Received Date:
10 December 2018 Revised Date:
06 January 2019 Available Online:
25 July 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21676187), and the China International Science and Technology Project (Nos. 2012DFG41980, 2016YFE0114900)
Abstract:
Tumor is one of the diseases with the highest mortality rate in the world. In view of the high risk and high mortality of tumor, researchers around the world are committed to develop more accurate and rapid diagnostic strategies and more effective treatments to fight tumor. Gradually, integrated optical diagnosis and treatment technologies for tumors have emerged. Fluoroboron fluorescein (BODIPY) has been widely used in tumor phototherapy because of its excellent optical properties. In this paper, BODIPY and its derivatives are introduced in detail as photosensitizers, photothermal transformants, and contrast agents in the diagnosis and treatment of tumors (photodynamic therapy, photothermal therapy, photoacoustic imaging) and integration of diagnosis and treatment. The effects of different BODIPY structures and their derivatives in tumor diagnosis and treatment were evaluated systematically. This is of great significance for the rational design of near-infrared BODIPY materials with high singlet oxygen quantum yield, high photothermal conversion, and good light stability and solubility.
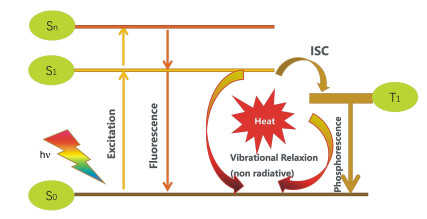
Figure 1.
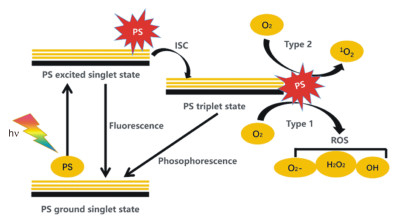
Mechanism diagram of photosensitizers in PDT
A photosensitizer (PS) can be activated from its ground state to a short-lived excited singlet state (PSEs) by light. Then, either the excited PS may decay back to the ground state by emitting fluorescence, or it can undergo intersystem crossing whereby the spin of its excited electron inverts to form a relatively long-lived triplet state (PSEt). The triplet excited PS can also decay back to the ground state by emitting phosphorescence, but most importantly it can directly interact with surrounding substrates (e.g., cell membrane or other biomolecules) to form radicals, which then react with O2 to produce reactive oxygen species (ROS), such as superoxide anion radicals (O2-), hydroxyl radicals (.OH), and hydrogen peroxides (H2O2, type Ⅰ reaction). Alternatively, the energy of the excited PS can be directly transferred to 3O2 (itself a triplet in the ground state) to form 1O2 (type Ⅱ reaction)[6]
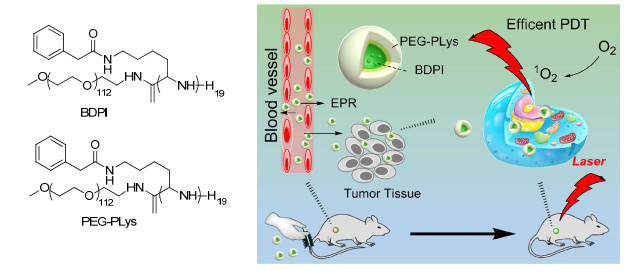
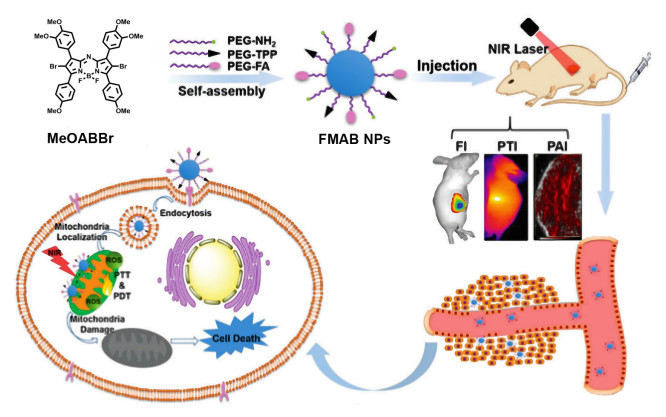
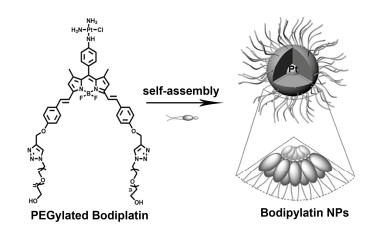
Figure 4.
Structures of BDPI, peptide PEG-PLys, and the schematic illustration of high ROS generation micelles PEG-PLys@BDPI nanoparticles for PDT in vivo
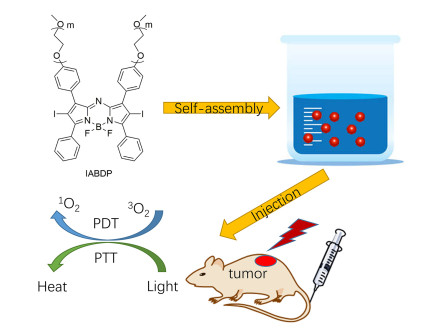
Figure 15.
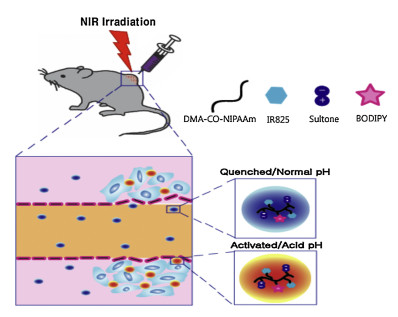
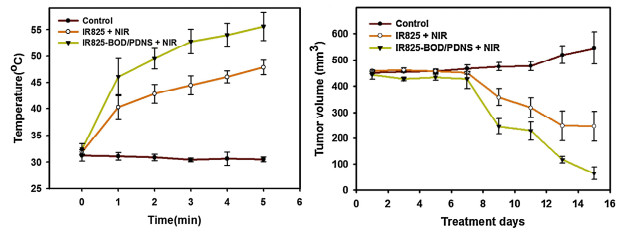
Schematic illustration for the preparation of zwitterionic IR825/BOD-PDNS nanoparticles activated in response to near infrared (NIR) light exposure
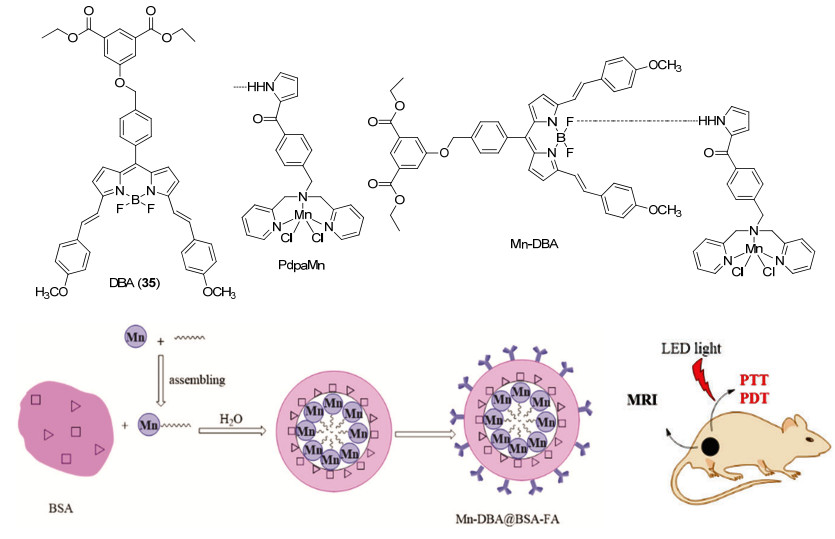
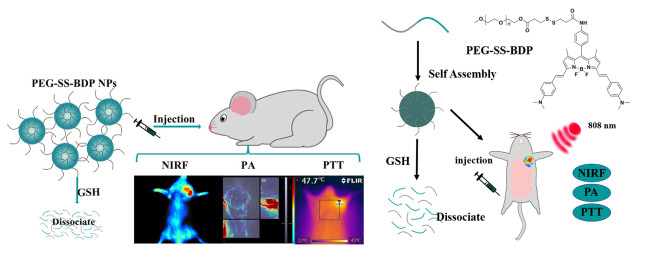
Figure 20.
NIR fluorescence imaging and photothermal imaging-guided synergistic PTT/PDT phototherapy (a) and the relationship between time and DPBF, time and temperature differences (b)
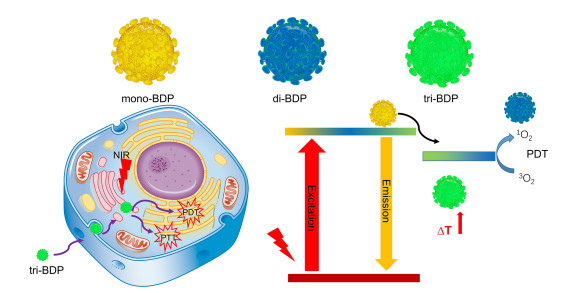
Figure 24.
Nanoparticles of mono-BDP, di-BDP, tri-BDP and cooperative phototherapy of tri-BDP-NPs against tumor cells through PTT/PDT treatments under light exposure
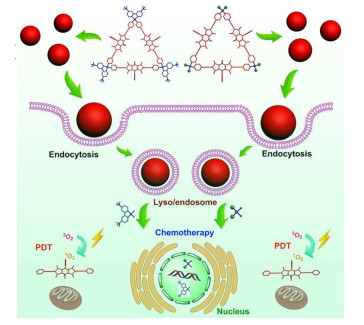
Figure 29.
Schematically illustrating the fabrication of lysosome-targeting BODIPY NP and their use in lysosomal PAI and pH-activatable PDT under NIR-light
Durantini, A. M.; Heredia, D. A.; Durantini, J. E.; Durantini, E. N. Eur. J. Med. Chem. 2018, 144, 651. doi: 10.1016/j.ejmech.2017.12.068
[9]
Zhang, J.; Jiang, C.; Figureueiro Longo, J. P.; Azevedo, R. B.; Zhang, H.; Muehlmann, L. A. Acta Pharm. Sin. B2018, 8, 137. doi: 10.1016/j.apsb.2017.09.003
Lu, W. L.; Lan, Y. Q.; Xiao, K. J.; Xu, Q. M.; Qu, L. L.; Chen, Q. Y.; Huang, T.; Gao, J.; Zhao, Y. J. Mater. Chem. B2017, 5, 1275. doi: 10.1039/C6TB02575G
Zhou, J.; Zhang, Y.; Yu, G.; Crawley, M. R.; Fulong, C. R. P.; Friedman, A. E.; Sengupta, S.; Sun, J.; Li, Q.; Huang, F.; Cook, T. R. J. Am. Chem. Soc. 2018, 140, 7730. doi: 10.1021/jacs.8b04929
图 1
光敏剂工作原理
Figure 1
Mechanism diagram of photosensitizers in PDT
A photosensitizer (PS) can be activated from its ground state to a short-lived excited singlet state (PSEs) by light. Then, either the excited PS may decay back to the ground state by emitting fluorescence, or it can undergo intersystem crossing whereby the spin of its excited electron inverts to form a relatively long-lived triplet state (PSEt). The triplet excited PS can also decay back to the ground state by emitting phosphorescence, but most importantly it can directly interact with surrounding substrates (e.g., cell membrane or other biomolecules) to form radicals, which then react with O2 to produce reactive oxygen species (ROS), such as superoxide anion radicals (O2-), hydroxyl radicals (.OH), and hydrogen peroxides (H2O2, type Ⅰ reaction). Alternatively, the energy of the excited PS can be directly transferred to 3O2 (itself a triplet in the ground state) to form 1O2 (type Ⅱ reaction)[6]
Figure 4
Structures of BDPI, peptide PEG-PLys, and the schematic illustration of high ROS generation micelles PEG-PLys@BDPI nanoparticles for PDT in vivo
Figure 15
Schematic illustration for the preparation of zwitterionic IR825/BOD-PDNS nanoparticles activated in response to near infrared (NIR) light exposure
Figure 20
NIR fluorescence imaging and photothermal imaging-guided synergistic PTT/PDT phototherapy (a) and the relationship between time and DPBF, time and temperature differences (b)
Figure 24
Nanoparticles of mono-BDP, di-BDP, tri-BDP and cooperative phototherapy of tri-BDP-NPs against tumor cells through PTT/PDT treatments under light exposure
Figure 29
Schematically illustrating the fabrication of lysosome-targeting BODIPY NP and their use in lysosomal PAI and pH-activatable PDT under NIR-light

 下载:
下载:






















 下载:
下载:




































 下载:
下载:
