

图式 1.
可能的机理
Scheme 1.
Proposed mechanism

含氧杂环化合物是分子的环状结构中含有一个或多个氧原子的有机化合物, 它们是自然界中一类种类较多且存在广泛的化合物, 也是许多具有生物活性天然和非天然化合物骨架中非常重要的结构单元[1~4]; 由于它们在生物活性方面表现出了独特的优势, 所以在医药、农药、功能材料和化工产品中, 都有该类化合物及其衍生物的存在.
氧杂环丙烷(俗称环氧乙烷)就是一类环内含有氧原子的三元杂环化合物, 它不仅存在于一些有机分子和天然分子中, 而且也是制备一些药物分子和天然产物的重要中间体[5~8].更为重要的是, 通过它及其衍生物与各种含有不同不饱和键的化合物发生的环加成反应就可以制备出种类更加丰富的含氧杂环化合物, 许多科研工作者在此方面已经做出了一定的贡献.本文将针对近10年来氧杂环丙烷与含有C=C, C≡C, C=N, C≡N和C=O等不饱和键的化合物发生的环加成反应进行了综述, 重点讨论了[3+2], [3+3], [4+3], [3+2+1]和[1+3]等环加成反应和研究进展.
氧杂环丙烷参与的[3+2]环加成一直受到了广泛的关注和重视, 且许多化学工作者对该类反应仍在不懈地探索开发中.他们利用氧杂环丙烷通过环内C—C键或C—O键的断裂分别与烯烃、炔烃、亚胺、腈类化合物、醛类化合物、异氰酸酯及CO2等含有不饱和键的化合物发生环加成来制备呋喃、噁唑烷、噁唑啉、噁唑烷酮和环状碳酸酯等衍生物.
呋喃衍生物, 包括四氢呋喃和2, 5-二氢呋喃衍生物, 是一类常见的含氧杂环化合物, 它们广泛地存在于天然产物和药物中.自1998年, Yamamoto等[9]首次报道了环氧乙烷衍生物与烯烃环加成合成四氢呋喃衍生物的反应之后, 许多课题组均对其与烯烃或炔烃的环加成进行了研究, 而且该类反应正朝着不对称催化的方向发展.它们遵循的反应规律一般都是烯烃或炔烃在催化剂的作用下, 一个碳原子先对氧杂的一个碳原子进行亲核开环反应, 通常是苄基位碳原子优先被进攻的原则, 在这个过程中, 由于底物取代基的不同会发生C—C键断裂或者C—O键断裂, 最后再关环而得到反应产物.
2005年, Hilt课题组[10]在FeCl2催化作用下, 用苯基环氧乙烷与单边双取代烯烃发生的[3+2]环加成合成了四氢呋喃衍生物(Eq. 1).该催化体系使用锌粉作还原剂, 三乙胺作碱, 三苯基膦(PPh3)和氮杂环卡宾(NHC)作双组分配体, 乙腈作溶剂于60 ℃条件下进行.对底物广普性研究发现, 所考察的底物仅得到了60%以下的收率; 但是, 该反应具有良好的化学和区域选择性, 反应产物主要是由烯烃的端位碳原子对环氧烷烃的苄基位进行亲核开环然后再关环而得到的.值得注意的是, 在缺少FeCl2或者Zn粉的条件下, 反应几乎得不到产物, 当体系中缺少三乙胺时, 产物收率大大降低, 而且二分子苯基环氧乙烷的缩合产物成为了主产物.
|
|
(1) |
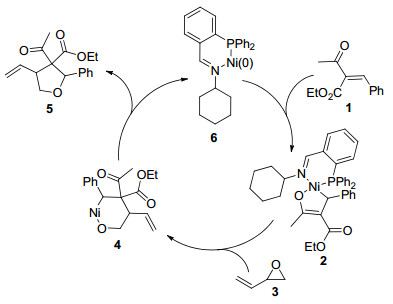
2011年, Matsubara课题组[11]报道了在配体L1的协助下, Ni(cod)2催化作用下乙烯基环氧乙烷与α, β-不饱和酮发生[3+2]环加成来制备四氢呋喃衍生物的反应(Eq. 2).该反应使用10 mol%的Ni(cod)2作催化剂, 20 mol%的L1作配体, 以甲苯为溶剂, 在30 ℃下进行.对底物广普性研究表明:当R3和R4均为甲基时, 反应没有得到环加成产物; 当R2为苯基、4-甲氧基苯基、4-三氟甲基苯基和1-萘基时, 反应取得51%~81%的收率; 当R2为正丙基时, 反应取得了71%的收率.机理研究认为:首先, 路易斯酸Ni(cod)2和配体L1络合形成的镍催化剂6的中心金属镍与不饱和酮1进行环化形成五元环中间体2, 接着, 该中间体与乙烯基环氧乙烷3发生亲核加成得到六元环中间体4, 这个过程中主要是酯基的羰基α碳对环氧乙烷中乙烯基所连的碳进行亲核开环发生C—O键断开再络合而得到的, 最后中间体4经过还原消除得到目标产物5, 同时镍催化剂6再生(Scheme 1).
|
|
(2) |
2012年, Zhang课题组[12]报道了Ni(ClO4)2•6H2O催化下芳基环氧乙烷衍生的二酮或二酯通过环内C—C键断裂与吲哚类化合物发生[3+2]环加成合成1H-呋喃并[3, 4-b]吲哚衍生物的反应(Eq. 3).该反应在室温下以二氯甲烷(DCM)作溶剂, 4 MS作添加剂.底物普适性探究表明:该反应具有较高的化学选择性和区域选择性, 反应产物是由吲哚的C(3)原子对氧杂的苄基碳进行亲核开环接着发生C—C键断开再关环而形成的.当R1为缺电子基取代的苯基时, 反应收率优于其为供电子基取代的苯基; 对吲哚底物考察表明, 当R3为烷基, R4为烷基或者氢时, 反应取得66%~81%的收率.值得注意的是, 当R3=Me、R4=Boc时, 由于叔丁氧羰基(Boc)的吸电子效应, 该反应没有得到相应的产物.
|
|
(3) |
Hou课题组[12~15]使用Pd2(dba)3•CHCl3作催化剂, 在不同配体的作用下, 分别于2012、2017和2018年完成了三例乙烯基型环氧乙烷与不同吸电子官能团取代的烯烃的[3+2]环加成反应. 2012年, 他们[13]使用二茂铁衍生的噁唑啉L2作配体, 四氢呋喃(THF)作溶剂于室温条件下完成了1-乙烯基-1-甲基环氧乙烷与硝基烯发生的[3+2]环加成反应(Eq. 4).对硝基烯进行拓展表明:所考察的底物取得了47%~99%的收率, 当R为对位取代的苯基时, 电子效应对反应的非对映选择性和对映选择性没有影响, 且当R为供电子取代基时, 反应收率比较低; 当R为3-MeOC6H4时, 反应具有较高的收率和选择性, 而当R的苯环上带有邻位吸电子基时, 反应得到较低的非对映和对映选择性, 说明R官能团的立体位阻对产物选择性影响不大; 当R为呋喃基时, 反应取得了77%的收率和较高的选择性.此外, 当R为n-Pr时, 反应也可得到75%的收率, 但选择性比较低.
|
|
(4) |
2017年, 他们[14]使用5 mol%的L3作配体, 二氧六环作溶剂于15 ℃条件下完成了乙烯基型环氧乙烷与α, β-不饱和酮的不对称[3+2]环加成反应(Eq. 5).当R1和R2均为氢时, 对不饱和酮普适性探究表明:底物大都取得了≥70%的收率, 当R3为异丙基时, 产物收率仅为44%.值得注意的是, 芳基Ar苯环上取代基的电子效应对产物收率产生了一定的影响, 对位引入供电子基时, 降低了反应的收率和活性.当Ar为苯基, R3为甲基时, 乙烯基型环氧乙烷底物取得了69%~85%的收率.重要的是, 该反应所考察的大多数底物均取得了>90:10的非对映选择性和>95%的对映选择性.
|
|
(5) |
2018年, 他们[15]仍使用6 mol%的L3作配体, THF作溶剂于-25 ℃条件下完成了乙烯基型环氧乙烷与α, β-二取代硝基烯发生的[3+2]环加成反应(Eq. 6).对α, β-二取代硝基烯普适性探究表明:所考察的底物收率在45%~96%之间, 非对映选择性在20:1~1:1之间且对映选择性均≥86%.相较于之前的2篇报道, 该反应使用了三取代的烯烃且R2取代基位阻较大, 但是反应收率和对映选择性没有受到影响.
|
|
(6) |
Feng课题组[16, 17]利用他们自主开发的双氮氧化合物作配体, 在Ni催化剂的催化作用下, 完成了两例三取代的环氧乙烷与含C=C键类的化合物发生的[3+2]环加成. 2015年, 他们[16]使用10 mol%的L4-Ni(ClO4)2• 6H2O(物质的量之比为1.05:1)和15 mol%的LiNTf2作催化剂, 1, 1, 2, 2-四氯乙烷(TCE)作溶剂, 4 MS作添加剂, 实现了芳基环氧乙烷衍生的二酯通过环内C—C键断裂与吲哚类化合物经过[3+2]环加成合成呋喃衍生物的反应(Eq. 7).对底物考察发现不同取代基的吲哚均取得了78%~98%的收率、56%~91%的对映选择性和51:49~>95:5的非对映选择性.其中当Br在吲哚的C(4)或C(5)位置上时, 反应的非对映选择性相对较低.
2016年, 他们[17]使用10 mol%的L5-Ni(BF4)2•6H2O (物质的量之比为1.1:1)和LiNTf2作催化剂, 1, 2-二氯乙烷(DCE)作溶剂, 4 MS作添加剂, 在35 ℃条件下完成了环氧乙烷通过环内C—C键断裂与杂原子取代的烯烃发生催化不对称[3+2]环加成合成手性四氢呋喃衍生物的反应(Eq. 8).对环氧乙烷进行拓展表明:当R1为供电子基取代的苯基时, 反应取得了93%~99%的收率、>88%的对映选择性和>89/11的非对映选择性; 对于大多数吸电子基取代的苯基, 结果也是如此; 但是, 当R1为2-F或3-F取代的苯基时, 即使加入2 equiv.的正丁基乙烯基醚或延长反应时间, 反应也仅能得到66%~71%的收率.对烷基杂原子取代的烯烃扩展表明:反应可取得88%~99%的收率、86%~99%的对映选择性和73/27~91/9的非对映选择性.
|
|
(7) |
|
|
(8) |
2016年, Hilinski课题组[18]考察了Sc(OTf)3催化作用下环氧乙烷衍生物通过C—O键断裂与烯烃发生的[3+2]环加成反应(Eq. 9).对环氧乙烷广普性研究表明:产物主要是由烯烃的端位碳原子对环氧烷烃的苄基位进行亲核开环然后再关环而得到的, R1空间位阻对收率有较大的影响, 苯环邻位有取代基的芳基环氧乙烷收率低于苯环对位有取代基的环氧乙烷; 对烯烃广普性研究表明:当R3为甲基、R2为4-环己基取代的苯基时, 反应收率可达71%;但是, 当R2为其它官能团取代的苯基时, 反应收率均低于50%.该反应的优点是反应时间较短、条件温和、催化剂用量低且不需要额外的试剂或添加剂, 但是该反应收率较低且非对映选择性也不高.
|
|
(9) |
2009年, Pale课题组[19]报道了在Ag(Ⅰ)催化剂催化作用下炔基环氧乙烷通过分子内发生的[3+2]环加成合成呋喃衍生物的反应(Eq. 10).通过条件筛选确定以AgOTf/p-TsOH (5 mol%)作催化剂, CH2Cl2与MeOH (V:V=9:1)作混合溶剂, 室温条件下反应24 h为最优条件.对底物的考察表明大多数底物都取得了52%~92%的收率, 值得注意的是, 当R1R2为环戊基, R3为正己基时, 没有检测到相应的产物; 当R1R2为环己基, R3为苯基时, 反应收率仅为11%;当R3为H时, 也没有得到相应的产物.这是截止2009年报道的第一例关于Ag(Ⅰ)盐催化的合成多取代呋喃的cascade反应.
|
|
(10) |
2011年, Zhang课题组[20]在温和的条件下, 以Sc(OTf)3作催化剂, 实现了环氧乙烷通过环内C—C键选择性断裂与富电子炔烃发生[3+2]环加成合成2, 5-二氢呋喃衍生物的反应(Eq. 11).通过条件筛选发现以5 mol%的Sc(OTf)3作催化剂, 1, 2-二氯乙烷(DCE)作溶剂, 4 MS作添加剂, 室温条件下反应取得最优收率.对炔烃进行扩展表明反应大都取得了82%~98%的收率, 值得注意的是, 当R3和R4为4-MeOC6H4时, 反应取得中等的收率; 对环氧乙烷衍生物普适性的考察发现多数底物都取得了>85%的收率; 但当把两个羰基变为酯基时, 产物收率将降至60%以下.
|
|
(11) |
2012年, Mongin课题组[21]报道了利用2, 2-二氰基- 3-芳基环氧乙烷与炔烃化合物发生的[3+2]环加成合成2, 5-二氢呋喃衍生物的反应(Eq. 12).该反应是在甲苯回流条件下进行的.在苯乙炔和丙炔酸甲酯分别参与的反应中, 考察了氯化铟对反应结果的影响, 当使用苯乙炔作原料时, 氯化铟不仅可缩短反应时间, 而且能提高反应的区域选择性; 当使用丙炔酸甲酯作原料时, 氯化铟仅能缩短反应时间, 除了R1为甲氧基时, 反应的区域选择性提高外, 其它均降低.为了解释这些反应过程中的反应活性和区域选择性, 作者利用密度泛函(DFT)方法从理论上研究了2, 2-二氰基-3-苯基环氧乙烷衍生的羰基叶立德中间体分别与丙炔酸甲酯或丁-2-炔酸甲酯之间的热化学反应.
|
|
(12) |
2014年, Feng课题组[22]报道了在N, Nꞌ-二氧化物-Ni(Ⅱ)络合物催化作用下, 环氧乙烷通过C—C键断裂与炔烃发生[3+2]环加成合成手性呋喃衍生物的反应(Eq. 13).该反应以L6作配体, NiClO4•6H2O与LiNTf2作共催化剂, 1, 1, 2, 2-四氯乙烷作溶剂, 4 MS作添加剂, 于30 ℃条件下进行, 所考察的底物取得了89%~95%的对映选择性.对环氧乙烷普适性的研究表明, R1苯环上取代基的电子效应对反应收率的影响较大, 供电子基比吸电子基能得到更高的收率, 稠环和芳香杂环取代的环氧乙烷取得了81%~95%的收率, 所考察的炔烃底物均取得了85%~96%的收率.此外, 在此催化体系作用下, 烯烃与环氧乙烷也可以通过不对称[3+2]环加成制得具有光学活性的四氢呋喃衍生物.
|
|
(13) |
噁唑类衍生物具有良好的抗菌、消炎和杀灭植物病毒等功效, 在医药和农药等领域应用较广, 是新药研发的重要领域之一, 其合成方法的研究深受研究者的关注.经过努力, 人们已经研究出了多种合成方法, 其中, 包括氧杂环丙烷与含氮不饱和键化合物的环加成反应.
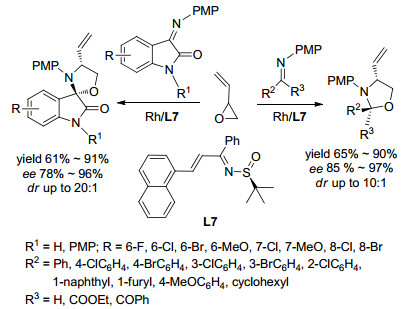
2012年, Du课题组[23]报道了在Rh和Ag催化剂催化作用下乙烯基环氧乙烷与亚胺发生[3+2]环加成合成噁唑烷衍生物的反应(Scheme 2).该反应使用[Rh-(C2H4)2Cl]2和AgOTf作共同催化剂, L7作配体, 乙酸乙酯作溶剂.对底物广普性探究发现:无论吲哚醌亚胺取代基R是供电子基还是吸电子基, 反应均得到了相应的产物, 其收率在61%~91%之间, 非对映选择性在10:1~20:1之间, 对映选择性在78%~96%之间.对于醛或酮衍生的亚胺, 反应收率在65%~90%之间, 对映选择性都大于85%, 非对映选择性可达10:1.
2013年, Zhang课题组[24]报道了Ni(ClO4)2•6H2O催化作用下环氧乙烷通过C—C键断裂与亚胺发生[3+2]环加成合成多取代的2, 4-反式噁唑烷衍生物的反应(Eq. 14).该反应在室温条件下, 以DCE作溶剂, 4 MS作添加剂, L8作配体.当R2和R3均为苯基时, 对环氧乙烷研究表明, 大多数底物都取得了80%~95%的收率和中等到优秀的非对映选择性.当Ar为苯基, R1为甲基时, 对亚胺研究表明, 如果R2为1-萘基时, 反应取得了95%的收率, 其它底物得到了70%~84%的收率.机理研究表明添加配体L8不仅能提高路易斯酸的催化活性, 而且也能提高反应的选择性.对照实验则表明, 在路易斯酸催化下, 环氧乙烷上的酮或者酯羰基对于提高环氧乙烷C—C键断裂的化学选择性是至关重要的.
|
|
(14) |
2017年, Guo课题组[25]在Pd催化剂催化作用下实现了乙烯基环氧乙烷与氨基磺酸酯衍生的环状亚胺的[3+2]环加成, 合成了噁唑烷衍生物的反应(Eq. 15).该反应以2.5 mol%的Pd2(dba)3•CHCl3作催化剂, 7.5 mol%的L9作配体, 5 mol%的四丁基溴化铵(TBAB)作添加剂, DCM作溶剂于室温条下进行.当R2为氢时, 对环状亚胺普适性的探究表明无论R1是富电子基还是缺电子基, 反应取得了86%~97%的收率和>20:1的非对映选择性.当R1为氢时, 对乙烯基环氧乙烷考察表明:如果R2为甲基时, 反应取得了92%的收率和5:3的非对映选择性; 如果R2为环己基时, 反应取得了37%的收率和>20:1的非对映选择性.
|
|
(15) |
2018年, Guo课题组[26]报道了Sc(OTf)3催化作用下, 氧杂环丙烷通过C—C键断裂与苯并唑类衍生物发生的[3+2]环加成反应(Eq. 16).该反应以10 mol%的Sc(OTf)3作催化剂, 4 MS作添加剂, DCE作溶剂于室温条件下进行.当Ar为苯基, R2为乙基, X为硫时, 对苯并唑类广普性研究表明, 所考察的底物均取得了82%~95%的收率.当R1为氢, X为硫时, 对氧杂环丙烷广普性研究表明, 所考察的底物取得了53%~96%的收率.并且Ar苯环上带有吸电子基的环氧乙烷收率明显低于苯环上带有供电子基的环氧乙烷.
|
|
(16) |
2018年, Vidal课题组[27]报道了在Yb(OTf)3的催化作用下利用环氧乙烷与烯酮亚胺的[3+2]环加成合成噁唑烷衍生物的反应(Eq. 17).该反应以20 mol%的Yb(OTf)3作催化剂, 4 MS作添加剂, DCE作溶剂于室温下进行.对底物广普性研究表明, 所考察的底物收率在47%~97%之间.但是, 本文未对Ar1和Ar2是邻位取代的苯基的情况进行考察, 对其是强吸电子基取代的苯基的情况也未进行考察.
|
|
(17) |
3-噁唑啉是一类相对罕见的化合物, 它的衍生物如2, 4, 5-三甲基-3-噁唑啉已被分离并当作食用香精应用到各种食物中.该类化合物已报道的合成方法较少, 主要包括1, 3-噁唑烷的氧化消除、脂肪醛与2-芳基-2-叠氮醇的Boyer反应等. 2015年, Zhong课题组[28, 29]分别在TfOH和SnCl4的催化作用下, 报道了芳基环氧乙烷与腈类化合物通过[3+2]环加成合成一系列3-噁唑啉衍生物的反应(Eq. 18).在TfOH催化体系中, 当R2为苯基时, 对环氧乙烷底物研究表明:如果Ar是供电子基或弱吸电子基取代的苯基时, 不论R1是甲基或乙基, 反应均可取得≥86%的收率; 如果Ar是强吸电子基取代的苯基或者R1是苄基时, 反应取得75%左右的收率.当Ar为苯基, R1是甲基时, 对腈类化合物的广普性研究表明, 芳香腈可以观察到同样的反应规律和结果, 而脂肪腈也可取得70%~80%的收率.对于SnCl4催化体系, 所有考察的底物收率均较TfOH体系低10%左右.推测的机理为:首先, H+或路易斯酸(LA)与环氧乙烷7的两个羰基配位形成六元环过渡态8, 然后, 通过环氧乙烷环内C—C键断裂生成羰基叶立德中间体10, 接着, 腈类化合物9的氮原子对中间体的碳原子进攻产生1, 5-偶极子11, 进而关环形成目标产物12 (Scheme 3).
|
|
(18) |
1, 3-二氧环戊烷衍生物可用作抗感染的心血管免疫剂.利用氧杂环丙烷通过C—C或C—O键断裂形成羰基叶立德, 再与醛类化合物反应生成1, 3-二氧环戊烷衍生物, 这是最直接和最经济合成1, 3-二氧环戊烷衍生物的方法.
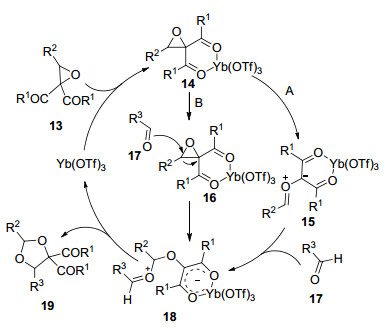
2011年, Zhang课题组[30]以Yb(OTf)3作催化剂, 实现了芳基环氧乙烷通过C—C键断裂与醛类化合物发生[3+2]环加成合成1, 3-二氧环戊烷衍生物的反应(Eq. 19).该反应催化剂用量为5 mol%, 以4 MS作添加剂, DCM作溶剂于室温条件下进行.对醛底物研究表明, 无论是吸电子基还是供电子基取代的芳香醛, 均取得了80%~97%的收率, 脂肪醛取得了90%~96%的收率.值得注意的是, 该反应没有检测到其它异构体产物.对环氧乙烷普适性研究表明, 所考察的底物均取得了大于82%的收率.机理推测认为:首先, Yb(OTf)3与环氧乙烷13的二个酮羰基进行络合得到六元环中间体14, 接着, 该中间体通过A或B两种途径促使环氧乙烷环内C—C键发生断裂形成中间体15或16, 然后, 中间体16直接变成中间体18, 进而形成产物19, 而中间体15再与醛发生反应即可得到六元环中间体18, 进而形成产物19 (Scheme 4).
|
|
(19) |
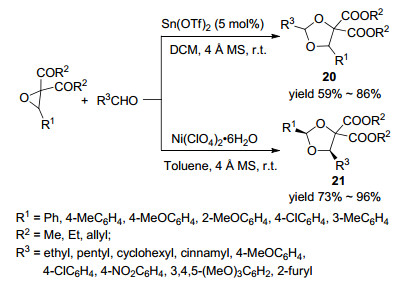
2012年, Zhang课题组[31]报道了路易斯酸催化作用下芳基环氧乙烷分别通过环内C—C键和C—O键断裂与醛发生的[3+2]环加成合成1, 3-二氧环戊烷衍生物的反应(Scheme 5).当该反应以Sn(OTf)2作催化剂, DCM作溶剂时, 环氧乙烷通过C—O键断裂形成产物20; 而以Ni(ClO4)2•6H2O作催化剂, 甲苯作溶剂时, 环氧乙烷则通过C—C键断裂而形成产物21.在相应条件下, 对环氧乙烷底物考察发现, 对于同样的原料来说, 产物21比产物20取得了更高的收率和非对映选择性; 然而, Ni(Ⅱ)催化的反应比SnⅡ)催化的反应速度慢很多.对醛进行普适性研究表明:可以得到同样的反应规律, 产物21比产物20取得了更高的收率和非对映选择性, 而且无论是带有供电子基或吸电子基的芳香醛还是脂肪族醛, 均取得了70%~96%的收率和1:1~>99:1的非对映选择性.
2014年, Feng课题组[32]采用N, Nꞌ-二氧化物-Gd(Ⅲ)络合物作催化剂, 实现了环氧乙烷通过C—C键断裂与芳香醛发生[3+2]环加成合成手性1, 3-二氧环戊烷衍生物的反应(Eq. 20).该反应使用Gd(OTf)3作催化剂, LiNTf2作辅助催化剂, L10作配体, 1, 2-二氯苯作溶剂, 4 MS作添加剂于0 ℃条件下进行.对环氧乙烷底物探究表明无论是吸电子基或供电子基取代的芳基、稠芳基还是杂芳基, 反应均获得了90%~99%的收率和77%~90%的对映选择性.但是, 如果R1是带有吸电子基的芳基, 则反应需要更高的温度.对醛类化合物的考察表明无论是吸电子基或供电子基取代的芳香醛还是杂芳香醛, 反应均取得了大于92%的收率和80%~91%的对映选择性.
烯基酰胺、氨基甲酸酯、氨基醇和氮杂环丙烷等作为原料都可以用来制备多种噁唑烷酮衍生物, 但是, 有些反应条件较为苛刻或需要经过多步反应才能得到目标产物.而通过氧杂环丙烷与异(硒)氰酸酯反应合成噁唑烷酮衍生物的方法仅需一步即可, 并且反应条件比较温和.
|
|
(20) |
2008年, Janssen课题组[33]在酶催化作用下完成了环氧乙烷与NaOCN发生[3+2]环加成合成手性5-官能团取代的噁唑烷酮衍生物的反应(Eq. 21).对底物普适性研究发现, 所有考察的12个底物中仅有5个底物得到了相应的产物, 收率在44%~77%之间, 对映选择性在69%~98%之间, 带有其它取代基的环氧乙烷收率非常低, 甚至没有得到相应的产物.这是截止到2008年, 第一例生物酶催化的环氧乙烷转化合成噁唑烷酮的反应.
|
|
(21) |
2010年, Phillips课题组[34]报道了Yb催化剂催化作用下2-环氧乙烷磷酸二乙酯与芳基异氰酸酯发生[3+2]环加成合成手性5-官能团取代的2-噁唑烷酮衍生物的反应(Eq. 22).该反应使用YbCl3•6H2O作催化剂, L11作配体, DCM作溶剂.对于4-取代的Ar来说, 反应产物得到的区域选择性均大于95%, 对映选择性均小于等于75%, 收率多数在60%左右.此外, 通过实验得出, 反应在低温下进行有利于该不对称诱导, 但不利于对位有取代基的芳基异氰酸酯参与反应时的区域选择性.
|
|
(22) |
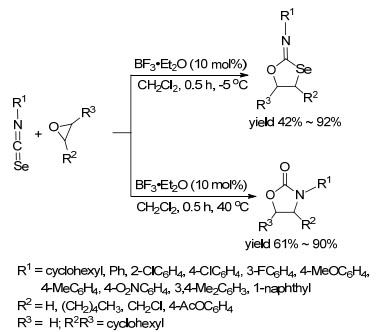
2012年, Punniyamurthy课题组[35]报道了在BF3• Et2O催化作用下环氧乙烷与异硒氰酸酯的[3+2]环加成反应(Scheme 6).该反应以DCM作溶剂, 反应过程中温度的作用比较大, 在-5 ℃下生成2-亚氨基-1, 3-氧杂硒烷衍生物, 在40 ℃下生成1, 3-噁唑烷酮衍生物.对异硒氰酸酯广普性研究表明, 带有供电子基的苯环、稠环或者杂芳环的芳基异硒氰酸酯容易发生反应, 并取得较高的收率, 且前者条件下的产物收率明显高于后者条件下的产物收率; 但当R1为4-NO2C6H4时, 两个条件下均没有发生环加成反应.对环氧乙烷进行底物扩展表明, 所考察的底物在两种条件下获得的产物均取得了62%~86%的收率.
2012年, Zhang课题组[36]报道了在无溶剂条件下, 以MgI2•(Et2O)n (30 mol%)作催化剂, 手性环氧乙烷与芳基异氰酸酯发生[3+2]环加成制备手性噁唑烷酮衍生物的反应(Eq. 23).对底物广普性研究表明:芳基异氰酸酯对环加成产物的收率没有明显的影响, 所得到的产物收率在88%~94%之间.此外, 该方法已被应用于抗菌药物利奈唑胺22的合成中.
2013年, North课题组[37]利用双核Al催化剂催化环氧乙烷与异氰酸酯的[3+2]环加成反应来制备噁唑烷酮衍生物(Eq. 24).该反应是在Cat. 1催化作用下, 以甲苯为溶剂于80 ℃条件下进行的.作者以苯基和脂肪族环氧化物为代表性实例, 分别考察了它们与多种芳香族异氰酸酯的反应, 结果表明, 氧化苯乙烯参与反应得到3, 4-噁唑烷酮23与3, 5-噁唑烷酮24的收率比例约为2:1, 相比之下, 脂肪族环氧乙烷与芳基异氰酸酯反应得到的产物主要以3, 5-噁唑烷酮为主, 但反应收率都不高且带有富电子基的苯基取代的异氰酸酯获得的产物收率高于带有缺电子基的苯基取代的异氰酸酯.假设的机理认为反应过程涉及到Al—O—Al键的断开和重新形成, 并且作者对这个假设使用单金属催化剂也进行了验证.
|
|
(23) |
|
|
(24) |
2014年, North课题组[38]在Ⅴ催化剂的催化作用下完成了环氧乙烷与异氰酸酯通过[3+2]环加成合成噁唑烷酮衍生物的反应(Eq. 25).该反应中Ⅴ催化剂Cat. 2和助催化剂四丁基溴化铵(TBAB)用量均为2 mol%, 甲苯作溶剂, 反应温度为80 ℃.与前一篇使用双核铝催化剂的报道相比, 该报道中催化剂量可以从5 mol%降至2 mol%, 反应时间从24 h减少到5 h.二者的另一区别是钒基催化剂使用了TBAB作助催化剂.对底物广普性考察表明, 不同取代基的环氧乙烷与芳香族异氰酸酯均可得到噁唑烷酮, 反应转化率在34%~98%之间, 而主要异构体产物收率在23%~89%之间.
|
|
(25) |
2014年, Nguyen课题组[39]报道了在Cr(Ⅲ)与PPh3O共同催化作用下环氧乙烷与异氰酸酯发生[3+2]环加成合成5-噁唑烷酮衍生物的反应(Eq. 26).该反应以Cat. 3作催化剂, DCM作溶剂于室温条件下进行.对底物广普性研究表明:烷基或芳基取代的环氧乙烷参与反应时, 反应得到了单一异构体产物且均取得了97%~100%的收率; 当R1为Ph时, 尽管反应产物总收率为99%, 但相比其它取代基, 主产物的选择性较低(主:副=63:36);对异氰酸酯的考察表明芳香族异氰酸酯取得了85%~97%的收率, 但是烷基异氰酸酯几乎没有得到相应的产物.
|
|
(26) |
环丙烷、氮杂环丙烷及氧杂环丙烷均可与氧杂环丙烷发生环加成反应来制备五元含氧杂环化合物.
2013年, Zhang课题组[40]报道了通过两种不同官能团取代的环氧乙烷发生[3+2]环加成来制备1, 3-二氧环戊烷的反应(Eq. 27).该反应以Ni(ClO4)2•6H2O作催化剂, 正戊烷作溶剂, 4 MS作添加剂于室温条件下进行.底物考察显示所考察的芳香族环氧乙烷得到了55%~75%的收率和大于20:1的非对映选择性.但当R1为4-MeOC6H4时, 环氧乙烷由于催化剂的作用不稳定, 生成的产物是复杂的混合物; 当R1为4-MeC6H4, R2为Me, R3为4-MeOC6H4时, 产物收率仅为15%, 而当R3为n-Bu时, 反应不能发生.推测的机理认为:在Ni(Ⅱ)催化剂催化作用下环氧乙烷25发生Meinwald重排形成醛中间体26, 而另一种环氧乙烷27两个酯基的羰基部分与Ni(ClO4)2•6H2O结合形成六元环中间体28, 最后, 两种中间体26与28发生环加成进而形成目标产物29 (Scheme 7).
|
|
(27) |
2015年, Banerjee课题组[41]报道了在InCl3催化作用下环丙烷与环氧乙烷发生[3+2]环加成合成四氢呋喃衍生物的反应(Eq. 28).该反应使用10 mol%的InCl3作催化剂, DCE作溶剂, 4 MS作添加剂.主要对带有富电子基的苯基取代的环丙烷进行了考察, 它们大都获得了中等的收率, 呋喃取代的环丙烷参与反应时, 没有得到目标产物.对环氧乙烷的考察表明带有4-甲氧基苯基的环氧乙烷作原料时, 反应取得了88%的收率, 而吸电子基取代的芳基环氧乙烷和脂肪族基团取代的环氧乙烷参与反应时均未得到目标产物.
|
|
(28) |
2016年, Banerjee课题组[42]报道了在三氟化硼乙醚催化作用下N-对甲苯磺酰基氮杂环丙烷与环氧乙烷在二氯甲烷中于室温条件下发生[3+2]环加成来合成噁唑烷衍生物的反应(Eq. 29).对环氧乙烷广普性研究发现:对于1, 1-双取代的环氧乙烷来说, 反应能得到总收率为69%~90%的顺反异构体两种产物, 且选择性总体不高, 其中, 顺反异构体产物的选择性是由1, 1-二取代环氧乙烷上甲基的顺式或反式选择性而导致的; 单取代环氧乙烷与1, 1-双取代环氧乙烷的反应规律相似.
|
|
(29) |
二氧化碳(CO2)是主要的温室气体, 随着可持续经济的发展, 控制二氧化碳的排放已成为全球性战略目标, 将其回收转化成高附加值的有机化合物是一个较好的方法.其中, 氧杂环丙烷与CO2反应制备环状碳酸酯就是一种有效的方法, 未来该类反应在可回收催化剂或无催化剂方面的研究仍然是热点和重点.
2010年, Kleij课题组[43]以TBAI作助催化剂, 实现了在锌络合物Cat. 4催化作用下环氧乙烷与CO2发生[3+2]环加成合成环状碳酸酯衍生物的反应(Eq. 30).对底物广普性考察表明单取代的环氧乙烷取得了65%~94%的收率, 双取代的环氧乙烷没有或微量得到相应的目标产物.
Zhang课题组[44~46]分别利用壳聚糖(CS)功能化的离子液体Cat. 5, L-色氨酸/KI催化体系Cat. 6和以硫脲为中心的双功能离子液体(TBILs) Cat. 7完成了环氧乙烷与CO2的环加成反应(Eq. 31).其中壳聚糖催化体系中CO2压力为2 MPa, 反应温度为120 ℃.所考察的底物取得了76%~99%的收率和大于94%的选择性.该催化剂作为一个可循环利用的生物聚合物负载的催化剂可以回收并重复使用5次而产物的选择性基本不变.催化机制可能为:壳聚糖在反应中发挥了双功能作用, 氢键辅助的环氧乙烷开环和亲核性叔胺诱导的CO2的活化.对L-色氨酸/KI催化体系普适性进行研究表明, 对于单取代环氧乙烷, 随着取代基的体积增大收率逐渐减少, 然而缩水甘油基苯基醚作原料时, 产物收率高达84%.双取代环氧乙烷比单取代环氧乙烷活性低, 需要更长的反应时间才能获得较高的收率.但对于环己烯衍生的环氧乙烷, 尽管延长反应时间, 收率也仅有19%, 这可能是由于其较高的空间位阻所导致的.该方法催化剂也容易再生. TBILs催化体系中反应压力为1.5 MPa, 温度为130 ℃, 所考察的底物都取得了81%~99%的收率和99%的区域选择性; 这个反应体系没有使用助催化剂和溶剂.机理探究提出:硫脲的两个仲胺官能团可以同时活化环氧乙烷和CO2, 且卤离子也起到了协同作用.
|
|
(30) |
|
|
(31) |
2016年, Yao课题组[47]使用双核铝配合物催化剂完成了环氧乙烷与CO2的环加成反应(Eq. 32).该反应催化剂Cat. 8用量为0.3 mol%, 助催化剂TBAB用量为0.9 mol%.广普性研究表明在100 kPa压力条件下, 单取代环氧乙烷的产物收率在60%~97%之间; 在1 MPa高压下, 位阻更大的双取代环氧乙烷取得了52%~90%的收率.
|
|
(32) |
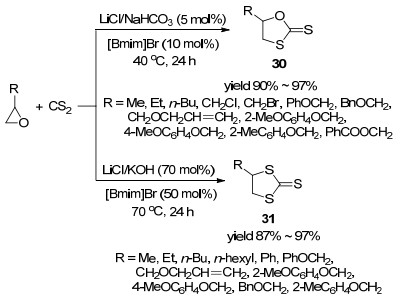
2017年, Shi等[48]报道了环氧乙烷与二硫化碳的[3+2]环加成反应(Scheme 8).条件筛选表明当以10 mol%的[Bmim]Br和5 mol%的LiCl作催化剂, 5 mol%的NaHCO3作添加剂, 40 ℃的条件下反应时, 产物以30为主.当使用50 mol%的[Bmim]Br和70 mol%的LiBr作催化剂, 70 mol%的KOH作添加剂, 70 ℃的条件下反应时, 产物以31为主.反应广普性考察表明对于两个催化体系, 无论是脂肪族的环氧乙烷还是芳基环氧乙烷, 产物30或31均可取得优秀的收率.催化剂循环利用实验显示, 催化剂循环使用3次后, 催化剂活性和产物收率都没有降低.
2015年, Selander课题组[49]报道了AlCl3或InX3 (X=Cl, Br)催化作用下环氧乙烷与芳香醛衍生的硝酮发生的[3+3]环加成反应(Eq. 33).该反应使用5 mol%的AlCl3作催化剂, 乙腈为溶剂于40 ℃条件下进行.当R1为苯基, R2为氢时, 对硝酮普适性考察发现:不同芳基取代的N-甲基硝酮可取得60%~97%的收率, 与N-甲基硝酮相比, 多数N-苄基硝酮得到相对较低的收率.另外, 选择N-甲基硝酮对环氧乙烷考察表明:当R2为氢, R1为脂肪基时, 反应取得了71%~97%的收率, 如果R1为芳香基时, 反应取得了≤65%的收率; 而当R1和R2都不为氢时, 反应取得了≥80%的收率.
|
|
(33) |
2017年, Ghorai课题组[50]在双催化剂体系作用下, 实现了苯基环氧乙烷与N-甲基硝酮的[3+3]环加成反应(Eq. 34).该反应在LiClO4和Bu4NBF4的共同催化作用下, 以水为溶剂于80 ℃下进行.相较于Selander课题组[49]的报道, 该反应使用无毒无污染的水作溶剂.对普适性研究表明所考察的产物收率在63%~85%之间, 且带有供电子基的芳基底物取得的收率比带有吸电子基的芳基底物的高.
|
|
(34) |
硝酮类化合物作为1, 3-偶极子, 与氧杂环丙烷发生环加成可制备一系列二噁嗪衍生物.上述报道中未使用昂贵的催化剂, 反应条件比较温和, 但是到目前为止均未涉及到手性化合物合成方面的研究.
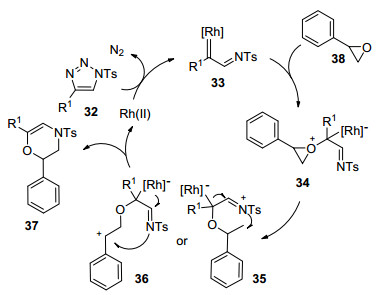
2014年, Chen课题组[51]报道了Rh(Ⅱ)催化剂催化作用下环氧乙烷与N-对甲苯磺酰基-1, 2, 3-三唑通过[3+3]环加成合成3, 4-二氢-2H-1, 4-噁嗪衍生物的反应(Eq. 35).该反应分别使用Rh2(S-NTTL)4 (Cat. 9)和Rh2(S-PTV)4 (Cat. 10)作催化剂, 在DCE溶剂中于120 ℃条件下进行.使用氧化苯乙烯作原料, Rh2(S-NTTL)4作催化剂, 对唑类化合物研究表明R1为吸电子基取代的苯基比其为供电子基取代的苯基时, 反应效果要好, 但其收率也低于60%;当R1为脂肪基或杂芳基时, 反应获得了34%~51%的收率.当R1为4-(甲氧羰基)苯基, R2为H或Me, Rh2(S-PTV)4作催化剂时, 对环氧乙烷考察发现R2取代基有明显的位阻效应, 且所考察的底物收率都不高于51%.值得注意的是, 当原料为脂肪族环氧乙烷时, 反应没有检测到相应的目标产物, 这可能是因为中间体的开环比较困难导致.机理推测为:在Rh催化剂的催化作用下, N-对甲苯磺酰基-1, 2, 3-三唑(32)首先脱掉N2而产生α-亚氨基铑(Ⅱ)卡宾中间体33, 然后, 环氧乙烷38的氧原子对卡宾碳进行亲核进攻得到中间体35或36, 进而产生目标产物37 (Scheme 9).
|
|
(35) |
2016年, Lee课题组[52]在Rh(Ⅱ)和Mg(Ot-Bu)2的共同催化作用下, 采用一锅法将缩水甘油与N-磺酰基- 1, 2, 3-三唑通过[3+3]环加成合成了噁嗪类衍生物(Eq. 36).该反应在Rh2(t-BuCO2)4和Mg(Ot-Bu)2催化下, 以甲苯为溶剂于80 ℃条件下进行.所考察的底物取得了48%~76%的收率, 且选择性在9/1~5/1之间.
|
|
(36) |
2011年, Zhang课题组[53]报道了在Au催化剂催化作用下, 2-炔基-2-酰基环氧乙烷与硝酮发生[4+3]环加成制备呋喃并七元杂环衍生物的反应(Eq. 37).该反应在氮气保护下, 以[AuL12][SbF6]作催化剂, DCE作溶剂于室温条件下进行.对环氧乙烷普适性研究表明:当R1和R2均为芳基, 不论R3是芳基还是脂肪基时, 反应得到了71%~98%的收率; 而当R2和R3均为芳基, R1为甲基时, 反应得到了64%~69%的收率.对硝酮广普性考察表明:当R4和R5为各种芳基或杂芳基时, 反应可得到79%~97%的收率, 作者未对R4和R5为脂肪基的情况进行考察.
|
|
(37) |
2016年, Liu课题组[54]报道了在Au催化剂的催化作用下, 丙炔酸叔丁酯与环氧乙烷发生[4+3]环加成合成1, 4-二氧杂环庚烷衍生物的反应(Eq. 38).该反应以IPrAuCl和AgSbF6作共催化剂, 以DCE作溶剂, 4 MS作添加剂于35 ℃条件下进行.对丙炔酸叔丁酯研究显示, 无论R是芳基还是烷基, 反应得到了55%~75%的收率.对环氧乙烷考察发现, 无论是单取代还是双取代的环氧乙烷, 反应取得了55%~70%的收率.
|
|
(38) |
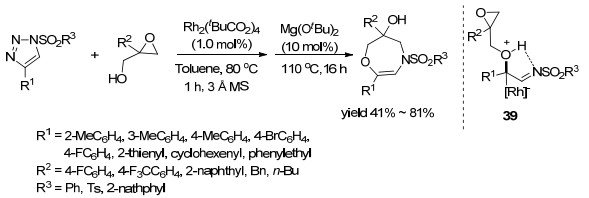
2016年, Lee课题组[52]在Rh2(t-BuCO2)4/Mg(Ot-Bu)2催化体系催化作用下, 采用一锅法通过环氧乙烷与N-磺酰基-1, 2, 3-三唑的[4+3]环加成合成了1, 4-氧氮杂环庚烷衍生物(Scheme 10).反应过程是:首先, Rh催化剂催化N-磺酰基-1, 2, 3-三唑与缩水甘油反应形成中间体39, 接着, Mg(OtBu)2催化该中间体39发生高区域选择性的分子内开环反应, 而且对环氧乙烷部分开环的区域选择性主要取决于缩水甘油环上的R2取代基, 当R2不是氢时, 反应才生成1, 4-氧氮杂环庚烷, 而当R2是氢时, 产物则是1, 4-噁嗪.对三氮唑研究表明, 不论R1和R2是芳基还是烷基, 反应仅能得到41%~81%的收率.
2014年, Jia课题组[55]报道了在Co2(CO)8/LiCl催化作用下, 环氧乙烷、亚胺与CO通过发生三组分[3+2+1]环加成合成1, 3-噁嗪烷-4-酮衍生物的反应(Eq. 39).该反应以1, 4-二氧六环作溶剂, CO的压力为5515.8 kPa, 在70 ℃的条件下进行.以甲胺与苯甲醛衍生的亚胺作原料, 对环氧乙烷广普性考察表明:当R1=H, R2=Me或R1=R2=Me时, 产物收率分别为96%和82%;但当R1=H、R2 =Ph或原料为环己烯衍生的环氧乙烷时, 反应都没有得到目标产物.当R1=H、R2=Me或R1= R2=Me时, 对亚胺广普性研究表明:所考察的N-甲基芳香族亚胺可取得78%~98%的收率, 但当R3 =4-BrC6H4时, 收率仅为50%, 当R3=t-Bu时, 反应仅得到了微量的产物.
|
|
(39) |
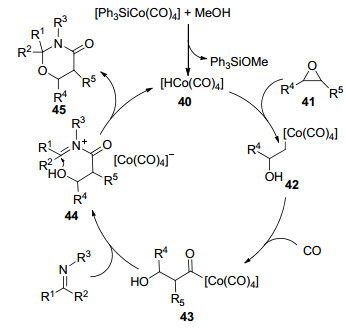
2014年, Sun课题组[56]在[HCo(CO)4]的催化作用下, 实现了通过环氧乙烷、亚胺与CO的三组份[3+2+1]环加成来合成1, 3-噁嗪烷-4-酮的反应(Eq. 40).该反应以甲苯作溶剂, 在50 ℃和6 MPa CO的条件下进行, 其中催化剂[HCo(CO)4]是由[Ph3SiCo(CO)4]和MeOH经过原位反应生成的.以环氧乙烷为基准, 对亚胺广普性研究表明:脂肪醛比芳香族醛衍生的亚胺取得的收率要低, 而且由于位阻作用, N-乙基和N-苄基亚胺比N-甲基亚胺取得的收率要低; 接着对环氧乙烷广普性研究表明:甲基和苯基环氧乙烷都取得了71%~90%的收率, 但环己烯衍生的环氧乙烷取得了较低的收率.机理推测为:首先, [HCo(CO)4]对环氧乙烷41进行亲核开环得到中间体42, 随后插入CO而形成中间体43, 接着, 亚胺对酰基钴物质中间体43进行进攻得到中间体44(速控步骤), 进而生成产物45并释放出催化剂(Scheme 11).
|
|
(40) |
2016年, Khalaj课题组[57]通过三组分高区域选择性[3+2+1]环加成合成了1, 4-氧硫杂环己烷衍生物(Eq. 41).该反应以Bu4NOAc作催化剂, N, N-二甲基甲酰胺(DMF)作溶剂于45 ℃条件下进行.对底物广普性考察表明:环烯烃衍生的氧杂收率差别很大, 环己烯衍生的氧杂取得了96%的收率, 环庚烯衍生的氧杂仅取得了49%的收率, 而环戊烯和环辛烯衍生的氧杂则没有得到相应的目标产物, 其它无论是烷基取代还是烯丙基、丙烯酸酯基取代的环氧乙烷均取得了84%~96%的收率; 苯基取代的氧杂也可以取得良好的收率.
|
|
(41) |
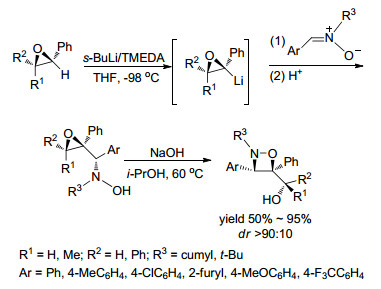
2006年, Florio课题组[58]报道了通过环氧乙烷与硝酮的环加成来合成1, 2-氧氮杂环丁烷的反应(Scheme 12).该反应的过程是:首先, 在s-BuLi和四甲基乙二胺(TMEDA)的共同作用下, 以THF作溶剂, 在-98 ℃下两个底物反应得到β, γ-环氧羟胺中间体; 然后, 体系以异丙醇为溶剂, 加入氢氧化钠水溶液, 在60 ℃条件下反应4~6 h得到最终产物.该反应主要针对硝酮Ar部分进行了考察, 多数考察的底物仅能取得中等的收率, 但都可取得优秀的非对映选择性.
2011年, Zhang课题组[59]在Rh催化剂催化作用下, 实现了通过1-(1-炔基)环氧乙烷基酮环内C—C键断裂发生的串联杂环化和[4+1]环加成合成了呋喃并[3, 4-b]呋喃-3-(2H)-酮的反应(Eq. 42).该反应采用[Rh-(COD)Cl]2作催化剂, L13或L14作配体, 它们的用量均为5 mol%, 反应以DCE作溶剂, CO压力为101 kPa, 反应温度为70 ℃.对普适性考察发现R1为芳基时, 取得的收率比烷基的高; R2为芳基或苯乙烯基, 而R3为芳基、烯基或烷基, 得到了58%~98%的收率.
|
|
(42) |
2017年, Guo课题组[25]报道了在Pd催化剂催化作用下2-芳基-2-乙烯基环氧乙烷与氨基磺酸酯衍生的环状亚胺通过[5+2]环加成合成1, 3-氧氮杂环庚烷衍生物的反应(Eq. 43).该反应采用2.5 mol%的Pd2(dba)3• CHCl3作催化剂, 7.5 mol%的L9作配体, 10 mol%的TBAB作添加剂, DCM作溶剂于室温条件下进行.对氨基磺酸酯衍生的环状亚胺广普性研究表明:当R为供电子基时, 反应得到了54%~96%的收率和良好的区域选择性; 而当R为吸电子基时, 反应得到了相对较低的收率(35%~83%).值得注意的是, 当R为6-OMe时, 反应的收率仅为54%.对环氧乙烷研究表明:不论Ar是供电子基还是吸电子基取代的苯基, 多数反应仅可得到中等的收率和良好的区域选择性.但是, 当Ar分别为2-FC6H4或3-ClC6H4时, 即使反应时间延长至48 h, 产物收率也仅为25%和48%.
|
|
(43) |
综上所述, 经过化学科研工作者的不懈努力, 氧杂环丙烷已经被广泛地应用于多种类型的环加成反应中来合成各种各样的含氧杂环化合物, 比如噁嗪烷、呋喃、噁唑烷酮、噁唑啉和内酯等.而且这些方法得到了不断的创新和改进, 比如反应底物结构的不断拓宽、催化剂易得和反应条件逐渐变温和等, 同时它们也可应用于更广泛的含氧杂环类化合物的合成中.但是, 随着手性科学的不断发展, 该领域仍有许多问题需要进一步研究和解决, 诸如:如何获得更高的产物收率, 如何提高目标产物的化学选择性、区域选择性和对映选择性.因此, 寻找更廉价易得的催化剂, 尤其是廉价易得的手性催化剂, 在发展高对映选择性的反应来提高反应收率、选择性和获得手性化合物方面仍然是研究的热点和重点.相信, 随着有机化学的不断发展, 化学研究者将会开发出更多的化合物与氧杂环丙烷发生环加成来构建种类更加丰富的含氧杂环化合物.
Armbrust, K. W.; Beaver, M. G.; Jamison, T. F. J. Am. Chem. Soc. 2015, 137, 6941. doi: 10.1021/jacs.5b03570
Hugelshofer, C. L.; Magauer, T. J. Am. Chem. Soc. 2016, 138, 6420. doi: 10.1021/jacs.6b03720
Tang, M.-C.; Zou, Y.; Watanabe, K.; Walsh, C. T.; Tang, Y. Chem. Rev. 2017, 117, 5226. doi: 10.1021/acs.chemrev.6b00478
Liu, K.; Khan, I.; Cheng, J.; Hsueh, Y. J.; Zhang, Y. J. ACS Catal. 2018, 8, 11600. doi: 10.1021/acscatal.8b03582
Nielsen, D. K.; Doyle, A. G. Angew. Chem., Int. Ed. 2011, 50, 6056. doi: 10.1002/anie.v50.27
Ilardi, E. A.; Njardarson, J. T. J. Org. Chem. 2013, 78, 9533. doi: 10.1021/jo401776s
He, J.; Ling, J.; Chiu, P. Chem. Rev. 2014, 114, 8037. doi: 10.1021/cr400709j
Ma, C.; Huang, Y.; Zhao, Y. ACS Catal. 2016, 6, 6408. doi: 10.1021/acscatal.6b01845
Shim, J.-G.; Yamamoto, Y. J. Org. Chem. 1998, 63, 3067. doi: 10.1021/jo972317w
Hilt, G.; Bolze, P.; Kieltsch, I. Chem. Commun. 2005, 1996.
Sako, S.; Kurahashi, T.; Matsubara, S. Chem. Lett. 2011, 40, 808. doi: 10.1246/cl.2011.808
Zhang, J.; Chen, Z.; Wu, H. H.; Zhang, J. Chem. Commun. 2012, 48, 1817 doi: 10.1039/c2cc16918e
Wu, W. Q.; Ding, C. H.; Hou, X. L. Synlett 2012, 1035. https://www.researchgate.net/publication/264602119_ChemInform_Abstract_Pd-Catalyzed_Diastereo-_and_Enantioselective_3_2-Cycloaddition_Reaction_of_Vinyl_Epoxide_with_Nitroalkenes
Suo, J. J.; Du, J.; Liu, Q. R.; Chen, D.; Ding, C. H.; Peng, Q.; Hou, X. L. Org. Lett. 2017, 19, 6658. doi: 10.1021/acs.orglett.7b03386
Du, J.; Jiang, Y.-J.; Suo, J.-J.; Wu, W.-Q.; Liu, X.-Y.; Chen, D.; Ding, C.-H.; Wei, Y.; Hou, X.-L. Chem. Commun. 2018, 54, 13143. doi: 10.1039/C8CC07996J
Chen, W.; Xia, Y.; Lin, L.; Yuan, X.; Guo, S.; Liu, X.; Feng, X. Chem.-Eur. J. 2015, 21, 15104. doi: 10.1002/chem.201502448
Yuan, X.; Lin, L.; Chen, W.; Wu, W.; Liu, X.; Feng, X. J. Org. Chem. 2016, 81, 1237. doi: 10.1021/acs.joc.5b02524
Shuler, W. G.; Combee, L. A.; Falk, I. D.; Hilinski, M. K. Eur. J. Org. Chem. 2016, 3335. https://www.researchgate.net/publication/304145435_Intermolecular_Electrophilic_Addition_of_Epoxides_to_Alkenes_32_Cycloadditions_Catalyzed_by_Lewis_Acids_Intermolecular_Electrophilic_Addition_of_Epoxides_to_Alkenes_32_Cycloadditions_Catalyzed_by_Lewi
Blanc, A.; Tenbrink, K.; Weibel, J. M.; Pale, P. J. Org. Chem. 2009, 74, 4360. doi: 10.1021/jo900483m
Liu, R.; Zhang, M.; Zhang, J. Chem. Commun. 2011, 47, 12870. doi: 10.1039/c1cc15669a
Bentabed-Ababsa, G.; Hamza-Reguig, S.; Derdour, A.; Domingo, L. R.; Sáez, J. A.; Roisnel, T.; Mongin, F. Org. Biomol. Chem. 2012, 10, 8434. doi: 10.1039/c2ob26442k
Chen, W.; Fu, X.; Lin, L.; Yuan, X.; Luo, W.; Feng, J.; Feng, X. Chem. Commun. 2014, 50, 11480. doi: 10.1039/C4CC04182H
Liu, Z.; Feng, X.; Du, H. Org. Lett. 2012, 14, 3154. doi: 10.1021/ol301248d
Zhang, J.; Xiao, Y.; Zhang, J. Adv. Synth. Catal. 2013, 355, 2793. doi: 10.1002/adsc.v355.14/15
Wu, Y.; Yuan, C.; Wang, C.; Mao, B.; Jia, H.; Gao, X.; Guo, H. Org. Lett. 2017, 19, 6268. doi: 10.1021/acs.orglett.7b02704
Zhang, S.-S.; Wang, D.-C.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M. Org. Lett. 2018, 20, 8026. doi: 10.1021/acs.orglett.8b03615
Alajarin, M.; Ba on, D.; Egea, A.; Marín-Luna, M.; Orenes, R. A.; Vidal, A. Org. Chem. Front. 2018, 5, 2020. doi: 10.1039/C8QO00255J
Zhou, H.; Zeng, X.; Xie, Y.; Zhong, G. Synlett 2015, 26, 1693. doi: 10.1055/s-00000083
Zhou, H.; Zeng, X.; Ding, L.; Xie, Y.; Zhong, G. Org. Lett. 2015, 17, 238. doi: 10.1021/ol503288e
Chen, Z.; Wei, L.; Zhang, J. Org. Lett. 2011, 13, 1170. doi: 10.1021/ol2000292
Chen, Z.; Tian, Z.; Zhang, J.; Ma, J.; Zhang, J. Chem.-Eur. J. 2012, 18, 8591. doi: 10.1002/chem.v18.28
Chen, W.; Lin, L.; Cai, Y.; Xia, Y.; Cao, W.; Liu, X.; Feng, X. Chem. Commun. 2014, 50, 2161. doi: 10.1039/c3cc48606k
Elenkov, M. M.; Tang, L.; Meetsma, A.; Hauer, B.; Janssen, D. B. Org. Lett. 2008, 10, 2417. doi: 10.1021/ol800698t
Barros, M. T.; Phillips, A. M. F. Tetrahedron:Asymmetry 2010, 21, 2746. doi: 10.1016/j.tetasy.2010.10.028
Mani, S.; Punniyamurthy, T. RSC Adv. 2012, 2, 2736. doi: 10.1039/c2ra00042c
Zhang, X.; Zhao, C.; Gu, Y. J. Heterocycl. Chem. 2012, 49, 1143. doi: 10.1002/jhet.970
Baronsky, T.; Beattie, C.; Harrington, R. W.; Irfan, R.; North, M.; Osende, J. G.; Young, C. ACS Catal. 2013, 3, 790. doi: 10.1021/cs4001046
Beattie, C.; North, M. RSC Adv. 2014, 4, 31345. doi: 10.1039/C4RA04427D
Paddock, R. L.; Adhikari, D.; Lord, R. L.; Baik, M. H.; Nguyen, S. T. Chem. Commun. 2014, 50, 15187. doi: 10.1039/C4CC07421A
Chen, Z.; Xiao, Y.; Zhang, J. Eur. J. Org. Chem. 2013, 4748.
Kumar Pandey, A.; Ghosh, A.; Banerjee, P. Eur. J. Org. Chem. 2015, 2517. https://www.researchgate.net/publication/272524965_ChemInform_Abstract_Lewis-Acid-Catalyzed_Tandem_Meinwald_RearrangementIntermolecular_3_2-Cycloaddition_of_Epoxides_with_Donor-Acceptor_Cyclopropanes_Synthesis_of_Functionalized_Tetrahydrofurans
Pandey, A. K.; Banerjee, P. Asian J. Org. Chem. 2016, 5, 360. doi: 10.1002/ajoc.v5.3
Decortes, A.; Belmonte, M. M.; Benet-Buchholz, J.; Kleij, A. W. Chem. Commun. 2010, 46, 4580. doi: 10.1039/c000493f
Sun, J.; Wang, J.; Cheng, W.; Zhang, J.; Li, X.; Zhang, S.; She, Y. Green Chem. 2012, 14, 654. doi: 10.1039/c2gc16335g
Yang, Z.; Sun, J.; Cheng, W.; Wang, J.; Li, Q.; Zhang, S. Catal. Commun. 2014, 44, 6. doi: 10.1016/j.catcom.2013.07.025
Xu, F.; Cheng, W.; Yao, X.; Sun, J.; Sun, W.; Zhang, S. Catal. Lett. 2017, 147, 1654. doi: 10.1007/s10562-017-2081-x
Gao, P.; Zhao, Z.; Chen, L.; Yuan, D.; Yao, Y. Organometallics 2016, 35, 1707. doi: 10.1021/acs.organomet.6b00153
Mei, C.; Li, X.; Liu, L.; Cao, C.; Pang, G.; Shi, Y. Tetrahedron 2017, 73, 5706. doi: 10.1016/j.tet.2017.08.009
Pathipati, S. R.; Singh, V.; Eriksson, L.; Selander, N. Org. Lett. 2015, 17, 4506. doi: 10.1021/acs.orglett.5b02195
Wani, I. A.; Sayyad, M.; Ghorai, M. K. Chem. Commun. 2017, 53, 4386. doi: 10.1039/C7CC01033H
Ma, X.; Pan, S.; Wang, H.; Chen, W. Org. Lett. 2014, 16, 4554. doi: 10.1021/ol5021042
Ko, Y. O.; Jeon, H. J.; Jung, D. J.; Kim, U. B.; Lee, S. G. Org. Lett. 2016, 18, 6432. doi: 10.1021/acs.orglett.6b03328
Wang, T.; Zhang, J. Chem.-Eur. J. 2011, 17, 86. doi: 10.1002/chem.201002395
Sahani, R. L.; Liu, R. S. Chem. Commun. 2016, 52, 7482. doi: 10.1039/C6CC02124G
Zhang, Y.; Ji, J.; Zhang, X.; Lin, S.; Pan, Q.; Jia, L. Org. Lett. 2014, 16, 2130. doi: 10.1021/ol500549c
Liu, L.; Sun, H. Angew. Chem., Int. Ed. 2014, 53, 9865. doi: 10.1002/anie.201403998
Khalaj, M.; Ghazanfarpour-Darjani, M. Monatsh. Chem. 2016, 147, 2043. doi: 10.1007/s00706-016-1845-0
Capriati, V.; Florio, S.; Luisi, R.; Salomone, A.; Cuocci, C. Org. Lett. 2006, 8, 3923. doi: 10.1021/ol061256y
Wang, T.; Wang, C.-H.; Zhang, J. Chem. Commun. 2011, 47, 5578. doi: 10.1039/c0cc05650b

图式 2 铑催化乙烯基环氧乙烷与亚胺的[3+2]环加成反应
Scheme 2 Rhodium-catalyzed [3+2] cycloadditions of vinyl epoxides with imines

图式 5 路易斯酸催化氧杂环丙烷与醛的[3+2]环加成反应
Scheme 5 LA-Catalyzed[3+2] cycloaddition of oxiranes with aldehydes

图式 6 BF3•Et2O催化环氧乙烷与异硒氰酸酯的[3+2]环加成反
Scheme 6 BF3•Et2O-catalyzed[3+2] cycloaddition of oxiranes with isoselenocyanates

图式 8 [Bmim]Br/LiCl(Br)催化环氧乙烷与二硫化碳的[3+2]环加成反应
Scheme 8 [Bmim]Br/LiCl(Br)-catalyzed[3+2] cycloaddition of epoxides with carbon disulfide

图式 10 Rh2(t-BuCO2)4/Mg(Ot-Bu)2催化环氧乙烷与N-磺酰基-1, 2, 3-三唑的[4+3]环加成反应
Scheme 10 Rh2(t-BuCO2)4/Mg(Ot-Bu)2-catalyzed [4+3] cycloaddition of epoxides with N-sulfonyl-1, 2, 3-triazoles

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:












































 下载:
下载:






 下载:
下载: