戊烯基侧链的存在增加了黄酮类化合物的亲脂性和生物导向性, 使其在生物体内更易透过细胞膜到达作用目标, 大大提高了黄酮类化合物的药用价值[3 . 8-异戊烯基槲皮素-3-甲醚(1 )是从藏药小叶莲(Sinopodophylli Fructus )中分离出来具有良好抑制乳腺癌细胞作用的天然产物[4 . 8-异戊烯基槲皮素- 3, 7, 3', 4'-四甲醚(2 )是从Phebaliun Dentatum Smith (Ruteceae)中分离出来的天然产物[5 . Artochamin C (3 )是8-异戊烯基木犀草素的衍生物, 它是从Artocarpus Chama 根部分离的对乳腺癌MCF-7、卵巢癌1A-9和人结肠癌HCT-8等多种人类癌细胞具有良好抑制作用的天然产物[6 .关于这3种天然产物的全合成未见报道.
8-异戊烯基黄酮类化合物合成的关键步骤是黄酮A环上8位异戊烯基的引入. Wang等[7 将黄酮在路易斯酸的作用下与2-甲基-3-丁烯-2-醇偶联, 可得6位、8位或6, 8-二异戊烯基衍生物的混合物. Zeng等[8 将黄酮在CH3 OH/KOH条件下直接用溴代异戊烯在6位和8位分别引入异戊烯基基团.上述2种方法在引入异戊烯基基团时无法控制其选择性, 得到的8-异戊烯基产物收率低且难分离.后来有人采用Claisen重排的方法在黄酮结构的8位上引入异戊烯基基团[9 , 它是将黄酮A环上5位的O -异戊烯基团在弱碱性条件下加热重排到酚羟基的对位(8位)或邻位(6位)成为C -异戊烯基基团.但异戊烯基重排到酚羟基邻位和对位的几率很接近, 仍存在选择性较差的问题[10 .据报道采用β -双酮类稀土铕络合物催化剂Eu(fod)3 催化时, 对位产物的产率有了很大提高, 但稀土金属价格昂贵, 且存在毒性和不稳定性的问题[11 .总之, 现有文献报道的关于合成8位异戊烯基黄酮类化合物所采用的8位引入异戊烯基的方法都存在一定的局限性[12 .
近几年来, 有报道称利用微波加热条件能大大提高烯丙基芳基醚发生Claisen重排反应时生成对位产物的比例, 同时还可缩短反应时间[13 14 .因而我们尝试利用微波加热技术在黄酮类天然产物的8位上引入C -异戊烯基并取得了良好的效果, 以微波促进的Claisen重排反应为关键步骤, 完成了具有生物活性天然产物8-异戊烯基槲皮素-3-甲醚(1 )、8-异戊烯基槲皮素-3, 7, 3', 4'-四甲醚(2 )和Artochamin C (3 )的全合成.
1.
结果与讨论
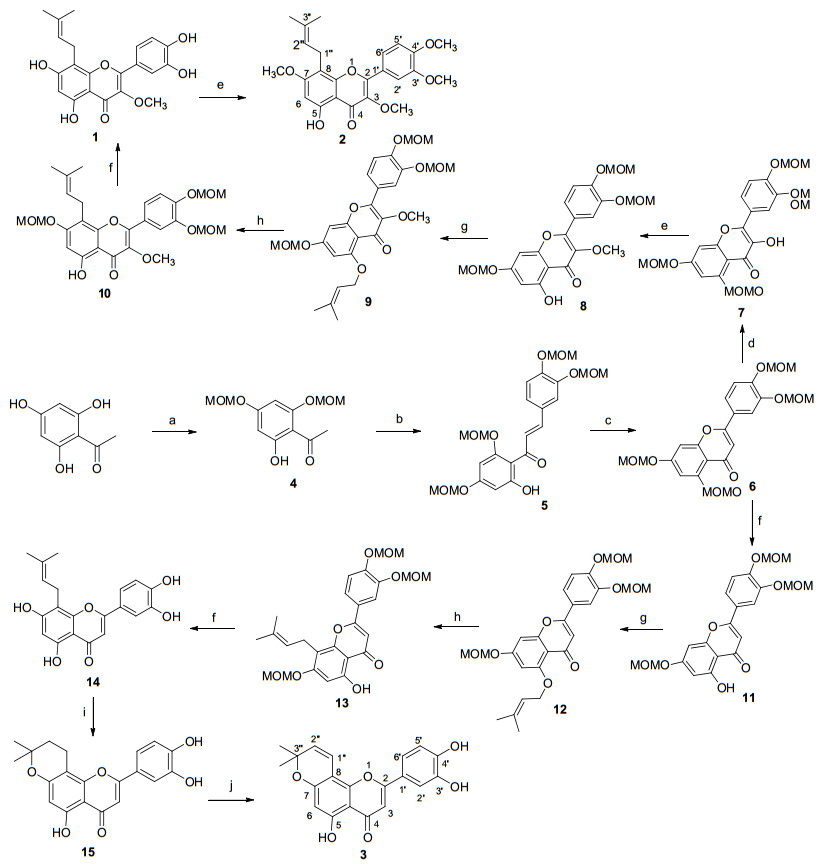
异戊烯基黄酮类天然产物8-异戊烯基槲皮素-3-甲醚(1 )、8-异戊烯基槲皮素-3, 7, 3', 4'-四甲醚(2 )和Artochamin C (3 )的全合成路线见Scheme 1 4 .化合物4 与同样经过氯甲基甲醚保护的3, 4-二甲氧甲基苯甲醛在10 mL 10% KOH溶液与10 mL乙醇的混合溶液中发生羟醛缩合反应得查尔酮类化合物5 .化合物5 以二甲基亚砜(DMSO)为溶剂, 在I2 催化下环合可得黄酮类化合物6 .化合物7 的合成则采用经我们实验室改进过后的过氧丙酮(DMDO)氧化法[15 , 即在化合物6 的二氯甲烷与丙酮的混合溶剂中间歇式滴加过硫酸氢钾复合盐水溶液对其双键进行环氧化反应, 这不仅简化了合成过氧丙酮的步骤, 还能大大提高了环氧化反应的选择性, 增加了生成黄酮醇化合物的产率.将化合物7 的3位羟基在硫酸二甲酯的作用下经O -甲氧基化反应后直接滤除碱性固体, 再在滤液中再滴加微弱的盐酸溶液即可得到选择性脱除5位保护基后的化合物8 .
图式 1
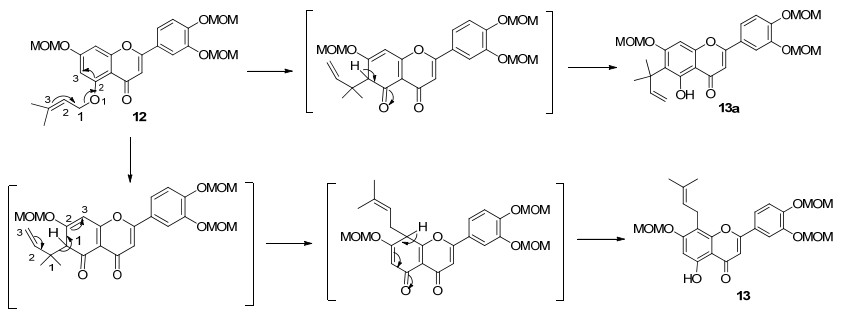
将化合物8 在碳酸钾和无水丙酮中与异戊烯基溴作用生成5-O -异戊烯基化产物9 .化合物9 在微波促进的条件下经Claisen重排反应得主要产物8-异戊烯基黄酮类化合物10 .化合物10 在稀盐酸的甲醇溶液中回流可完全脱去保护基得天然产物8-异戊烯基槲皮素-3-甲醚(1 ).化合物1 再采用硫酸二甲酯在碱性条件下经O -甲基化反应得另一天然产物8-异戊烯基槲皮素-3, 7, 3', 4'-四甲醚(2 ).将化合物6 在温和酸性条件下, 选择性脱去5-位保护基团可得化合物11 , 化合物11 在碱性条件下经5-O -异戊烯基化反应可得化合物12 .在微波作用下化合物12 先经过一次σ [3,3]重排得到主要为13a 的重排的产物, 随着温度和时间的增加, 一次重排的中间体会再经过一次σ [3,3]重排得到化合物13 , 发生Claisen重排反应过程如下所示(Scheme 2 13 在稀盐酸的甲醇溶液中回流可完全脱去保护基可得天然产物8-异戊烯基槲皮素14 .化合物14 在甲酸催化下, 异戊烯基侧链与邻位酚羟基发生分子内环化得化合物15 , 然后经二氯二氰基苯醌(DDQ)脱氢反应得天然产物Artochamin C (3 ).所合成天然产物1 , 2 和3 的波谱数据与文献报道相符[4 6 .
图式 2
对于关键步骤Claisen重排反应, 在本实验对影响重排反应的因素与传统的油浴加热方法进行了对比, 重点探索反应温度、反应时间、微波辐射功率等因素对产物的影响.
1.1
反应温度对产物的影响
对于微波促进的Claisen重排反应, 以化合物12 为原料, 当反应时间3 h, 反应温度小于150 ℃时, 基本不发生重排反应.随着温度的增加, 发生一次重排的产物13a 不断增多, 当温度为200 ℃时达到峰值.随着温度的继续增加, 6-重排的产物13a 再经过一次σ [3,3]重排得到8-重排化合物13 , 当温度为220 ℃时得到的对位化合物13 收率最多, 所以该反应的最佳温度为220 ℃.在最佳温度下, 与微波促进的Claisen重排反应得到的产物相比, 通过普通油浴加热得到的邻位产物13a 偏多, 对位产物13 偏低, 即微波反应能促进第二次重排的发生.
1.2
反应时间对产物的影响
在以N , N -二乙基苯胺为溶剂, 在最佳温度微波加热220 ℃下进行反应, 用薄层色谱(TLC)追踪反应全过程, 反应时间为3 h时可得到较好的收率, 随着反应时间的延长对位重排产物并不会得到显著的提高, 反而会出现其它副产物.如果缩短反应时间, Claisen重排则进行的不彻底, 原料的转化率降低, 主要生成6-重排产物13a , 得到的对位重排产物较少.而采用传统油浴方法的重排反应则需要24 h才能完成, 且生成副产物13a 较多.
1.3
微波功率对反应的影响
对于微波加热下的Claisen重排反应, 微波辐射功率对反应速率有着重要的影响.辐射功率越低, 微波的热效能就会越差, 反应速率较慢.微波功率加大时, 分子振动能得到提高, 引起的热效应就会加速反应的进行, 从而大大的缩短反应时间.但如果微波功率太大, 反应产率增加的并不明显.通过实验我们发现该反应的最佳微波功率为700 W.
2.
结论
8-异戊烯基槲皮素-3-甲醚(1 )、8-异戊烯基槲皮素- 3, 7, 3', 4'-四甲醚(2 )和Artochamin C (3 )是3种具有抗肿瘤、抗炎抗菌等生物活性的异戊烯基黄酮类天然产物.我们从2, 4, 6-三羟基苯乙酮出发, 经10~12步反应, 以16%~23%的总收率首次全合成上述3种天然产物.对于关键步骤8-异戊烯基黄酮骨架的合成, 我们采用微波促进方法对5-O -异戊烯基黄酮进行二次Claisen重排进行.在以N , N -二乙基苯胺为溶剂, 控制微波功率为700 W, 反应温度为220 ℃的条件下, 反应3 h得到产率最高的8-重排产物.
3.
实验部分
3.1
仪器与试剂
熔点在XRC-1型显微熔点仪上测定(温度计未校正); NMR用Bruker Am-400型核磁共振仪测定(溶剂CDCl3 或DMSO-d 6 , TMS为内标); ESI或EI-MS用Agilent 1100液-质联用仪或ZAB-HS型仪测定记录; 微波反应器型号为XH-MC-1(北京祥鹄科技发展有限公司产品).薄层和柱层析用硅胶均为青岛海洋化工厂产品; 所用试剂和溶剂为化学纯和分析纯.
2-羟基-4, 6-二甲氧甲基苯乙酮(4 )和2'-羟基- 3, 4, 4', 6'-四甲氧甲基查尔酮(5 )按参考文献报道的方法合成[14 .
3.2
3', 4', 5, 7-四甲氧甲基黄酮(6)的合成
将化合物5 (5.0 g, 10.8 mmol)溶入60 mL DMSO中, 加入I2 1.9 g, 100 ℃搅拌24 h, 冷却至常温后倒入冰水中, 再加入饱和Na2 S2 O3 溶液, 用二氯甲烷萃取水相, 得到的有机相减压旋干, 得到的固体用二氯甲烷/石油醚(V :V =1:10)重结晶, 得黄色固体6 3.4 g, 产率72%. m.p. 125~126 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 7.58 (d, J =2.2 Hz, 1H, 6'-H), 7.47 (dd, J =8.6, 2.2 Hz, 1H, 5'-H), 7.20 (d, J =8.7 Hz, 1H, 2'-H), 6.79 (d, J =2.3 Hz, 1H, 8-H), 6.70 (d, J =2.3 Hz, 1H, 6-H), 6.53 (s, 1H, 3-H), 5.29 (s, 2H, CH2 O), 5.25 (s, 2H, CH2 O), 5.21 (s, 2H, CH2 O), 3.51 (s, 3H, OCH3 ), 3.50 (s, 3H, OCH3 ), 3.47 (2s, 6H, 2OCH3 );
13 C NMR (100 MHz, CDCl3 ) δ : 177.29, 161.03, 160.50, 159.18, 158.01, 149.83, 147.15, 125.37, 120.77, 115.89, 114.16, 108.06, 101.79, 97.09, 95.46, 95.44, 95.42, 94.94, 94.16, 56.50, 56.41, 56.32, 56.30; ESI-MS m /z : 463 [M+H]+ .
3.3
3-羟基-3', 4'5, 7-四甲氧甲基黄酮(7)的合成
将化合物6 (3.5 g, 7.5 mmol)溶入丙酮(90 mL)二氯甲烷(120 mL)的混合溶液中, 加入100 mL含碳酸钠(14 g)和碳酸氢钠(7 g)的缓冲溶液, 在冰水浴0 ℃下搅拌.再逐滴加入过硫酸氢钾(60 g, 200 mL水)溶液.常温反应24 h, 用二氯甲烷萃取, 在有机相中加入少量对羟基苯甲酸(15 mg), 搅拌30 min, 将溶液旋干, 经硅胶柱层析分离纯化(石油醚/乙酸乙酯, V :V =3:1)得黄色固体7 2.79 g, 产率77%. m.p. 143~145 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 7.99 (d, J =1.8 Hz, 1H, 6'-H), 7.77 (dd, J =8.7, 1.9 Hz, 1H, 5'-H), 7.22 (d, J =8.7 Hz, 1H, 2'-H), 6.77 (d, J =1.9 Hz, 1H, 8-H), 6.68 (d, J =1.9 Hz, 1H, 6-H), 5.31 (s, 2H, CH2 O), 5.26 (s, 2H, CH2 O), 5.25 (s, 2H, CH2 O), 5.20 (s, 2H, CH2 O), 3.50 (2s, 6H, 2OCH3 ), 3.47 (s, 3H, OCH3 ), 3.46 (s, 3H, OCH3 ); 13 C NMR (100 MHz, CDCl3 ) δ : 166.54, 156.26, 152.98, 152.55, 143.30, 141.51, 136.78, 132.40, 119.85, 116.87, 110.69, 110.65, 95.32, 91.33, 90.28, 89.95, 89.77, 89.01, 51.30, 51.23, 51.11, 50.99; EI-MS m /z : 478 [M]+ .
3.4
3-甲氧基-3', 4', 7-四甲氧甲基-5-羟基黄酮(8)的合成
将化合物7 (1.5 g, 3.1 mmol)溶入50 mL丙酮中, 加入3 g无水碳酸钾室温搅拌30 min, 再加入硫酸二甲酯1.2 mL, 搅拌2 h.过滤出去碳酸钾固体, 旋除溶剂, 再将固体溶入20 mL乙醇溶液中, 滴加1 mL 3%的HCl常温搅拌10 min后倒入冰水中, 用乙酸乙酯萃取.旋除溶剂, 用石油醚/乙酸乙酯(V :V =1:2)重结晶, 得黄色固体8 1.2 g, 产率77%. m.p. 154~155 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 7.96 (d, J =1.9 Hz, 1H, 6'-H), 7.73 (dd, J =8.7, 1.9 Hz, 1H, 5'-H), 7.26 (d, J =8.7 Hz, 1H, 2'-H), 6.60 (d, J =1.9 Hz, 1H, 8-H), 6.44 (d, J =1.9 Hz, 1H, 6-H), 5.32 (s, 2H, CH2 O), 5.30 (s, 2H, CH2 O), 5.23 (s, 2H, CH2 O), 3.87 (s, 3H, 3-OCH3 ), 3.55 (s, 3H, OCH3 ), 3.53 (s, 3H, OCH3 ), 3.49 (s, 3H, OCH3 ); 13 C NMR (100 MHz, CDCl3 ) δ : 178.78, 162.82, 161.81, 156.47, 155.53, 149.63, 146.71, 139.13, 124.23, 123.37, 117.16, 115.68, 106.61, 99.57, 95.64, 94.97, 94.14, 94.06, 60.15, 56.35, 56.29; EI-MS m /z : 448 [M]+ .
3.5
3-甲氧基-3', 4', 7-四甲氧甲基-5-O -异戊烯基黄酮(9)的合成
将化合物8 (0.5 g, 1.12 mmol)溶入20 mL丙酮中, 加入2 g无水碳酸钾室温搅拌1 h, 再加入异戊烯基溴0.75 mL, 升温至45 ℃搅拌2 h, 过滤出去碳酸钾, 旋除溶剂, 用乙酸乙酯/石油醚(V :V =1:2)重结晶, 得白色固体9 0.52 g, 产率89%. m.p. 76~77 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 7.96 (d, J =1.9 Hz, 1H, 6'-H), 7.72 (dd, J =8.7, 2.0 Hz, 1H, 5'-H), 7.25 (d, J =8.1 Hz, 1H, 2'-H), 6.69 (d, J =2.1 Hz, 1H, 8-H), 6.43 (d, J =1.9 Hz, 1H, 6-H), 5.59 (t, J =6.2 Hz, 1H, 2" -H), 5.31 (s, 2H, CH2 O), 5.30 (s, 2H, CH2 O), 5.25 (s, 2H, CH2 O), 4.69 (d, J =6.5 Hz, 2H, 1" -H), 3.88 (s, 3H, 3-OCH3 ), 3.55 (s, 3H, OCH3 ), 3.53 (s, 3H, OCH3 ), 3.51 (s, 3H, OCH3 ), 1.79 (s, 3H, 3" -CH3 ), 1.76 (s, 3H, 3" -CH3 ); 13 C NMR (100 MHz, CDCl3 )
δ : 172.98, 160.16, 159.27, 157.43, 151.22, 148.06, 145.67, 140.26, 136.57, 123.89, 122.01, 118.43, 116.04, 114.71, 109.38, 96.90, 94.68, 94.31, 94.04, 93.28, 65.57, 59.00, 55.42, 55.34, 55.31, 24.79, 17.41; ESI-MS m /z : 517 [M+H]+ .
3.6
3-甲氧基-3', 4', 7-四甲氧甲基-8-异戊烯基黄酮(10)的合成
50 mL双口圆底烧瓶中加入化合物9 (200 mg, 0. 39 mmol), 10 mL新制备的N , N -二乙基苯胺, 置于微波反应器中, 温度为220 ℃, 功率为700 W, 在N2 保护下加热反应3 h.反应结束后, 将反应液倒入水中, 用二氯甲烷(10 mL×3)萃取, 有机相用5%的稀盐酸(10 mL×3)洗涤, 无水MgSO4 干燥, 过滤, 旋除溶剂, 经硅胶柱层析(石油醚:乙酸乙酯, V :V =5:1)分离纯化, 得黄色固体10 156 mg, 产率77%. m.p. 179~180 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 12.63 (s, 1H, 5-OH), 8.10 (d, J =1.9 Hz, 1H, 6'-H), 7.83 (dd, J =8.7, 1.9 Hz, 1H, 5'-H), 7.34 (s, 1H, 2'-H), 6.63 (s, 1H, 6-H), 5.38 (s, 2H, CH2 O), 5.34 (s, 2H, CH2 O), 5.31 (s, 2H, CH2 O), 5.27 (t, J =6.7 Hz, 1H, 2" -H), 3.93 (s, 3H, OCH3 ), 3.60 (s, 3H, OCH3 ), 3.52 (s, 3H, OCH3 ), 3.57 (d, J =7.1 Hz, 2H, 1" -H), 1.83 (s, 3H, 3" -CH3 ), 1.71 (s, 3H, 3" -CH3 ); 13 C NMR (100 MHz, CDCl3 ) δ : 178.21, 159.19, 159.00, 154.44, 152.52, 148.61, 145.85, 137.99, 131.00, 123.75, 122.42, 121.11, 116.34, 114.76, 107.59, 105.36, 96.48, 94.75, 94.05, 93.27, 59.17, 55.42, 55.28, 55.25, 24.69, 20.84, 16.92; ESI-MS m /z : 517 [M+H]+ ; HR-ESI- MS calcd for C27 H33 O10
[M+H]+ 517.2068, found 517.2064.
3.7
5-羟基-3', 4', 7-三甲氧甲基黄酮(11)的合成
将化合物6 (1.2 g, 2.6 mmol)溶入20 mL乙醇溶液中, 滴加3 mL 3%的HCl常温搅拌30 min后倒入冰水, 用乙酸乙酯萃取.旋除溶剂, 用乙酸乙酯/石油醚(V : V =1:2)重结晶, 得黄色固体11 0.98 g, 产率90%. m.p. 108~109 ℃. 1 H NMR (400 MHz, CDCl3 ) δ : 12.67 (s, 1H, 5-OH), 7.60 (d, J =2.0 Hz, 1H, 6'-H), 7.48 (dd, J =8.6, 2.0 Hz, 1H, 5'-H), 7.21 (d, J =8.8 Hz, 1H, 2'-H), 6.60 (d, J =2.0 Hz, 1H, 8-H), 6.54 (s, 1H, 3-H), 6.42 (d, J =2.0 Hz, 1H, 6-H), 5.26 (s, 2H, OCH2 O), 5.25 (s, 2H, OCH2 O), 5.19 (s, 2H, OCH2 O), 3.49 (s, 3H, OCH3 ), 3.47 (s, 3H, OCH3 ), 3.44 (s, 3H, OCH3 );
13 C NMR (100 MHz, CDCl3 ) δ : 182.50, 163.82, 162.93, 162.66, 162.02, 150.48, 149.39, 147.38, 125.18, 121.30, 116.02, 114.54, 105.11, 100.12, 95.64, 95.05, 94.40, 94.18, 56.47; ESI-MS m /z : 419 [M+H]+ .
3.8
3', 4', 7-三甲氧甲基-5-O -异戊烯基黄酮(12)的合成
将化合物11 (0.5 g, 1.20 mmol)溶入20 mL丙酮中, 加入2 g无水碳酸钾室温搅拌1 h, 再加入异戊烯基溴0.75 mL, 升温至50 ℃搅拌2 h, 过滤出去碳酸钾, 旋除溶剂, 用乙酸乙酯/石油醚(V :V =1:2)重结晶, 得白色固体12 0.54 g, 产率92%. m.p. 130~131 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 7.56 (d, J =2.1 Hz, 1H, 6'-H), 7.45 (dd, J =8.6, 2.1 Hz, 1H, 5'-H), 7.18 (d, J =11.5 Hz, 1H, 2'-H), 6.68 (d, J =2.2 Hz, 1H, 8-H), 6.54 (s, 1H), 6.40 (d, J =2.2 Hz, 1H, 6-H), 5.50 (t, J =6.3 Hz, 1H, 2" -H), 5.24 (s, 2H, OCH2 O), 5.24 (s, 2H, OCH2 O), 5.19 (s, 2H, OCH2 O), 4.63 (d, J =6.3 Hz, 2H, 1" -H), 3.48 (s, 3H, OCH3 ), 3.46 (s, 3H, OCH3 ), 3.46 (s, 3H, OCH3 ), 1.72 (s, 3H, 3" -CH3 ), 1.69 (s, 3H, 3" -CH3 ); 13 C NMR (100 MHz, CDCl3 ) δ : 177.43, 161.19, 160.26, 160.06, 159.45, 149.80, 147.19, 137.41, 125.55, 120.78, 119.42, 115.98, 114.25, 110.10, 108.20, 98.36, 95.54, 95.00, 94.21, 92.53, 66.57, 56.41, 56.37, 56.34, 25.77, 18.37; ESI-MS m /z : 487 [M+H]+ .
3.9
3', 4', 7-三甲氧甲基-8-异戊烯基黄酮(13)与3', 4', 7-三甲氧甲基-6-(1" , 1" -二甲基烯丙基)黄酮(13a)的合成
在50 mL双口圆底烧瓶中加入化合物12 (200 mg, 0.41 mmol), 10 mL新制备的N , N -二乙基苯胺, 置于微波反应器中, 温度为220 ℃, 功率为700 W, 在N2 保护下加热反应3 h.反应结束后, 将反应液倒入水中, 用二氯甲烷(10 mL×3)萃取, 有机相旋干, 加入无水甲醇10 mL使其溶解, 再加入3 mol•L-1 的盐酸1 mL, 70 ℃下加热回流, TLC追踪反应全过程, 待原料点消失后停止反应, 加水淬灭, 乙酸乙酯(10 mL×3)萃取, 合并有机层, 无水MgSO4 干燥, 过滤, 旋除溶剂, 经硅胶柱层析(石油醚/乙酸乙酯, V :V =3:1)分离纯化, 最先得到为化合物13 , 黄绿色固体103 mg, 产率为82%. m.p. 237~239 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 12.65 (s, 1H, 5-OH), 7.66 (d, J =1.9 Hz, 1H, 6'-H), 7.49 (dd, J =8.6, 1.9 Hz, 1H, 5'-H), 7.19 (d, J =2.5 Hz, 1H, 2'-H), 6.53 (s, 2H, 3-H, 6-H), 5.25 (s, 2H, OCH2 O), 5.22 (s, 2H, OCH2 O), 5.20 (s, 2H, OCH2 O), 5.18 (t, J =6.0 Hz, 1H, 2" -H), 3.48 (d, J =4.3 Hz, 2H, 1" -H), 3.48 (s, 3H, OCH3 ), 3.47 (s, 3H, OCH3 ), 3.41 (s, 3H, OCH3 ), 1.74 (s, 3H, 3" -CH3 ), 1.60 (s, 3H, 3" -CH3 ); 13 C NMR (100 MHz, CDCl3 ) δ : 182.95, 163.65, 160.35, 160.13, 154.45, 150.42, 147.46, 132.07, 125.61, 122.12, 121.24, 116.07, 114.83, 108.80, 105.82, 104.67, 97.89, 95.76, 95.07, 94.26, 56.48, 56.37, 56.34, 25.74, 22.01, 17.97; ESI-MS m /z : 487 [M+H]+ ; HR-ESI-MS calcd for C26 H31 O9
[M+H]+ 487.1963, found 487.1958.
然后得到化合物13a , 黄绿色固体12 mg, 产率10%. m.p. 231~233 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 13.57 (s, 1H, 5-OH), 7.57 (d, J =1.9 Hz, 1H, 6'-H), 7.4 (dd, J =8.3, 2.1 Hz, 1H, 5'-H), 7.19 (d, J =4.7 Hz, 1H, 2'-H), 6.65 (s, 1H, 8-H), 6.52 (s, 1H, 3-H), 6.22 (dd, J =8.7, 7.6 Hz, 1H, 2" -H), 5.24 (s, 2H, OCH2 O), 5.24 (s, 2H, OCH2 O), 5.14 (s, 2H, OCH2 O), 4.78 (dd, J =5.8, 3.2 Hz, 2H, 3" -H), 3.48 (s, 3H, OCH3 ), 3.46 (s, 3H, OCH3 ), 3.43 (s, 3H, OCH3 ), 1.55 (s, 6H, 1" -2CH3 ); 13 C NMR (100 MHz, CDCl3 ) δ : 181.94, 161.13, 159.88, 154.94, 149.58, 146.34, 131.03, 124.16, 121.09, 120.24, 115.03, 113.53, 107.76, 105.86, 104.08, 96.87, 94.63, 94.03, 93.07, 92.13, 55.44, 55.34, 28.00, 24.70, 20.98, 16.94; ESI-MS m /z : 487 [M+H]+ .
3.10
8-异戊烯基木犀草素(14)的合成
在50 mL单口圆底烧瓶中加入化合物13 (100 mg, 0.19 mmol)和10 mL甲醇, 使其溶解后再加入3 mol•L-1 的盐酸1 mL, 65 ℃下加热回流, TLC追踪反应全过程, 待原料点消失后停止反应, 加水淬灭, 乙酸乙酯(10 mL×3)萃取, 合并有机层, 无水MgSO4 干燥, 过滤, 旋除溶剂, 经硅胶柱层析(石油醚/乙酸乙酯, V :V =4:1)分离纯化, 得到黄绿色固体14 78 mg, 产率为78%. m.p. 223~225 ℃; 1 H NMR (400 MHz, DMSO-d 6 ) δ : 12.92, 10.78, 9.98, 9.43 (4s, 4H, 4OH), 7.42 (s, 1H, 2'-H), 7.41 (d, J =8.9 Hz, 1H, 6'-H), 6.91 (d, J =8.2 Hz, 1H, 5'-H), 6.66 (s, 1H, 3-H), 6.29 (s, 1H, 6-H), 5.22 (t, J =6.4 Hz, 1H, 2" -H), 3.44 (d, J =6.3 Hz, 2H, 1" -H), 1.77 (s, 3H, 3" - CH3 ), 1.64 (s, 3H, 3" -CH3 ); 13 C NMR (100 MHz, DMSO- d 6 ) δ : 182.45, 164.29, 162.02, 159.52, 154.89, 150.08, 146.26, 131.50, 122.91, 122.39, 119.27, 116.43, 113.91, 106.54, 104.09, 103.08, 98.81, 25.94, 21.83, 18.28; ESI-MS m /z : 355 [M+H]+ .
3.11
3', 4', 5-三羟基-6" , 6" -二甲基-4" , 5" -二氢吡喃[2" , 3" :7, 8]-黄酮(15)的合成
将化合物14 (180 mg, 0.51 mmol)溶入45 mL甲醇溶液中, 加入15 mL硫酸水溶液(20%)回流搅拌2 h, 将反应液倒入冰水中, 用乙酸乙酯萃取.旋除溶剂, 采用柱层析分析提纯(石油醚/乙酸乙酯, V :V =5:1)得黄色固体15 152 mg, 产率84%. m.p. 207~209 ℃; 1 H NMR (400 MHz, DMSO-d 6 ) δ : 13.36 (s, 1H, 5-OH), 7.45 (d, J =2.0 Hz, 1H, 6'-H), 7.41 (s, 1H, 2'-H), 6.92 (d, J =8.2 Hz, 1H, 5'-H), 6.69 (s, 1H, 3-H), 6.49 (s, 1H, 6-H), 2.63 (t, J =6.7 Hz, 2H, 1" -H), 1.84 (t, J =6.7 Hz, 2H, 2" -H), 1.35 (s, 6H, 3" -2CH3 ); 13 C NMR (100 MHz, DMSO-d 6 ) δ : 182.28, 164.53, 160.23, 158.84, 155.38, 150.21, 146.20, 122.03, 119.47, 116.44, 113.87, 105.23, 103.87, 103.12, 95.03, 76.66, 31.37, 26.85, 16.17; ESI-MS m /z : 355 [M+H]+ ; HR- ESI-MS calcd for C20 H18 O6 Na [M+Na]+ 377.0996, found 377.0973.
3.12
8-异戊烯基槲皮素-3-甲醚(1)的合成
在50 mL单口圆底烧瓶中加入化合物10 (100 mg, 0. 19 mmol)和无水甲醇10 mL, 加热使其溶解后加入3 mol•L-1 的盐酸1 mL, 65 ℃下加热回流, TLC追踪反应全过程, 待原料点消失后停止反应, 加水淬灭, 乙酸乙酯(10 mL×3)萃取, 合并有机层, 无水MgSO4 干燥, 过滤, 旋除溶剂, 经硅胶柱层析(石油醚/乙酸乙酯, V : V =3:1)分离纯化, 得到黄绿色固体1 65 mg, 产率为89%, m.p. 232~233 ℃; 1 H NMR (400 MHz, DMSO-d 6 ) δ : 12.67 (s, 1H, 5-OH), 10.81 (s, 1H, OH), 9.83 (s, 1H, OH), 9.39 (s, 1H, OH), 7.60 (s, 1H, 2'-H), 7.47 (d, J =8.5 Hz, 1H, 6'-H), 6.94 (d, J =8.4 Hz, 1H, 5'-H), 6.31 (s, 1H, 6-H), 5.18 (t, J =6.0 Hz, 1H, 2" -H), 3.81 (s, 3H, 3-OCH3 ), 3.43 (d, J =6.3 Hz, 2H, 1" -H), 1.75 (s, 3H, 3" -CH3 ), 1.65 (s, 3H, 3" -CH3 ); 13 C NMR (100 MHz, DMSO-d 6 ) δ : 178.61, 161.91, 159.27, 155.99, 154.01, 149.12, 145.76, 137.96, 131.52, 122.85, 121.61, 120.87, 116.16, 115.98, 106.31, 104.61, 98.59, 60.11, 25.92, 21.70, 18.27; ESI-MS m /z : 385 [M+H]+ ; HR-ESI-MS calcd for C21 H20 O7 Na [M+Na]+ 407.1101, found 407.1168.
3.13
8-异戊烯基槲皮素-3, 4, 3', 4'-四甲醚(2)的合成
将化合物1 (120 mg, 0. 31 mmol)溶入20 mL丙酮中, 加入2 g无水碳酸钾室温搅拌30 min, 再加入硫酸二甲酯0.5 mL, 搅拌2 h.过滤出去碳酸钾固体, 旋除溶剂, 用乙酸乙酯/石油醚(V :V =1:2)重结晶, 得黄色固体2 96 mg, 产率72%. m.p. 106~107 ℃; 1 H NMR (400 MHz, CDCl3 ) δ : 7.73 (d, J =9.2 Hz, 1H, 6'-H), 7.67 (s, 1H, 2'-H), 6.93 (d, J =8.6, 1H, 5'-H), 6.32 (s, 1H, 6-H), 5.15 (t, J =6.4 Hz, 1H, 2" -H), 3.90 (s, 3H, OCH3 ), 3.87 (s, 3H, OCH3 ), 3.83 (s, 3H, OCH3 ), 3.80 (s, 3H, OCH3 ), 3.42 (d, J =5.8 Hz, 2H, 1" -H), 1.70 (s, 3H, 3" -CH3 ), 1.60 (s, 3H, 3" -CH3 ); 13 C NMR (100 MHz, CDCl3 ) δ : 178.12, 161.64, 159.33, 154.60, 152.33, 150.24, 147.72, 137.62, 131.21, 122.34, 121.20, 121.15, 110.14, 109.83, 106.64, 104.47, 93.79, 59.12, 55.06, 54.97, 54.88, 24.72, 20.60, 16.93; EI-MS m /z : 426 [M]+ ; HR-ESI-MS calcd for C24 H27 O7 [M+H]+ 427.1751, found 427.1752.
3.14
Artochamin C (3)的合成
在50 mL单口烧瓶中加入化合物15 (120 mg, 0.34 mmol), DDQ (110 mg)和15 mL干燥的1, 4-二氧六环, 在氮气保护下加热至110 ℃搅拌24 h, 反应液冷却至常温后过滤, 减压旋干.经硅胶柱层析(石油醚/乙酸乙酯, V :V =6:1)分离纯化, 得黄色固体3 80 mg, 产率67%. m.p. 251~253 ℃; 1 H NMR (400 MHz, DMSO-d 6 ) δ : 13.14 (s, 1H, 5-OH), 7.50 (s, 1H, 2'-H), 7.49 (d, J =4.0 Hz, 1H, 6'-H), 6.93 (d, J =9.0 Hz, 1H, 5'-H), 6.86 (d, J =10.0 Hz, 1H, 1" -H), 6.78 (s, 1H, 3-H), 6.25 (s, 1H, 6-H), 5.85 (d, J =10.0 Hz, 1H, 2" -H), 1.48 (s, 6H, 3" -2CH3 ); 13 C NMR (100 MHz, DMSO-d 6 )
δ : 182.39, 164.27, 161.38, 159.12, 151.67, 150.32, 146.25, 128.62, 121.79, 119.57, 116.63, 114.66, 113.76, 105.04, 103.39, 101.43, 99.74, 78.62, 28.20; EI-MS m /z : 352 [M]+ ; HR-ESI-MS calcd for C20 H16 O6 Na [M+Na]+ 375.0839, found 375.0897.
辅助材料(Supporting Information) 所合成的化合物的1 H NMR, 13 C NMR和MS谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/ )上下载.

 下载:
下载:

 下载:
下载:



 下载:
下载:
