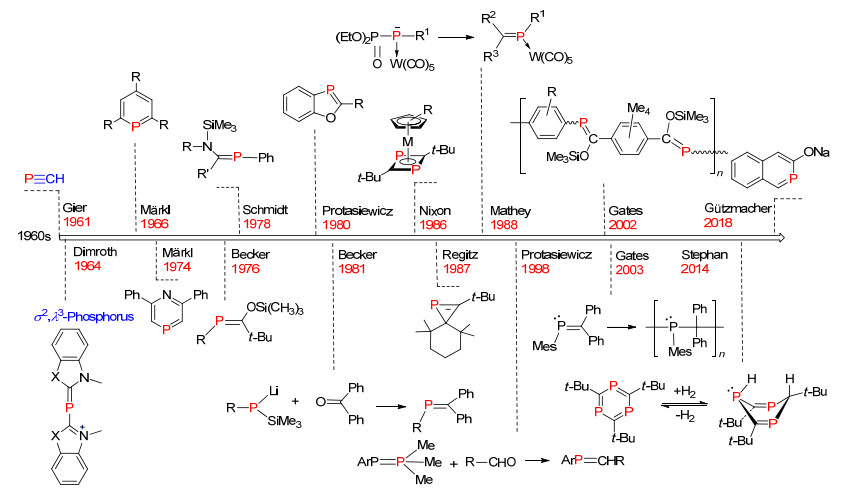
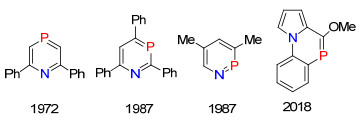
图 1.
膦烯化合物发展的历史轴
Figure 1.
Historically developing timeline of phosphaalkenes

目前有机磷化学的研究主要围绕着三价三配位(σ3, λ3)和五价四配位(σ4, λ5)的磷化合物, 研究领域主要集中在医药、农药、阻燃剂、光电材料和均相催化等领域中.而含膦烯结构(RP=CR'2)低配位磷的研究进展较为缓慢, 其主要研究尚处在理论及合成方法的研究阶段, 而应用性研究较少[1].纵观膦烯化合物研究的发展, 1964年, Dimroth等[2]报道了首例σ2, λ3-磷衍生物的合成, 发现磷元素的三价二配位结构.直到1966年, Märkl[3]合成得到磷杂苯化合物, σ2, λ3-低配位有机磷化学才逐渐引起化学家们的重视.在过去的近半个世纪里, Märkl, Becker, Mathey, Gates, Nixon和Protasiewicz等科学家的不懈努力下, 膦烯化合物的研究取得了长足的发展(图 1).迄今为止, 未见有关于系统介绍膦烯化合物的综述文章发表[1].为了引起更多科学家对膦烯化合物的重视, 系统总结了该类化合物的合成方法及性质的研究进展.
膦烯化合物主要可分为开链型和杂环型两种结构类型, 其中开链型的膦烯聚合物被广泛应用于电子材料中, 而杂环型膦烯化合物主要被应用于均相催化和光电材料中.其中五元磷杂环化合物主要包括:磷杂环戊二烯、磷杂呋喃、磷杂噻吩、磷杂吲哚及衍生物等[4~7].六元杂环化合物主要包括磷杂苯、多磷杂苯和氮磷杂苯及衍生物等[8~10].将从小分子链型膦烯化合物、链型膦烯聚合物、杂环膦烯化合物和其它膦烯化合物等分类介绍膦烯化合物的合成方法(图 2).
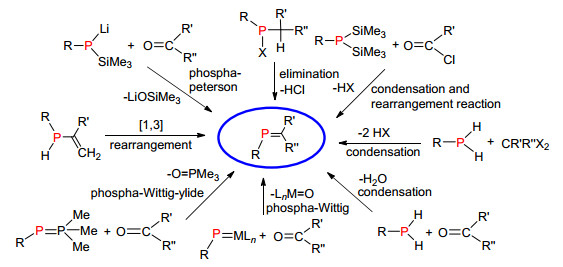
小分子链型膦烯化合物是整个膦烯化合物研究的基础, 对P=C双键化学性质的初步认识主要来自于对小分子链膦烯化合物的研究, 其合成方法也为膦烯聚合物和膦烯杂环化合物的合成提供了参考和借鉴.这部分的研究主要集中在二十世纪七八十年代, 而最近二十年的研究主要是在该基础上对合成方法的优化及应用研究.链型膦烯化合物的主要合成方法概括如下:重排反应、1, 2消除反应、“phospha-Peterson”反应、分子间缩合反应和“phospha-Wittig”反应(图 3).
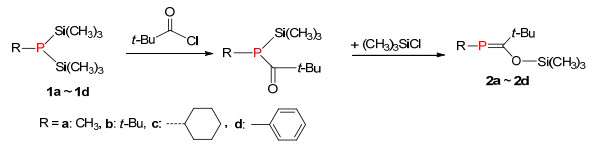
1976年, Becker等[11]第一次报道了稳定的膦烯化合物2 (Scheme 1), 奠定了整个膦烯化合物发展的里程碑, 该反应被命名为“Becker”反应.其反应过程如下:首先双三甲基硅膦化合物1与酰氯反应得到酰基三甲基硅基膦化合物, 由于硅原子与氧原子的强亲和力, 导致发生[1, 3]硅迁移反应(硅迁移到氧)形成P=C化合物2a~2d.由于存在较大位阻的三甲硅烷氧基和叔丁基, 这类含P=C键的化合物表现出较好的稳定性.后来这个方法被广泛应用于合成多种类型的膦烯化合物[12]和链型膦烯聚合物.
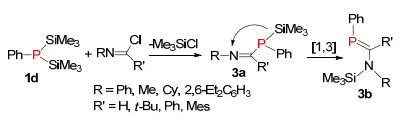
1978年, Schmidt等[13]扩展了硅迁移的方法, 他们通过双三甲基硅膦化物1d与氯代亚胺反应, 形成P—C单键中间体3a, 该中间体容易发生[1, 3]硅迁移反应(硅迁移到氮上), 形成P=C双键结构3b (Scheme 2).
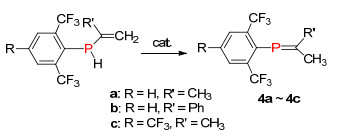
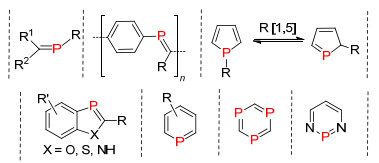
2004年, Gates课题组[14]通过碱催化的方法, 磷上的氢质子[1, 3]迁移到β碳上得到膦烯化合物4.整个反应操作简单, 但必须在磷原子上引入强的吸电子基团(Scheme 3).除了质子迁移到β碳上的方法, 还有质子[1, 3]迁移和[1, 5]迁移的方法, 并成功应用到磷杂吲哚化合物的合成中.
1978年, Bickelhaupt课题组[15]通过磷卤化物的消除合成膦烯化合物, 即在碱性条件下磷原子上的卤素与磷原子α位的氢发生1, 2-消除构建P=C双键骨架单元(Scheme 4).该方法的应用范围非常广泛, 不仅可以适用于开链型膦烯化合物的合成, 也适用于各类杂环型膦烯化合物的合成, 整个操作过程简便, 是一类相对通用的合成方法.
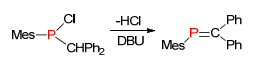
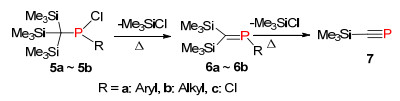
1981年, Issleib等[16]报道了化合物5在加热条件下, 发生1, 2-消除三甲基氯硅烷合成膦烯化合物6, 甚至可以进一步发生消除反应得到膦炔化合物7 (Scheme 5), 该方法同样也可以应用到一些杂环膦烯化合物的合成.
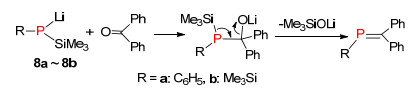
1981年, Becker等[17]报道了三甲基硅基膦的锂化物8与醛酮反应构建P=C结构的方法, 整个反应过程如下:首先磷负离子亲核进攻缺电子的羰基碳发生亲核加成反应, 然后分子内消除三甲基硅醇锂, 最终得到膦烯化合物.由于这种方法类似于Peterson反应, 所以称为“phospha-Peterson”反应(Scheme 6).
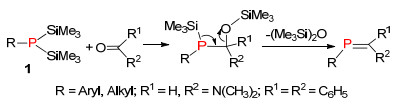
之后, Becker等[18]在1983年对该反应进一步研究和发展, 选用活性更低的双-三甲基硅基膦为原料, 与醛酮在酸或碱的催化条件下反应, 成功得到膦烯化合物.由于双三甲基硅基膦原料易得, 稳定性较好, 从而提高了整个反应的产率和实用性(Scheme 7).
Oehme等[19a]1980年报道了伯膦化合物9与醛或缩醛反应得到膦烯化合物, 反应过程中产生一分子水或两分子甲醇(Scheme 8).同年, Issleib等[19b]也报道了通过伯膦与醛基的缩合反应形成P=C双键结构.

1984年, Appel课题组[20]报道了伯膦化合物与卤代烷在强碱的条件发生缩合反应构建P=C双键, 首先分子间缩合脱去一分子卤化氢, 再进一步发生1, 2消除另一分子卤化氢得到膦烯化合物(Scheme 9, A). 2012年, Sasamori等[21]利用该方法成功合成了稳定的三磷杂[3]轴烯化合物10 (Scheme 9, B).
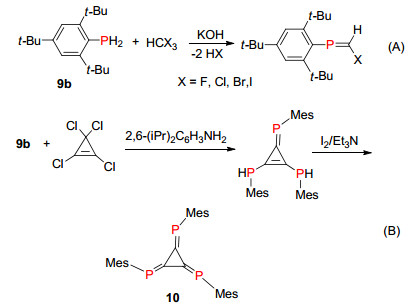
“phospha-Wittig”反应是另一种比较常用的膦烯化合物合成方法, 反应主要分为以下三大类:“phospha-Wittig-Horner”反应、“Phospha-Wittig ylide”反应和端基亚磷烯的Wittig反应.
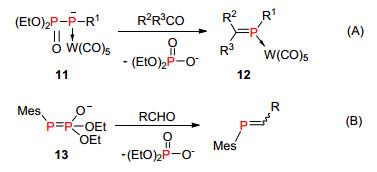
1988年, Mathey等[22]报道了类似的磷-磷叶立德结构11, 结构中磷负离子与金属配位, 我们称之为“phospha-Wittig-Horner”试剂.该试剂可以与醛、酮、缩醛或缩酮类化合物较好地发生反应, 得到磷杂烯烃与金属络合的稳定产物12 (Scheme 10, A), 该反应具有很好的可操作性和选用范围. 2016年, Arkhypchuk等[23]首次报道了无金属配位的“phospha-Wittig-Horner”试剂13, 该试剂与醛反应得到裸露的膦烯化合物(Scheme 10, B).
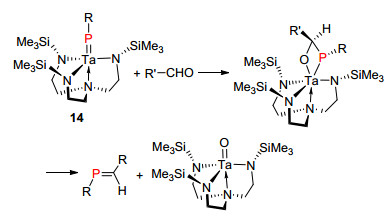
1993年, Schrock等[24]报道了端基亚磷烯配合物14作为“phospha-Wittig”试剂用于膦烯化合物的合成, 该合物只能与醛发生反应(Scheme 11).
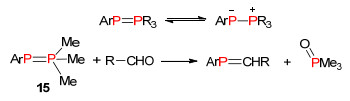
1998年, Protasiewicz课题组[25a]报道以二氯化膦、三烷基膦和锌为原料合成了“phospha-Wittig-ylide”试剂15.该试剂成功地与醛反应得到膦烯化合物.由于“phospha-Wittig-ylide”试剂没有金属参与配位, 所以反应生成的P=C双键结构也是没有金属配位的裸露的结构. 2011年, 段征课题组[25b]也应用该方法合成磷杂菲化合物(Scheme 12).
具有π共轭骨架结构的聚对苯乙炔是一类非常重要的电子材料, 被广泛应用到非线性光学材料、发光二极管材料、掺杂后导电材料以及太阳能电池材料等领域中.把低配位P=C双键引入到聚合物的主链中, 是非常具有挑战性的前沿研究.相比于C=C双键, 有着相似结构的P=C双键往往表现出独特的性质, 聚对苯磷杂乙烯(PPP)作为聚对苯乙炔(PPV)的类似物也开始引起化学家们的重视.近十几年来, 为了开发和研究聚对苯磷杂乙烯(PPP)的物理和化学等性质, 化学家们做出了不懈的努力, 取得一些可喜的进展.
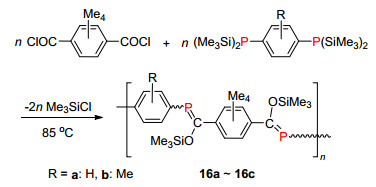
2002年, Gates课题组[26]利用[1, 3]硅迁移的方法(即Becher反应), 首次合成了含有P=C双键顺反混合(E/Z≈1)的聚合物(Scheme 13).他们选择对苯二(双-三甲基硅磷基)和对苯二甲酰氯为原料, 通过简单加热得到π共轭骨架的中等聚合度(Xn=5~21)的聚合物16a~16c (Scheme 13), 并对其光学性质进行了测试(最大吸收波长为328~338 nm). 2006年, 他们又通过控制空间位阻和反应时间来调节顺反异构化的比例, 选择性得到单一构型的E-聚合物, 发现全E-型聚合物的溶解度较差, 其紫外最大吸收波长发生了较大的红移(从338 nm红移至394 nm)[27].
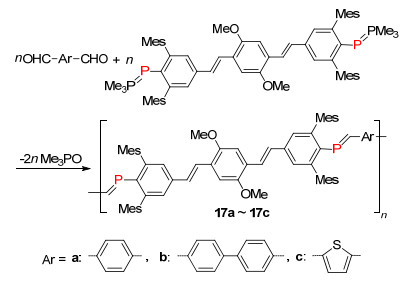
2004年, Protasiewicz课题组[28]利用“phospha-Wittig“反应合成大位阻稳定的含P=C双键聚合物, 聚合度Xn在4~7左右, 分子量Mn为5000~7500左右, 该聚合物表现出优异的光电性质, 并有望替代PPVs聚合物应用到电子材料器件中(Scheme 14).
将低配位磷引入到芳香结构中一直以来都是非常具有挑战性的研究课题.目前对于膦烯化合物的研究主要集中在杂环化合物的研究, 其中包括磷杂环戊二烯和磷杂苯结构等.这些化合物广泛应用于均相催化和功能材料等领域中.下面将从磷杂环丙烯、磷杂环丁二烯、磷杂环戊二烯和磷杂苯等分类综述杂环膦烯化合物的研究进展.
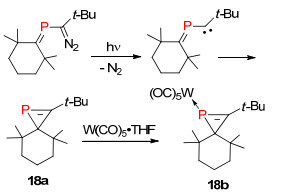
λ3-磷杂环丙烯是最小的杂环膦烯化合物, 由于其较大的结构张力, 因此表现出非常高的反应活性, 另外2位上的取代基容易迁移到1位磷上形成热力学更稳定的1H-磷杂环丙烯化合物. Regitz课题组[29]在1987年报道了利用α位卡宾碳发生分子内插入反应形成含有P=C双键的磷杂环丙烯骨架结构化合物18a (Scheme 15).
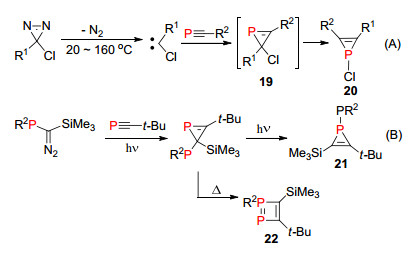
1990年, Heydt课题组[30]报道了利用膦炔化合物与氯代卡宾反应形成含P=C双键的磷杂环丙烯中间体19, 这个中间体化合物上2位Cl原子非常容易发生迁移, 得到热力学稳定的1-氯-磷杂环丙烯化合物20 (Scheme 16, A). 1999年, Bertrand等[31]也利用类似的方法合成了1H-磷杂环丙烯化合物21和化合物22, 并对其反应性质进行了深入研究(Scheme 16, B).
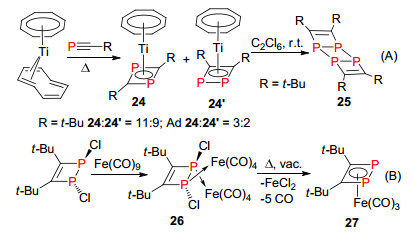
λ3-磷杂环丁二烯是含有P=C双键的四元环结构, 主要有两种类型的磷杂环丁二烯的结构, 即1, 3-二磷杂环丁二烯和1, 2-二磷杂环丁二烯.早在1986年, Nixon等[32]通过膦炔化合物与金属配合物反应, 首次报道了两分子膦炔头对尾的二聚体1, 3-二磷杂环丁二烯与金属的络合物23 (Scheme 17), 从而开启了对磷杂环丁二烯化合物的研究.该方法已经成为这类化合物的经典合成方法, 就目前为止, 还未见其它合成方法的进一步报道.
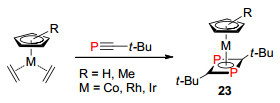
1995年, Binger等[33]同样利用膦炔[2+2]环加成反应得到部分头对头的二聚体1, 2-二磷杂环丁二烯络合物24.该化合物很容易继续发生[2+2]环加成得到1, 4-磷环丁烷化合物25 (Scheme 18, A). Zenneck等[34, 35]在1999年报道了1, 2-二氯-1, 2-丁二烯-1, 2-η-(Fe(CO)4)2络合物26在真空加热条件下合成1, 2-二磷杂环丁二烯与铁金属的络合物27 (Scheme 18, B).
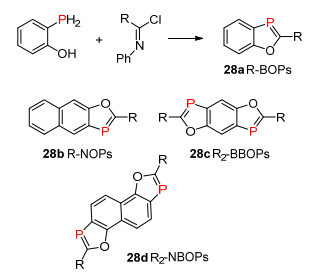
λ3-磷杂环戊二烯化合物是含有P=C双键的磷杂五元环结构, 在磷杂环化合物中具有非常重要的地位, 主要应用于光电材料领域中.其中研究比较多的结构类型主要有单磷杂环戊二烯、3-磷杂吲哚、3-磷杂苯并呋喃或3-磷杂苯并噻吩等化合物.其中磷杂苯并呋喃是为数不多的含有非常稳定的P=C双键结构的化合物, 该类化合物表现出较好的光电性质, 是非常有开发前景的一类化合物. 1980年, Heinicke等[36]首次报道了邻位羟基的伯膦化合物与氯代亚胺反应得到3-磷杂苯并呋喃化合物28a (Scheme 19), 并由Protasiewicz等[5]继续发展, 同时对该类化合物28a~28d的光电性质进行了深入研究.
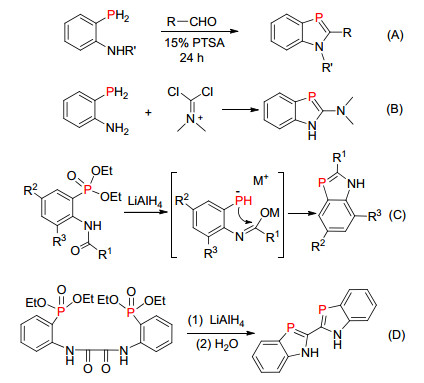
除了运用“phospha-Peterson”反应合成磷杂吲哚的方法外, 科学家们也发展了其它的合成方法.比如在酸的催化下邻位氨基的苯基膦与醛首先发生缩合反应, 然后被过量的的醛氧化得到磷杂吲哚化合物(Scheme 20, A)[37].邻位氨基的苯基膦和氯代亚胺(或二氯代亚胺盐)化合物分子间的反应(Scheme 20, B)[38].邻位为酰胺基的磷酸脂化合物通过氢化铝锂还原, 发生分子内的关环反应(Scheme 20, C)[39], 其反应机理为磷酸酯和酰胺分别被氢化铝锂还原成磷氢负离子和亚胺醇负离子, 然后磷负离子中心进攻缺电子的碳, 再消除一分子氢氧根形成磷杂吲哚负离子, 最后质子化形成含P=C双键的磷杂吲哚化合物. 2018年, 段征课题组[40]利用这个方法成功合成了2, 2'-双磷杂吲哚化合物(Scheme 20, D).
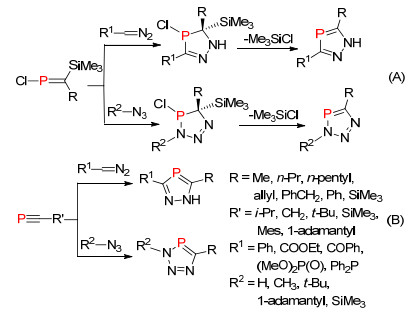
二氮杂或三氮杂磷杂环戊二烯是另一类研究较多的氮磷杂五元环结构, Schmidpet课题组[41a]和Regitz课题组[41c]分别报道了这两类化合物的合成方法.目前为止这类氮磷杂五元环的合成只有两种合成方法的报道.一种是氯代膦烯化合物与三甲基硅基重叠化物或叠氮化物的[2+3]环加成, 然后消除一分子三甲基氯硅烷得到目标化合物(Scheme 21, A)[42]; 另一种是膦炔化合物与重叠化物或叠氮化物的环加成反应(Scheme 21, B)[43].
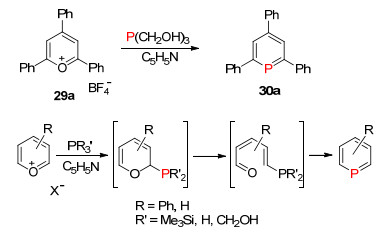
λ3-磷杂苯及衍生物是具有膦烯结构的平面芳香性的六元杂环结构, 是低配位磷杂环化合物中最具有代表性的化合物, 也是被研究最多的一类低配位有机磷化合物, 主要应用于均相催化和功能材料中.第一个磷杂苯化合物2, 4, 6-三苯基磷杂苯(30a)是Märkl[44]在1966年通过嗡盐29a与膦试剂P(CH2OH)3发生磷氧交换的方法合成得到, 而且后续研究发现P(SiMe3)3和PH3等膦试剂与氧盐或硫盐也能很好地发生与磷的交换反应, 为低配位磷杂苯化学的发展奠定了良好的基础(Scheme 22)[45].
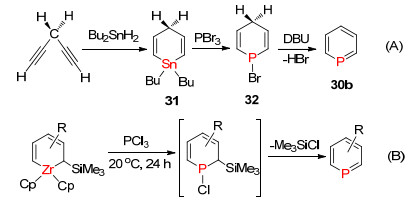
1971年, Ashe Ⅲ课题组[46]首次报道了卤化磷与金属交换的方法合成磷杂苯母核结构, 其反应过程如下:首先是1, 4-二炔化合物和二丁基锡烷发生环加成反应形成环状的二丁基锡杂1, 4-二烯化合物31, 再与PBr3发生P/Sn交换反应形成环状1, 4-二烯的磷卤化合物32, 之后在碱性条件下消除一分子卤化氢得到磷杂苯化合物30b (Scheme 23, A).后来也发展了一些其它金属的交换合成磷杂苯衍生物的方法, 例如Mathey等[47]在1996年报道的P/Ti交换合成磷杂苯和Whitby等[48]在2004年报道的P/Zr交换的合成磷杂苯(Scheme 23, B).
膦炔是低配位有机磷化学中非常重要的一类化合物, 其反应活性较高, 所以常常可作为合成其它有机磷化合物的反应试剂.其中膦炔很容易和二烯类化合物发生[4+2]环加成反应, 进一步合成多种类型的磷杂苯化合物[49].比如Märkl和Mathey等[50, 51]先后在1982年报道的膦炔与吡喃酮[4+2]和逆[4+2]环加成反应合成磷杂苯(Scheme 24, A), Regitz等[52]在1988年报道的膦炔与环戊二烯酮反应合成磷杂苯(Scheme 24, B), Grützmacher等[53]在2018年报道了膦炔与2, 3-二氮杂萘反应得到磷杂萘化合物33 (Scheme 24, C). 2015年, Nishibayashi[54]报道了铁催化的磷炔与炔的[2+2+2]环加成反应, 得到一系列磷杂苯衍生物34 (Scheme 24, D).

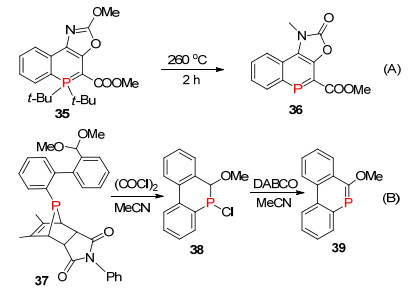
2017年, 段征课题组[55]以叶立德结构的λ5-磷杂苯为原料, 在甲苯溶剂中260 ℃高温条件下成功从σ2, λ3-磷杂苯化合物35降解成低配位的σ2, λ3-磷杂苯36 (Scheme 25, A).在2015年, 他们[56]已经报道了7-磷杂降冰片烯衍生物37为原料, 反应经过λ5-磷盐过渡态后降解成λ3-磷卤化合物38, 然后在碱性条件下1, 2-消除形成相对稳定的α位甲氧基的磷杂苯化合物39 (Scheme 25, B).
除了上述介绍的开链型、聚合物型和杂环型膦烯化合物, 还有部分比较特殊的膦烯化合物, 它们同样包含有低配位的P=C双键结构单元.如氮磷杂苯化合物和多膦杂苯化合物等.这些化合物往往表现出更高的反应活性, 其合成与分离更具有挑战性.目前为止, 关于这类化合物的报道非常少, 也一直处在对其理论和结构的研究阶段, 尚未有关于它们的应用研究报道.
氮磷杂苯是同时含有sp2-N与sp2-P的芳香六元杂环化合物, 往往表现出比磷杂苯更高的反应活性.目前为止, 已经报道了邻位、间位和对位的全部氮磷杂苯化合物, 关于该类化合物的研究主要是在二十世纪八十年代由Märkl和Mathey等完成, 之后一直没有新的研究进展报道[57].一直到2018年, 段征课题组[58]报道了另一种sp2杂化氮的稠环氮磷杂苯化合物, 由于α位甲氧基的作用, 这个化合物表现处较好的稳定性(图 4).
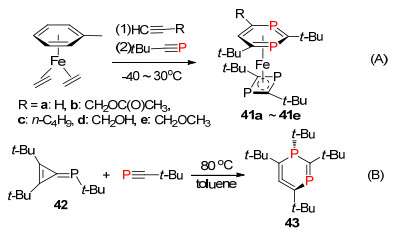
多磷杂苯是包含多个sp2-P的磷杂苯化合物, 主要包括间位和对位二磷杂苯化合物, 以及含有三个sp2-P的1, 3, 5-三磷杂苯化合物, 它们都具有两个或两个以上的P=C双键单元结构.最早于1976年, Kobayashi等[59]报道了第一个多磷杂苯化合物, 它们利用逆Diels- Alder反应成功合成2, 3, 5, 6-四-三氟甲基-1, 4-二-磷杂苯化合物40, 同时也研究了该类化合物[4+2]环加成的反应性质(Scheme 26).

Zenneck课题组[60]在1995年报道了通过一分子炔与两分子膦炔发生[2+2+2]环加成反应, 得到首例1, 3-二-磷杂苯化合物41 (Scheme 27, A). 2001年, Regitz等[61]利用亚磷基环丙烯42与膦炔反应合成2, 4, 5, 6-取代-1, 3-二-磷杂苯化合物43 (Scheme 27, B).
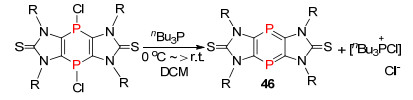
1995年, Binger课题组[62]报道了膦炔发生[2+2+2]环加成反应, 形成1, 3, 5-三-磷杂苯化合物44和Dewar-1, 3, 5-三-磷杂苯化合物45 (Scheme 28), 后来也有许多关于这类化合物的理论研究与合成研究的报道, 迄今为止, 所报道的合成方法都是通过膦炔的环加成反应得到.
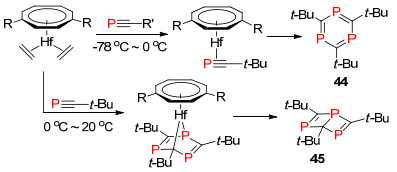
2017年, Nyulászi和Streubel等[63]报道了利用PBu3还原P—Cl键的方法, 以较高产率合成得到1, 4-二-磷杂苯衍生物46, 并进一步研究了其部分反应性能(Scheme 29).
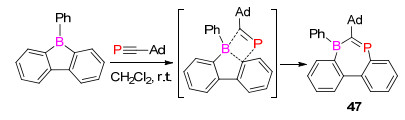
2018年, Martin课题组[64]报道了磷炔化合物直接插入到B—C键中, 形成七元环的1, 3-硼杂膦烯化合物47 (Scheme 30).
磷是一个具有“多面性”的元素, 主要表现以下两个方面: (1)配位数的多面性, 即磷原子中心主要有低配位(σ1与σ2)、中等配位(σ3与σ4)和高配位(σ5与σ6); (2)化学性质的多面性, 即高配位的磷和硅类似[4], 中等配位的磷和氮类似, 低配位的磷又和碳类似.低配位磷化学中, 膦烯化合物与碳的相似程度非常高, 甚至化学家们对这类磷化合物定义为“碳的复制品”. σ2, λ3-膦烯化合物是具有P=C双键的基本结构单元, 除了表现出C=C双键类似的性质之外, 由于其更低的键能和极化的性质等, 往往也表现出一些独特的化学性质.理论计算和实验研究均表明:膦烯化合物的磷中心一般为缺电子中心(C-—P+), 很容易受到亲核试剂进攻而发生加成反应(如:与质子化试剂或锂试剂发生加成反应等), 而且这类化合物一般难以发生季磷盐化反应.如果是极性反转(C+—P-)的磷杂烯化合物的季磷盐化反应很容易进行.下面简要分析和总结主要膦烯化合物的反应性质和配位化学性质的研究现状.
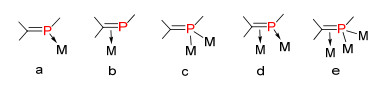
根据目前的文献报道, 低配位膦烯化合物基本上可以有三种配位方式: (a)磷原子上的孤对电子与一个金属形成σ方向的配位键(磷提供两个电子).这种配位的方式是最常见的, 基本不改变P=C双键的化学性质和结构.因此常用这种配位方式保护磷上的孤对电子, 避免发生进一步反应, 从而可以分离和研究膦烯化合物的化学性质等. (b) P=C双键的π电子与金属配位: π轨道的电子提供给金属, 然后金属上的电子又反馈回去占用π*反键轨道. (c)磷原子上的孤对电子与两个金属中心配位, 形成四位配位磷杂烯化合物.这种c的配位方式恰好可以说明, 低配位磷化合物有向中等配转变的倾向.另外, 这三种配位方式又可以相互组合, 形成新的金属配位化合物.如图 5所示, d是a和b配位方式的组合, e是b和c配位方式的组合.这些配位方式在一定条件下也是可以相互转化的, 而且配位方式的不同往往也表现出不同的性质[1].
膦烯化合物既是σ电子供体也是π电子受体, 可以较好地调谐金属的中心价态, 是非常有代表性的有机磷配体之一, 在均相催化中发挥着非常重要的作用.其中二亚膦基环丁烯48作为螯合配体与金属形成络合物, 在Pd和Pt金属催化反应中, 有非常优异的催化效应. 2001年Yoshifuji等[65]首次应用这类配体络合物成功催化1, 3-丁二烯与苯胺发生1, 4加成反应(Scheme 31, A), 苯乙酮与三取代基硅烷发生酮的脱氢甲硅烷基化反应(Scheme 31, B)[66]. 2002年, Yoshifuji等[67]又利用这个络合物催化丙烯醇与苯胺或活化的亚甲基发生烷基化反应(Scheme 31, C). 2004年, Ozawa等[68]报道了该催化剂下, 烯丙基醚的选择性去烯丙基化的反应(Scheme 31, D).
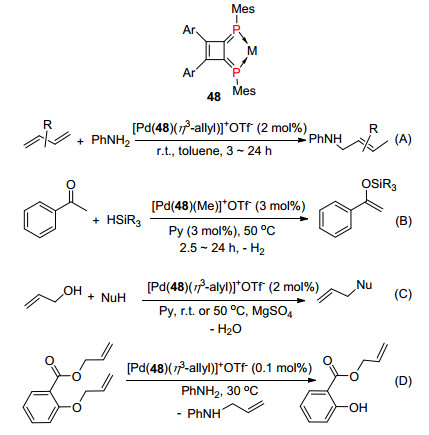
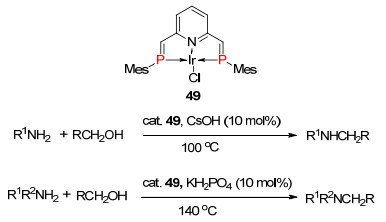
2013年, Ozawa课题组[69]报道了1, 6-二磷杂乙烯基吡啶与铱的三齿络合物49催化一级氨和二级氨与醇的N-烷基化反应, 产率高达90%以上(Scheme 32).
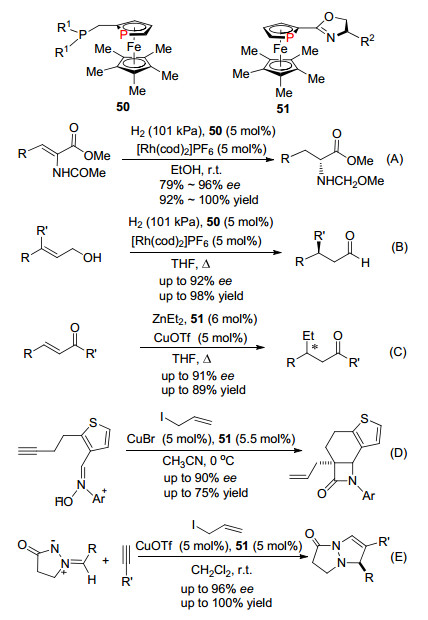
Fu等先后发展了手性的磷杂二茂铁化合物, 其作为手性配体应用于一系列手性催化反应.例如1998和2001年报道了化合物50作为手性配体Rh催化乙烯的氢化还原反应(Scheme 33, A)[70]和双键异构化的反应(Scheme 33, B)[71]; 2002和2003年又报道了化合物51作为手性配体, 廉价金属铜催化的加成反应(Scheme 33, C)[72]和环化反应(Scheme 33, D, E)[73].
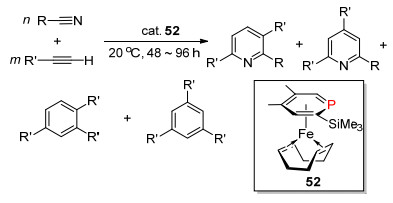
1996年, Zenneck等[74]利用2-三甲基硅-4, 5-二甲基磷杂苯为配体的η6-金属铁络合物(COD) 52作为催化剂, 催化缺电子的炔(DMAD, 二甲基乙炔二羧酸酯)发生[2+2+2]的三聚环加成反应形成苯衍生物, 或是炔与腈基衍生物发生三聚环加成反应形成吡啶衍生物(Scheme 34). 1998年, 他们[75]又利用磷杂苯为配体的η6-金属铁的1, 4-二氮杂-1, 3-丁二烯(DAD)络合物为催化剂, 催化1, 3-丁二烯的[4+2]和[4+4]的环加成反应. 2002年, Kozlowski等[76]利用零价镍磷杂苯络合物催化分子内的[4+2]环加成反应, 同时也发现反应速率受配体的电子和空间结构的影响非常显著.
就反应性质而言, 膦烯化合物与全碳的烯烃化合物是非常相似的, 烯烃化合物所能发生的许多经典反应, 膦烯化合物同样能够发生, 如金属催化的氢化还原反应[77], 与过氧化物的环氧化反应[78], 卡宾的环加成反应[79], 1, 3-或1, 5-迁移反应[80], Diels-Alder反应[81]和电环化反应[82]等.
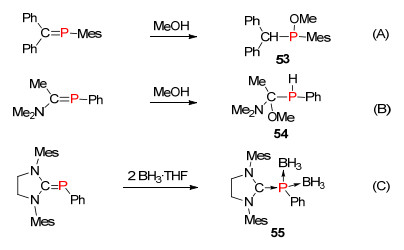
不仅如此, 由于P=C双键结构的键能低和极化的性质等, 膦烯化合物往往表现出非常高的反应活性, 更容易与质子试剂(如水分子或醇类化合物等)发生加成反应.根据极化性质的不同, 与质子试剂反应可得到不同的磷化学产物, 一般极化的磷碳双键(C-—P+)或反极化的磷碳双键(C+—P-)的膦烯化合物发生质子化反应, 分别形成C—H产物53 (Scheme 35, A)或P—H (极性反转)产物54 (Scheme 35, B)[83].当极性翻转的磷杂烯的极化强度非常大时, 可以在磷原子中心配位两个硼烷而形成碳卡宾化合物55 (Scheme 35, C)[84].
另一方面, P=C双键单元又是一个缺电子的体系, 容易与富电子的烯烃或炔烃发生环加成反应, 甚至自身可以在较为温和的条件下发生[2+1]或[2+2]环加成反应形成二聚体[85].关于膦烯化合物的环加成反应的报道较多, 如磷杂烯化合物与富电子的炔发生[2+2]环加成(Scheme 36, A)[86], 与极化分子的[2+3]环加成反应或与1, 3-丁二烯的[2+4]环加成(Scheme 36, B)[87], 及[2+8]环加成反应(Scheme 36, C)等[88].该类环加成反应都遵循Woodward-Horffmann规则[1].
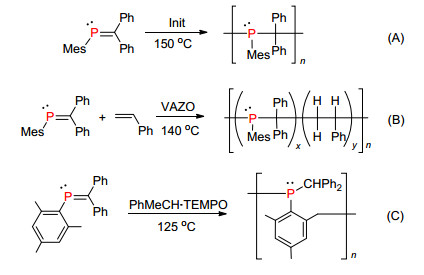
2003年, Gates课题组[89]进一步研究发现膦烯化合物与烯烃化合物类似, 在自由基引发剂和加热条件下会发生自由基聚合反应形成聚合物(Scheme 37, A), 而且也能与苯乙烯共聚形成共聚物(Scheme 37, B)[90]. 2013年Gates等[91]发现这类化合物在特定的条件下能够活化邻位苯甲基的氢得到1, 4-聚合的产物(Scheme 37, C).
2014年, Stephan等[92]报道了1, 3, 5-三磷杂苯化合物具有裂解简单氢分子的能力, 在4 bar氢气压力下与110 ℃条件下与氢气发生加成反应形成船式磷杂二烯化合物56a, 该化合物磷上的氢原子也容易发生翻转形成化合物56b (Scheme 38).

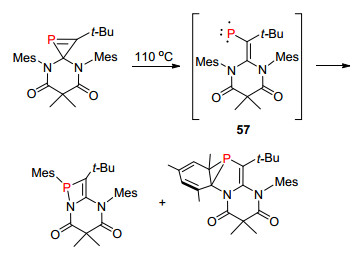
2017年, Stephan课题组[93]对2H-磷杂环丙烯化合物的化学性质进行更深入的研究, 研究发现这类化合物由于张力的原因, 在一定条件下容易通过热力学重排反应形成高活性的亚膦烯中间体57, 并利用该中间体合成一些结构更为复杂的化合物(Scheme 39).
综上所述, 许多膦烯化合物与烯烃的反应性质表现出非常高的相似性, 除了由于P=C双键所表现出的特有性质外, 基本的反应性质与全碳化合物类似, 所以相较于狭义的全碳“有机化学”, 将低配位磷化学命名为“有机磷-化学”(Phospha-organic chemistry)是非常有道理的.[1]
在膦烯化合物中, 虽然磷与碳的化学性质非常相似, 但是又不全同于碳化学, 它们也表现出磷自身所具有的独特的化学性质.也正因为P=C双键的特殊电子属性, 它们在均相催化或有机功能材料等领域都有较好的应用价值.近十几年来, 有机磷化学的发展涉及到了越来越多的交叉学科领域, 其中膦烯化合物的研究也不例外, 其研究主要朝着催化和光电性质等应用型研究的领域发展, 也从原来的功能小分子领域朝着多功能高分子领域发展.迄今为止, 我们对低配位膦烯化合物的认识还远远不够, 还有许多方面的应用有待开发, 需要更多磷化学工作者们的坚持和努力.相信在不久的将来, 膦烯化合物将在各个化学、材料和催化等领域中的发展发挥其特殊的作用.
(a) Appel, R.; Knoll, F.; Ruppert, I. Angew. Chem. 1981, 93, 771.
(b) Appel, R.; Knoll, F. Adv. Inorg. Chem. 1989, 33, 259.
(c) Markovski, L. N.; Romanenko, V. D. Tetrahedron 1989, 45, 6019.
(d) Regitz, M.; Binger, P. Angew. Chem. 1988, 100, 1541.
(e) Mathey F. Angew. Chem., Int. Ed. 2003, 42, 1578.
(f) Gates, D. P. New Aspects in Phosphorus Chemistry V. Springer, Berlin, Heidelberg, 2005, pp. 107~126.
(g) Russell, C. A. Coord. Chem. Rev. 2015, 297, 146.
(h) Regulska, E.; Romero-Nieto, C. Dalton Trans. 2018, 47, 10344.
(i) Tokarz, P.; Zagórski, P. M. Chem. Heterocycl. Compd. 2017, 53, 858.
(j) Francois, M.; Duan, Z. Sci. Sin.: Chim. 2010, 40, 888(in Chinese).
(Francois, M., 段征, 中国科学: 化学, 2010, 40, 888.)
(a) Dimroth, K.; Hoffmann, P. Angew. Chem., Int. Ed. 1964, 3, 384.
(b) Allmann R. Angew. Chem., Int. Ed. 1965, 4, 150.
Märkl, G. Angew. Chem., Int. Ed. 1966, 5, 846.
(a) Keglevich, G.; Farkas, R.; Ludányi, K.; Kudar, V.; Hanusz, M.; Simon, K. Heteroat. Chem. 2005, 16, 104.
(b) Deschamps, E.; Ricard, L.; Mathey, F. Organometallics 2001, 20, 1499.
(c) Markovskii, L. N.; Romanenko, V. D.; Ruban, A. V.; Iksanova, S. V. Zhur. Obshch. Khim. 1982, 52, 2796.
(d) Aitken, R. A. Sci. Synth. 2001, 10, 809.
(e) Hu, Z.; Tian, R.; Zhao, K.; Liu, Y.; Duan, Z.; Mathey, F. Org. Lett. 2017, 19, 5004.
(a) Washington, M. P.; Gudimetla, V. B.; Laughlin, F. L.; Deligonul, N.; He, S.; Payton, J. L.; Protasiewicz, J. D. J. Am. Chem. Soc. 2010, 132, 4566.
(b) Washington, M. P.; Payton, J. L.; Simpson, M. C.; Protasiewicz, J. D. Organometallics 2011, 30, 1975.
(c) Laughlin, F. L.; Rheingold, A. L.; Deligonul, N.; Laughlin, B. J.; Smith, R. C.; Higham, L. J.; Protasiewicz, J. D. Dalton Trans. 2012, 41, 12016.
(d) Laughlin, F. L.; Deligonul, N.; Rheingold, A. L.; Golen, J. A.; Laughlin, B. J.; Smith, R. C.; Protasiewicz, J. D. Organometallics 2013, 32, 7116.
(a) Ashe Ⅲ, A. J.; Fang, X. Chem. Commun. 1999, 14, 1283.
(b) Worch, J. C.; Hellemann, E.; Pros, G.; Gayathri, C.; Pintauer, T.; Gil, R. R.; Noonan, K. J. Organometallics 2015, 34, 5366.
(c) Qiu, Y.; Worch, J. C.; Chirdon, D. N.; Kaur, A.; Maurer, A. B.; Amsterdam, S.; Noonan, K. J. Chem. Eur. J. 2014, 20, 7746.
(d) Issleib, K.; Vollmer, R. Tetrahedron Lett. 1980, 21, 3483.
(e) Worch, J. C.; Hellemann, E.; Pros, G.; Gayathri, C.; Pintauer, T.; Gil, R. R.; Noonan, K. J. Organometallics 2015, 34, 5366.
(a) Zhang, L., Yu, W.; Liu, C.; Xu, Y.; Duan, Z.; Mathey, F. Organometallics 2015, 34, 5697.
(b) Aluri, B. R.; Kindermann, M. K.; Jones, P. G.; Heinicke, J. Chem. Eur. J. 2008, 14, 4328.
(c) Aluri, B. R.; Burck, S.; Gudat, D.; Niemeyer, M.; Holloczki, O.; Nyulaszi, L.; Heinicke, J. Chem. Eur. J. 2009, 15, 12263.
(a) Le Floch, P. Phosphorus-Carbon Heterocycl. Chem. 2001, 485.
(b) Le Floch, P.; Mathey, F. J. Chem. Soc., Chem. Commun. 1993, 16, 1295.
(c) Mao, Y.; Mathey, F. Chem. Eur. J. 2011, 17, 10745.
(a) Tabellion, F.; Peters, C.; Fischbeck, U.; Regitz, M.; Preuss, F. Chem. Eur. J. 2000, 6, 4558.
(b) Tabellion, F.; Nachbauer, A.; Leininger, S.; Peters, C.; Preuss, F.; Regitz, M. Angew. Chem., Int. Ed. 1998, 37, 1233.
(c) Böhm, D., Knoch, F.; Kummer, S.; Schmidt, U.; Zenneck, U. Angew. Chem., Int. Ed. 1995, 34, 198.
(d) Kobayashi, Y.; Hamana, H.; Fujino, S.; Ohsawa, A.; Kumadaki, I. J. Am. Chem. Soc. 1980, 102, 252.
(e) RKoner, A.; Pfeifer, G.; Kelemen, Z.; Schnakenburg, G.; Nyulászi, L.; Sasamori, T.; Streubel, R. Angew. Chem. 2017, 129, 9359.
(a) Märkl, G.; Matthes, D. Angew. Chem., Int. Ed. 1972, 11, 1019.
(b) Märkl, G.; Dorfmeister, G. Tetrahedron Lett. 1987, 28, 1093.
(b) Bourdieu, C.; Foucaud, A. Tetrahedron Lett. 1987, 28, 4673.
(c) Märkl, G.; Dietl, S.; Ziegler, M. L.; Nuber, B. Angew. Chem., Int. Ed. 1988, 27, 389.
(d) Appel, V. R.; Poppe, M. Angew. Chem. 1989, 101, 70.
(e) Avarvari, N.; Le Floch, P.; Mathey, F. J. Am. Chem. Soc. 1996, 118, 11978.
(f) Märkl, G.; Dorges, C. Angew. Chem. 1991, 103, 82.
(g) Avarvari, N.; Floch, P. L.; Ricard, L.; Mathey, F. Organometallics 1997, 16, 4089.
(h) Frison, G.; Sevin, A.; Avarvari, N.; Mathey, F.; Floch, P. L. J. Org. Chem. 1999, 64, 5524.
Becker, G. Z. Anorg. Allg. Chem. 1976, 423, 242. doi: 10.1002/(ISSN)1521-3749
(a) Becker, G. Mundt, O. Z. Anorg. Allg. Chem. 1978, 443, 53.
(b) Becker, G.; RUssler, M.; Uhl, W. Z. Anorg. Allg. Chem. 1981, 473, 7.
Issleib, K.; Schmidt, H.; Meyer, H. J. Organomet. Chem. 1978, 160, 47. doi: 10.1016/S0022-328X(00)91197-5
Yam, M.; Tsang, C. W.; Gates, D. P. Inorg. Chem. 2004, 43, 3719. doi: 10.1021/ic049796j
Klebach, T. C.; Lourens, R.; Bickelhaupt, F. J. Am. Chem. Soc. 1978, 100, 4886. doi: 10.1021/ja00483a041
Issleib, K.; Schmidt, H.; Wirkner, C. Z. Anorg. Allg. Chem. 1981, 473, 85. doi: 10.1002/(ISSN)1521-3749
Becker, G.; Uhl, W.; Wessely, H. J. Z. Anorg. Allg. Chem. 1981, 479, 41. doi: 10.1002/(ISSN)1521-3749
(a) Becker, G.; Becker, W.; Mundt, O. Phosphorus Sulfur 1983, 14, 267.
(b) Romanenko, V. D.; Ruban, A. V.; Chernega, A. N.; Povolotskii, M. I.; Antipin, M. Y.; Struchkov, Y. T.; Markovskii, L. N. Zh. Obsh. Khim. 1990, 59, 1718.
(c) Romanenko, V. D.; Ruban, A. V.; Povolotskii, M. I.; Polyachenko, L. K.; Markovskii, L. N. Zh. Obsh. Khim. 1986, 56, 1186.
(a) Oehme, H.; Leissring, E.; Meyer, H. Tetrahedron Lett. 1980, 21, 1141.
(b) Issleib, K.; Vollmer, R. Tetrahedron Lett. 1980, 21, 3483.
(a) Appel, R.; Casser, C.; Immenkeppel, M.; Knoch, F. Angew. Chem., Int. Ed. 1984, 23, 895.
(b) Appel, R.; Immenkeppel, M. Z. Anorg. Allg. Chem. 1987, 553, 7.
Miyake, H.; Sasamori, T.; Tokitoh, N. Angew. Chem. 2012, 124, 3514. doi: 10.1002/ange.v124.14
(a) Marinetti, A.; Mathey, F. Angew. Chem., Int. Ed. 1988, 27, 1382.
(b) Le Floch, P.; Marinetti, A.; Ricard, L.; Mathey, F. J. Am. Chem. Soc. 1990, 112, 2407.
(c) Le Floch, P.; Mathey, F. Synlett 1990, 171.
(d) Marinetti, A.; Le Floch, P.; Mathey, F. Organometallics 1991, 10, 1190.
Esfandiarfard, K.; Arkhypchuk, A. I.; Orthaber, A.; Ott, S. Dalton Trans. 2016, 45, 2201. doi: 10.1039/C5DT03686K
(a) Cummins, C. C.; Schrock, R. R.; Davis, W. M. Angew. Chem., Int. Ed. 1993, 32, 756.
(b) Breen, T. L.; Stephan, D. W. J. Am. Chem. Soc. 1995, 117, 11914.
(d) Masuda, J. D.; Jantunen, K. C.; Ozerov, O. V.; Noonan, K. J.; Gates, D. P.; Scott, B. L.; Kiplinger, J. L. J. Am. Chem. Soc. 2008, 130, 2408.
(a) Shah, S.; Protasiewicz, J. D. Chem. Commun. 1998, 1585.
(b) Wang, H.; Zhao, W.; Zhou, Y.; Duan, Z.; Mathey, F. Eur. J. Inorg. Chem. 2011, 4585.
Wright, V. A.; Gates, D. P. Angew. Chem. 2002, 114, 2495. doi: 10.1002/(ISSN)1521-3757
Wright, V. A.; Patrick, B. O.; Schneider, C.; Gates, D. P. J. Am. Chem. Soc. 2006, 128, 8836. doi: 10.1021/ja060816l
Smith, R. C.; Protasiewicz, J. D. J. Am. Chem. Soc. 2004, 126, 2268. doi: 10.1021/ja0394683
Wagner, O.; Maas, G.; Regitz, M. Angew. Chem. 1987, 99, 1328. doi: 10.1002/(ISSN)1521-3757
Regitz, M.; Heydt, H.; Wagner, O.; Schnurr, W.; Ehle, M.; Hoffmann, J. Phosphorus, Sulfur Silicon Relat. Elem. 1990, 49, 341. doi: 10.1080/10426509008038975
Sanchez, M.; Réau, R.; Marsden, C. J.; Regitz, M.; Bertrand, G. Chem. Eur. J. 1999, 5, 274. doi: 10.1002/(ISSN)1521-3765
Hitchcock, P. B.; Maah, M. J.; Nixon, J. F. J. Chem. Soc., Chem. Commun. 1986, 737.
Binger, P.; Glaser, G.; Albus, S.; Krüger, C. Chem. Ber. 1995, 128, 1261. doi: 10.1002/(ISSN)1099-0682
Heinemann, F. W.; Kummer, S.; Seiss-Brandl, U.; Zenneck, U. Organometallics 1999, 18, 2021. doi: 10.1021/om980862x
Ito, S.; Sugiyama, H.; Yoshifuji, M. Chem. Commun. 2002, 1744.
(a) Heinicke, J.; Tzschach, A. Z. Chem. 1980, 20, 342.
(b) Heinicke, J.; Tzschach, A. Phosphorus Sulfur 1985, 25, 345.
(d) Tian, R.; Zhang, C.; Xu, Y.; Liu, C.; Duan, Z.; Mathey, F. Chem. Eur. J. 2017, 23, 13006.
Niaz, B.; Ghalib, M.; Jones, P. G.; Heinicke, J. W. Dalton Trans. 2013, 42, 9523. doi: 10.1039/c3dt50981h
(a) Issleib, K.; Vollmer, R. Z. Anorg. Allg. Chem. 1981, 481, 22.
(b) Bansal, R. K.; Gupta, N.; Heinicke, J.; Nikonov, G. N.; Saguitova, F.; Sharma, D. C. Synthesis 1999, 264.
(a) Aluri, B. R.; Niaz, B.; Kindermann, M. K.; Jones, P. G.; Heinicke, J. Dalton Trans. 2011, 40, 211.
(b) Ghalib, M.; Niaz, B.; Jones, P. G.; Heinicke, J. W. Heteroat. Chem. 2013, 24, 452.
(c) Heinicke, J.; Gupta, N.; Surana, A.; Peulecke, N.; Witt, B.; Steinhauser, K.; Jones, P. G. Tetrahedron 2001, 57, 9963.
(d) Bansal, R. K.; Gupta, N.; Heinicke, J.; Nikonov, G. N.; Saguitova, F.; Sharma, D. C. Synthesis 1999, 264.
Huang, H.; Luo, H.; Tao, G.; Cai, W.; Cao, J.; Duan, Z.; Mathey, F. Org. Lett. 2018, 20, 1027. doi: 10.1021/acs.orglett.7b03971
(a) Schmidpeter, A.; Willhalm, A. Angew. Chem., Int. Ed. 1984, 23, 903.
(b) Märkl, G.; Trötsch, I. Angew. Chem., Int. Ed. 1984, 23, 901.
(c) Rösch, W.; Regitz, M. Angew. Chem., Int. Ed. 1984, 96, 898.
(a) Chia, S. P.; Li, Y.; So, C. W. Organometallics 2013, 32, 5231.
(b) Chapyshev, S. V.; Anisimov, V. M. Chem. Heterocycl. Compd. 1997, 33, 587.
(c) Choong, S. L.; Nafady, A.; Stasch, A.; Bond, A. M.; Jones, C. Dalton Trans. 2013, 42, 7775.
(d) Sklorz, J. A.; Hoof, S.; Sommer, M. G.; Weißer, F.; Weber, M.; Wiecko, J.; Müller, C. Organometallics 2014, 33, 511.
(e) Takeda, H.; Ishitani, O. Coord. Chem. Rev., 2010, 254, 346.
(f) Lowe-Ma, C. K.; Nissan, R. A.; Wilson, W. S. J. Org. Chem. 1990, 55, 3755
(g) Sklorz, J. A.; Hoof, S.; Rades, N.; De Rycke, N.; Koenczoel, L.; Szieberth, D.; Mueller, C. Chem. Eur. J. 2015, 21, 11096.
(a) Chapyshev, S. V.; Bergsträsser, U.; Regitz, M. Khim. Geterotsikl. Soedin. 1996, 1, 67.
(b) Märkl, G.; Troetsch-Schaller, I.; Hölzl, W. Tetrahedron Lett. 1988, 29, 785.
(c) Yeung Lam Ko, Y. Y. C.; Carrié, R. J. Chem. Soc., Chem. Commun. 1984, 1640.
(d) Bansal, R. K.; Gupta, N. Sci. Synth. 2014, 46, 754.
(e) Sklorz, J. A.; Mueller, C. Eur. J. Inorg. Chem. 2016, 2016, 595.
(f) Fuchs, E. P. O.; Hermesdorf, M.; Schnurr, W.; Rösch, W.; Heydt, H.; Regitz, M.; Binger, P J. Organomet. Chem. 1988, 338, 329.
Märkl, G. Angew. Chem. 1966, 78, 907.
(a) Märkl, G.; Lieb, F.; Merz, A. Angew. Chem. 1967, 79, 475.
(b) Weemers, J. J.; van der Graaff, W. N.; Pidko, E. A.; Lutz, M.; Müller, C. Chem. Eur. J. 2013, 19, 8991.
Ashe Ⅲ, A. J.; Shu, P. J. Am. Chem. Soc. 1971, 93, 1804. doi: 10.1021/ja00736a052
Avarvari, N.; Le Floch, P.; Mathey, F. J. Am. Chem. Soc. 1996, 118, 11978. doi: 10.1021/ja962736v
Hunter, R. A.; Whitby, R. J.; Light, M. E.; Hursthouse, M. B. Tetrahedron Lett. 2004, 45, 7633. doi: 10.1016/j.tetlet.2004.08.098
(a) Habicht, M. H.; Wossidlo, F.; Bens, T.; Pidko, E. A.; Müller, C. Chem. Eur. J. 2018, 24, 944.
(b) Rösch, W.; Regitz, M. Z. Naturforsch. B 1986, 41, 931.
(c) Wrackmeyer, B.; Klaus, U. Organomet. Chem. 1996, 520, 211.
(d) Ko, Y. Y. Y. L.; Carrié, R. J. Chem. Soc., Chem. Commun. 1984, 1640.
(e) Fink, J.; Rösch, W.; Vogelbacher, U. J.; Regitz, M. Angew. Chem. 1986, 98, 265.
(f) Fink, J.; Rösch, W.; Vogelbacher, U. J.; Regitz, M. Angew. Chem., Int. Ed. 1986, 25, 280.
(g) Blatter, K.; Rösch, W.; Vogelbacher, U. J.; Fink, J.; Regitz, M. Angew. Chem., Int. Ed. 1987, 26, 85.
Märkl, G.; Jin, G. Y.; Silbereisen, E. Angew. Chem., Int. Ed. 1982, 21, 370.
(a) Wang, H.; Li, C.; Geng, D.; Chen, H.; Duan, Z.; Mathey, F. Chem. Eur. J. 2010, 16, 10659.
(b) Chen, H.; Li, J.; Wang, H.; Liu, H.; Duan, Z.; Mathey, F. Eur. J. Inorg. Chem. 2011, 1540.
Regitz, M.; Binger, P. Angew. Chem. 1988, 100, 1541. doi: 10.1002/(ISSN)1521-3757
Mei, Y.; Wu, D. J.; Borger, J. E.; Grützmacher, H. Angew. Chem., Int. Ed. 2018, 57, 5512. doi: 10.1002/anie.v57.19
(a) Nakajima, K.; Takata, S.; Sakata, K.; Nishibayashi, Y. Angew. Chem. 2015, 127, 7707.
(b) Nakajima, K.; Liang, W.; Nishibayashi, Y. Org. Lett. 2016, 18, 5006.
Zhang, L.; Yang, F.; Tao, G.; Qiu, L.; Duan, Z.; Mathey, F. Eur. J. Inorg. Chem. 2017, 2355.
(a) Wang, L.; Zhang, L.; Shi, H.; Duan, Z.; Mathey, F. Synlett 2013, 24, 2006.
(c) Wang, L.; Wang, Z.; Wang, Q.; Duan, Z.; Mathey, F. Dalton Trans. 2015, 44, 3717.
(d) Huang, H.; Wei, Z.; Wang, M.; Duan, Z.; Mathey, F. Eur. J. Org. Chem. 2017, 5724.
(a) Märkl, G.; Matthes, D. Angew. Chem., Int. Ed. 1972, 11, 1019.
(b) Märkl, G.; Dorfmeister, G. Tetrahedron Lett. 1987, 28, 1093.
(c) Bourdieu, C.; Foucaud, A. Tetrahedron Lett. 1987, 28, 4673.
(d) Markl, G.; Dietl, S.; Ziegler, M. L.; Nuber, B. Angew. Chem., Int. Ed. 1988, 27, 389.
(e) Appel, V. R.; Poppe, M. Angew. Chem. 1989, 101, 70.
Huang, H.; Wei, Z.; Hou, J.; Wang, R.; Tao, G.; Wang, M.; Mathey, F. Eur. J. Org. Chem. 2018, 2018, 2863. doi: 10.1002/ejoc.201800492
(a) Kobayashi, Y.; Kumadaki, I.; Ohsawa, A.; Hamana, H. Tetrahedron Lett. 1976, 17, 3715.
(b) Kobayashi, Y.; Kumakaki, I.; Oshaea, A.; Hamana, H. Tetrahedron Lett. 1977, 10, 867.
(c) Kobayashi, Y.; Hamana, H.; Fujino, S.; Ohsawa, A.; Kumadaki, I. J. Am. Chem. Soc. 1980, 102, 252.
Böhm, D.; Knoch, F.; Kummer, S.; Schmidt, U.; Zenneck, U. Angew. Chem., Int. Ed. 1995, 34, 198. doi: 10.1002/(ISSN)1521-3773
(a) Hofmann, M. A., Heydt, H., Regitz, M. Synthesis 2001, 0463.
(b) Hofmann, M. A.; Bergsträßer, U.; Reiß, G. J.; Nyulászi, L.; Regitz, M. Angew. Chem., Int. Ed. 2000, 39, 1261.
(a) Binger, P.; Stannek, J.; Gabor, B.; Mynott, R.; Bruckmann, J.; Krüger, C.; Leininger, S. Angew. Chem., Int. Ed. 1995, 34, 2227.
(b) Tabellion, F.; Nachbauer, A.; Leininger, S.; Peters, C.; Preuss, F.; Regitz, M. Angew. Chem., Int. Ed. 1998, 37, 1233.
(c) Tabellion, F.; Peters, C.; Fischbeck, U.; Regitz, M.; Preuss, F. Chem. Eur. J. 2000, 6, 4558.
(d). Heydt, H. New Aspects in Phosphorus Chemistry Ⅱ, Springer, Berlin, Heidelberg, 2003, pp. 215~249.
(e) Trincado, M.; Rosenthal, A. J.; Vogt, M.; Grützmacher, H. Eur. J. Inorg. Chem. 2014, 1599.
Koner, A.; Pfeifer, G.; Kelemen, Z.; Schnakenburg, G.; Nyulászi, L.; Sasamori, T.; Streubel, R. Angew. Chem., Int. Ed. 2017, 56, 9231. doi: 10.1002/anie.201704070
Yruegas, S.; Barnard, J. H.; Al-Furaiji, K.; Dutton, J. L.; Wilson, D. J.; Martin, C. D. Organometallics 2018, 37, 1515. doi: 10.1021/acs.organomet.8b00248
Minami, T.; Okamoto, H.; Ikeda, S.; Tanaka, R.; Ozawa, F.; Yoshifuji, M. Angew. Chem., Int. Ed. 2001, 40, 4501. doi: 10.1002/1521-3773(20011203)40:23<4501::AID-ANIE4501>3.0.CO;2-K
Ozawa, F.; Yamamoto, S.; Kawagishi, S.; Hiraoka, M.; Ikeda, S.; Minami, T.; Yoshifuji, M. Chem. Lett. 2001, 30, 972. doi: 10.1246/cl.2001.972
Ozawa, F.; Okamoto, H.; Kawagishi, S.; Yamamoto, S.; Minami, T.; Yoshifuji, M. J. Am. Chem. Soc. 2002, 124, 10968. doi: 10.1021/ja0274406
Murakami, H.; Minami, T.; Ozawa, F. J. Org. Chem. 2004, 69, 4482. doi: 10.1021/jo049732q
Chang, Y. H.; Nakajima, Y.; Ozawa, F. Organometallics 2013, 32, 2210. doi: 10.1021/om4000743
Qiao, S.; Fu, G. C. J. Org. Chem. 1998, 63, 4168. doi: 10.1021/jo980624b
Tanaka, K.; Fu, G. C. J. Org. Chem. 2001, 66, 8177. doi: 10.1021/jo010792v
Shintani, R.; Fu, G. C. Org. Lett. 2002, 4, 3699. doi: 10.1021/ol026651c
(a) Shintani, R.; Fu, G. C. Angew. Chem. 2003, 115, 4216.
(b) Shintani, R.; Fu, G. C. J. Am. Chem. Soc. 2003, 125, 10778.
Knoch, F.; Kremer, F.; Schmidt, U.; Zenneck, U.; Le Floch, P.; Mathey, F. Organometallics 1996, 15, 2713. doi: 10.1021/om950800w
Le Floch, P.; Knoch, F.; Kremer, F.; Mathey, F.; Scholz, J.; Scholz, W.; Zenneck, U. Eur. J. Inorg. Chem. 1998, 119. doi: 10.1002/%28SICI%291099-0682%28199801%291998%3A1%3C119%3A%3AAID-EJIC119%3E3.0.CO%3B2-X
DiMauro, E. F.; Kozlowski, M. C. J. Chem. Soc., Perkin Trans. 1 2002, 439. https://www.researchgate.net/publication/239230674_ChemInform_Abstract_Phosphabenzenes_as_Electron_Withdrawing_Phosphine_Ligands_in_Catalysis
de Vaumas, R.; Marinetti, A.; Mathey, F. J. Organomet. Chem. 1991, 413, 411. doi: 10.1016/0022-328X(91)80066-S
Bauer, S.; Marinetti, A.; Ricard, L.; Mathey, F. Angew. Chem., Int. Ed. 1990, 29, 1166. doi: 10.1002/(ISSN)1521-3773
(a) Schnurr, W.; Regitz, M. Tetrahedron Lett. 1989, 30, 395.
(b) Yoshifuji, M.; Yoshimura, H.; Toyota, K. Chem. Lett. 1990, 19, 827.
(a) Bachrach, S. M. J. Org. Chem. 1993, 58, 541.
(b) Appel, R.; Barth, V.; Halstenberg, M. Chem. Ber. 1982, 115, 1617.
Abbari, M.; Cosquer, P.; Tonnard, F.; Ko, Y. Y. C. Y. L.; Carrié, R. Tetrahedron 1991, 47, 71. doi: 10.1016/0040-4020(91)80009-Q
Marinetti, A.; Ricard, L.; Mathey, F. Organometallics 1990, 9, 788. doi: 10.1021/om00117a039
(a) Yoshifuji, M.; Toyota, K.; Inamoto, N. Tetrahedron Lett. 1985, 26, 1727.
(b) Meriem, A.; Majoral, J. P.; Revel, M.; Navech, J. Tetrahedron Lett. 1983, 24, 1975.
Arduengo Ⅲ, A. J.; Carmalt, C. J.; Clyburne, J. A. C.; Cowley, A. H.; Pyati, R. Chem. Commun. 1997, 981.
Bates, J. I., Gates, D. P. Chem. Eur. J. 2012, 18, 1674. doi: 10.1002/chem.v18.6
Marinetti, A.; Mathey, F. J. Chem. Soc. Chem. Commun. 1990, 153.
(a) Van Der Knaap, T. A.; Klebach, T. C.; Visser, F.; Lourens, R.; Bickelhaupt, F. Tetrahedron 1984, 40, 991.
(b) Allspach, T.; Regitz, M.; Becker, G.; Becker, W. Synthesis 1986, 31.
(c) Le Floch, P.; Mathey, F. Tetrahedron Lett. 1989, 30, 817.
(d) Le Floch, P.; Ricard, L.; Mathey, F. Polyhedron 1990, 9, 991.
(e) van der Knaap, T. A. Bickelhaupt, F. Tetrahedron 1983, 39, 3189.
Märkl, G.; Seidl, E.; Trötsch, I. Angew. Chem., Int. Ed. 1983, 22, 879. doi: 10.1002/anie.198308791
Tsang, C. W.; Yam, M.; Gates, D. P. J. Am. Chem. Soc. 2003, 125, 1480. doi: 10.1021/ja029120s
(a) Tsang, C. W.; Baharloo, B.; Riendl, D.; Yam, M.; Gates, D. P. Angew. Chem. 2004, 116, 5800.
(b) Dugal-Tessier, J.; Serin, S. C.; Castillo-Contreras, E. B.; Conrad, E. D.; Dake, G. R.; Gates, D. P. Chem. Eur. J. 2012, 18, 6349.
Siu, P. W.; Serin, S. C.; Krummenacher, I.; Hey, T. W.; Gates, D. P. Angew. Chem. 2013, 125, 7105. doi: 10.1002/ange.201301881
Longobardi, L. E.; Russell, C. A.; Green, M.; Townsend, N. S.; Wang, K.; Holmes, A. J.; Stephan, D. W. J. Am. Chem. Soc. 2014, 136, 13453. doi: 10.1021/ja5077525
(a) Liu, L. L.; Zhou, J.; Cao, L. L.; Andrews, R.; Falconer, R. L.; Russell, C. A.; Stephan, D. W. J. Am. Chem. Soc. 2017, 140, 147.
(b) Liu, L. L.; Zhou, J.; Andrews, R.; Stephan, D. W. J. Am. Chem. Soc. 2018, 140, 7466.

图式 1 运用[1, 3]硅迁移(硅迁移到氧)合成膦烯化合物
Scheme 1 Synthesis of phosphaalkenes by [1, 3] migration (Si→O)

图式 2 运用[1, 3]硅迁移(硅迁移到氮)合成膦烯化合物
Scheme 2 Synthesis of phosphaalkenes by [1, 3] migration (Si→N)

图式 3 运用[1, 3]质子迁移合成膦烯化合物
Scheme 3 Synthesis of phosphaalkenes by [1, 3] proton transfer

图式 4 运用1, 2消除卤化氢合成膦烯化合物
Scheme 4 Synthesis of phosphaalkenes by elimination of hydrogen halide

图式 5 运用1, 2-消除TMSCl合成膦烯化合物
Scheme 5 Synthesis of phosphaalkenes by elimination of TMSCl

图式 6 运用“phospha-Peterson”反应合成膦烯
Scheme 6 Synthesize of phosphaalkenes by "phospha-Peterson" reaction

图式 7 运用类“phospha-Peterson”反应合成膦烯
Scheme 7 Synthesis of phosphaalkenes by similar "phospha-Peterson" reaction

图式 8 运用伯膦与缩醛的缩合反应合成膦烯化合物
Scheme 8 Synthesis of phosphaalkenes by condensation reaction with acetal

图式 9 运用伯膦与卤代烷的缩合反应合成膦烯化合物
Scheme 9 Synthesis of phosphaalkenes by condensation reaction with halides

图式 10 运用“phospha-Wittig-Horner”反应合成膦烯化合物
Scheme 10 Synthesis of phosphaalkenes by "phospha-Wittig-Horner" reaction

图式 11 运用“亚膦烯wittig”反应合成膦烯化合物
Scheme 11 Synthesis of phosphaalkenes by "phosphinidene-Wittig" reaction

图式 12 运用“phospha-Wittig ylide”反应合成膦烯化合物
Scheme 12 Synthesis of phosphaalkenes by "phospha-Wittig-ylide " reaction

图式 13 运用[1, 3]硅迁移方法合成膦烯聚合物
Scheme 13 Synthesis of polymeric phosphaalkenes by [1, 3] migration

图式 14 运用“phospha-Wittig-Horner”方法合成膦烯聚合物
Scheme 14 Synthesis of polymeric phosphaalkenes by "pho-spha-Wittig-Horner" reaction

图式 15 运用卡宾插入P=C合成磷杂环丙烯
Scheme 15 Synthesis of phosphacyclopropene by carbene inserting P=C

图式 16 运用卡宾插入膦炔合成磷杂环丙烯
Scheme 16 Synthesis of phosphacyclopropene by carbene inserting P≡C

图式 17 膦炔[2+2]环加成反应合成1, 3-二磷杂环丁二烯
Scheme 17 Synthesis of 1, 3-diphosphabutadiene by phospha-alkyne [2+2] cycloaddition reaction

图式 24 运用膦炔[4+2]反应合成磷杂苯
Scheme 24 Synthesis of phosphinines by [4+2] cycloaddition reaction

图式 25 运用五配位磷降解成三配位磷杂苯
Scheme 25 Synthesis of phosphinines by degradation of λ5 into λ3-phosphorus

图式 26 运用逆Diels-Alder反应合成1, 4-二磷杂苯
Scheme 26 Synthesis of 1, 4-diphosphinine by re-Diels-Alder reaction

图式 28 运用[2+2+2]反应合成1, 3, 5-三磷杂苯
Scheme 28 Synthesis of 1, 3, 5-triphosphinines by [2+2+2] cycloaddition reaction

图式 29 运用PBu3还原合成1, 4-二磷杂苯
Scheme 29 Synthesis of 1, 4-diphosphinine by reduction with PBu3

图式 30 运用磷炔插入硼碳键合成膦烯化合物
Scheme 30 Synthesis of 1, 3-boraphosphaalkene by the insertion of P≡C into B—C bond

图式 31 二亚膦基环丁烯化合物在催化反应的应用
Scheme 31 Application of diphosphinidenecyclobutenes as catalyst

图式 32 二亚膦基吡啶化合物在催化反应的应用
Scheme 32 Application of 1, 6-diphosphaalkenylpyridine as catalyst

图式 38 1, 3, 5-三磷杂苯化合物断裂氢气小分子的反应
Scheme 38 Cleavage of hydrogen molecule by 1, 3, 5-triphos-phinine

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:

















 下载:
下载: