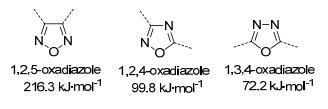
图 1.
呋咱与异呋咱的结构及生成焓[7]
Figure 1.
Structures, and enthalpies of formation of furazan and isofurazan

近几十年, 高能量密度材料的(HEDMS)的发展十分迅速, 追求高能量成为含能材料永恒的目标之一.随着武器弹药使用环境的日益苛刻, 在追求弹药高能量的同时, 对其安全性的要求也越来越高.如何实现高能与安全的平衡, 设计和合成出高能、高氧平衡、低感且环境友好新型含能材料一直是研究人员的不懈追求[1~6].
富氮五元杂环是常见的含能材料母体环, 其中, 1, 2, 5-噁二唑环(呋咱环)因有着优良的性能而得到深入研究: 1, 2, 5-呋咱环含有两个C=N和两个N—O键, 为其提供了高生成焓和高氧平衡值[8, 9], 呋咱的芳香型平面结构增加了其结构的稳定性, 近年来, 在这一领域取得了显著进展, 合成了一系列性能优异的含能化合物[9~15].异呋咱是呋咱的同分异构体, 涵盖1, 2, 4-噁二唑和1, 3, 4-噁二唑两种氮杂五元环, 但生成焓远低于呋咱[16], 先前很少将它们作为含能材料的母体单元进行研究. 2012年, 陈甫雪等[17]首次报道了以1, 2, 4-噁二唑为母体环的一系列含能化合物, 其中3-硝基-5-硝氨基- 1, 2, 4-噁二唑的理论密度为1.883 g·cm-1, 理论爆速9095 m·s-1, 理论爆压37.68 GPa, 而其摩擦感度仅为15 J, 与TNT相当, 是一种新型高能钝感含能材料.从此, 异呋咱类含能材料逐渐受到科研人员的关注并迅速发展, 成为含能材料合成领域的研究热点之一.
异呋咱环虽然生成焓较低, 但稳定性远高于呋咱, 可以以其作为母体结构, 再引入高能化学基团(如呋咱环、NH2、NO2、ONO2、N3、NHNO2)[18~19], 有效地提高化合物的生成焓, 通过氮杂骨架与含能基团的调控可获得性能优良的含能材料.依据这研究思路, 目前已合成部分性能优异的异呋咱含能化合物, 如理论爆速大于10000 m·s-1高能单质炸药DDAzF[20], 高能钝感耐热炸药TKX-55[21], 可作为高能增塑剂的5, 5'-乙硝酸酯- 3, 3'-联-1, 2, 4-噁二唑[22]等.
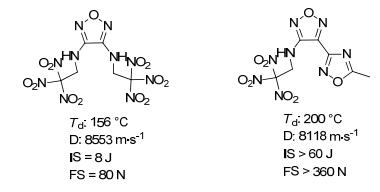
第二种研究思路是, 将异呋咱环作为降感基团引入已知高能含能化合物中, 与其芳香性环形成共轭体系, 有望实现含能材料高能与安全性的矛盾与统一.以3, 4-二(2, 2, 2-三硝基乙氨基)-呋咱[23]为例, 其爆速达8553 m·s-1, 撞击感度(IS)约为8 J, 分解温度156 ℃; 如将其单侧的2, 2, 2-三硝基乙氨基替换为5-甲基-1, 2, 4-噁二唑[24], 爆速为8118 m·s-1, 摩擦感度(FS)>360 N, IS>60 J, 其分解点温度升高, 机械感度大幅降低, 表明1, 2, 4-噁二唑是设计、合成不敏感含能材料的理想单元结构(图 2).
由于异呋咱含能化合物研究时间不长, 对异呋咱含能材料环母体合成方法和合成机理的综述还尚未见报道, 而这方面内容的总结对于异呋咱含能化合物的进一步发展很有必要.因此, 本文以目前合成的异呋咱含能化合物为研究背景, 着重介绍了1, 2, 4-噁二唑及1, 3, 4-噁二唑等氮杂五元环的构建方法, 探讨了环化反应机理; 全面概述了近年来异呋咱含能化合物的最新合成研究进展, 并对典型含能化合物的物化、爆轰性能进行了阐述, 以期为进一步开展相关研究工作提供参考.
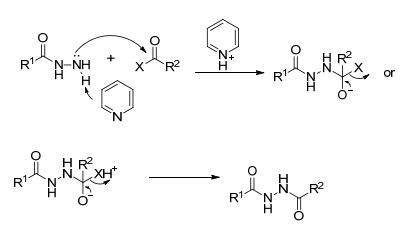
以偕胺肟与羧酸衍生物为底物合成1, 2, 4-噁二唑含能化合物是最常见的构建方法.反应过程可概括为:偕胺肟与活化的羧酸衍生物发生O-酰化反应得到O-酰化中间体, 然后进行脱水环化.
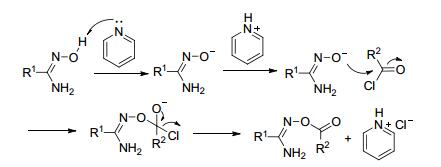
酰氯反应活性高, 酰氯与偕胺肟的O-酰化易于进行. Nishiwaki等[25]发现采用有机碱(吡啶、三乙胺)作为缚酸剂, 11种芳香族或脂肪酰氯与偕胺肟在室温下搅拌隔夜即可生成O-酰化中间体(Eq. 1).
|
|
(1) |
酰氯与偕胺肟O-酰化反应(以吡啶作缚酸剂为例)机理见如图 3.
相较于酰氯的O-酰化反应, 其O-酰化中间体的脱水环化是反应的决速步骤[26].主要的脱水方法有加热法、碱催化法、微波辐射法等.
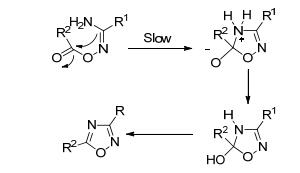
加热法是指O-酰化中间体在高温下(100 ℃以上)长时间反应脱水得到环化产物[27, 28], 这种方法选择性差, 反应时间长, 收率低.其可能的反应机理见图 4[29].
碱催化法是目前研究较多的方法, 碱催化可加快反应进程, 可能的反应机理见图 5.
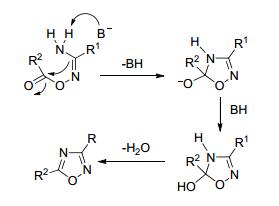
反应一般采用吡啶、三乙胺等有机弱碱作为催化剂. Shine等[30]采用15种脂肪族或苯基酰氯与5种苯基或芳香族偕胺肟在吡啶中回流下反应30 min, 即可得到1, 2, 4-噁二唑(Eq. 2).
|
|
(2) |
Gangloff等[31]首次报道了以O-酰化中间体作为底物, 在室温下THF/四丁基氟化铵(TBAF)体系中合成3, 5取代的1, 2, 4-噁二唑.环化反应收率受取代基影响较大, 延长反应时间不能提高收率(Eq. 3).
|
|
(3) |
Nishiwaki等[25]以(Z)-2-乙酰氧基-2-氨基-N-甲基乙酰胺为原料, 比较了吡啶、三乙胺、TBAF作为催化剂时环化产物的收率.用1 equiv.三乙胺(Eq. 4)和吡啶(Eq. 5)在80 ℃加热2 h, 1, 2, 4-噁二唑收率仅为12%和25%;用0.1 equiv. TBAF处理原料, 在室温下反应2 h即可以100%的收率得到产品(Eq. 6).
氟阴离子具有腐蚀性, 目前已提出了一些替代环化体系.如氮杂二环十一碳-7-烯(DBU)/DMSO[32]、四丁基氢氧化铵(TBAH)/DMSO[33]、KOH/DMSO等.这些体系的特点是反应时间短, 室温下即可完成环化反应.
微波合成是近十年来发展最快的方法, 其优点为反应时间更快, 产率更高, 副产物含量少. Kaboudin等[34]报道了在微波/MgO催化作用下, 10种不同的苯基偕胺肟与酰氯仅反应1 min就可以高收率地得到1, 2, 4-噁二唑(67%~91%) (Eq. 7).
|
|
(4) |
|
|
(5) |
|
|
(6) |
|
|
(7) |
在有机合成中, 微波技术使得反应热效应及加热效率大大增加, 但难以控制反应进度, 在含能化合物的合成中应用较少.
1990年Kiseleva等[35]首次报道了3, 3'-联-1, 2, 4-噁二唑-5, 5'-乙二酸二乙酯的合成(Eq. 8), 但该合成方法选择性较差, 操作较为复杂, 收率较低. 2017年黄晓川等[36]报道了它的改进方法(Eq. 9), 利用氯甲酰乙酸乙酯中高活性的氯甲酰基替代酯基, 在较低的温度下与二氨基乙二肟发生缩合环化反应, 与原反应相比, 降低了反应温度, 提高了反应的选择性.
|
|
(8) |
|
|
(9) |
3, 3'-联-1, 2, 4-噁二唑-5, 5'-乙二酸二乙酯是重要的含能化合物中间体, 它具有芳香共平面结构, 偶极矩小, 结构较为紧凑, 因此能有效提高化合物的密度及稳定性[37].此外, 利用乙酸乙酯这一反应活性位点, 通过硝化、氟化等反应, 可将一些高能基团引入酯的α-碳上.
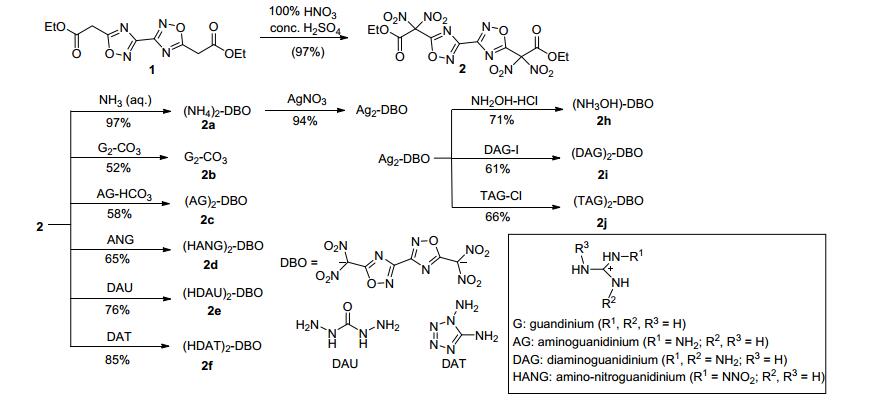
Klapötke课题组[7]以3, 3'-联-1, 2, 4-噁二唑-5, 5'-乙二酸二乙酯(1)为原料, 经硝化得到3, 3'-联-1, 2, 4-噁二唑- 5, 5'-二硝基乙二酸二乙酯(2), 在碱性条件下脱酯形成偕二硝基阴离子, 随后与含能阳离子配位得到10种含能离子盐(2a~2j).其中羟胺盐2h的理论密度达1.946 g·cm-3 (298 K), 理论爆速达8935 m·s-1, 而且其撞击感度为4 J, 摩擦感度为108 N, 是一种具有潜在应用价值的高能含能离子盐(图 6).
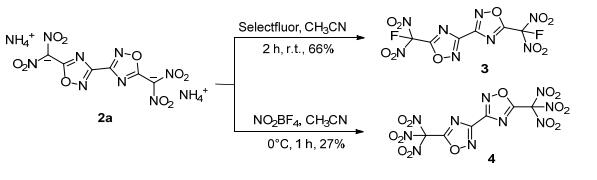
此外, 他们以铵盐为原料, 经氟化反应成功引入了氟二硝基甲基得到5, 5'-氟二硝基甲基-3, 3'-联-1, 2, 4-噁二唑(3) [38]; 经硝化反应, 引入三硝甲基得到5, 5'-三硝甲基-3, 3'-联-1, 2, 4-噁二唑(4)[39]. 3的计算密度为1.96 g· cm-3 (298 K), 理论爆速达8367 m·s-1, 撞击感度为10 J, 摩擦感度为192 N, 是一种潜在的不敏感含能材料; 4的计算密度为1.94 g·cm-3 (298 K), 理论爆速达8814 m· s-1, 氧平衡水平ΩCO2为7.3%, 一种综合性能优良的氧化剂(图 7).
Gregory等[40]首次报道了1, 2, 4-噁二唑-3, 5-二甲酸乙酯的合成, 采用氨基肟甲酸乙酯与氯甲酰乙酸乙酯在CHCl3中(吡啶催化)下首先发生O-酰化反应, 然后回流环化, 收率达99%以上(Eq. 10).
|
|
(10) |
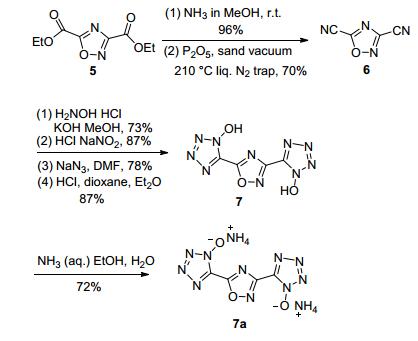
Pagoria等[41]采用这一反应前体侧链的酯基经酰氨化、脱水转变为氰基, 氰基再经肟化、氯化、叠氮化以及环化生成3, 5-二(2-羟基-四唑基)-1, 2, 4-噁二唑(7).该含能化合物显酸性, 在氨气的乙醇溶液中发生中和反应生成含能铵盐(7a), 但其含能离子盐的能量水平及稳定性均未见报道(图 8).
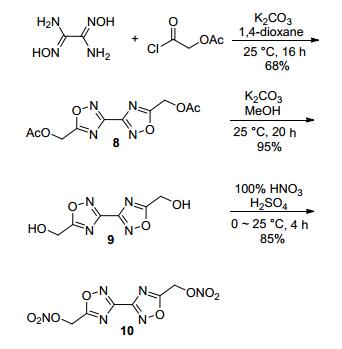
Sabatini等[22]以二氨基乙二肟和乙酰氧基乙酰氯为原料, 经脱水环化、水解、硝化反应得到化合物3, 3'-联- 1, 2, 4-噁二唑-5, 5'-二乙硝酸酯(10), 其理论密度达1.832 g·cm-3, 爆速8180 m·s-1, 熔点84.5 ℃, Tdec=183.5 ℃, IS为8.7 J, FS为282 N.
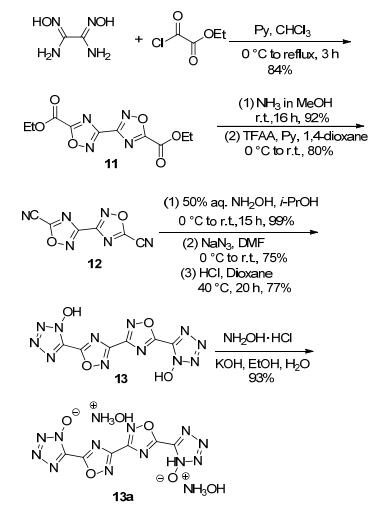
Pagoria等[41]利用二氨基乙二肟与草酰氯单乙酯脱水环化合成3, 3'-联-1, 2, 4-噁二唑-5, 5'-二甲酸乙酯(11), 经酰氨化、脱水反应将酯基变为氰基, 氰基再经肟化、氯化、叠氮化以及环化生成羟基四唑(13).该化合物在碱性条件下与盐酸羟胺进行离子交换获得羟胺含能离子盐(13a).
酯与偕胺肟可经O-酰化和脱水环化合成1, 2, 4-噁二唑.酯基反应活性较低, 且易于分解[42], 加热法制备1, 2, 4-噁二唑的收率很低.在碱催化下, 该反应收率可大大提高:在碱的存在下芳基肟的氧原子上快速形成共振稳定的阴离子, 帮助亲核进攻反应的进行.反应机理如图 11所示.
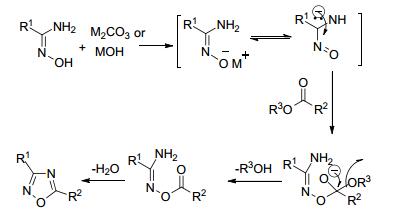
Srivastava等[43]利用K2CO3作为O-酰化反应的活化剂, 用2-羟基乙酸乙酯和不同取代基苯基偕胺肟在甲苯/DMF (V:V=9:1)的混合溶剂中回流8 h, 脱水环化得到1, 2, 4-噁二唑, 产率为35%~76% (Eq. 11).
|
|
(11) |
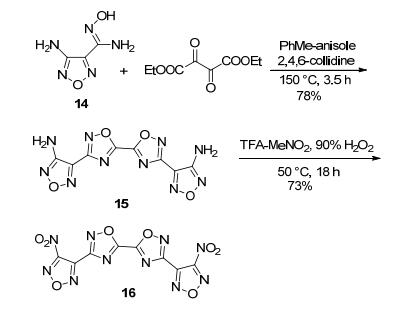
每一个呋咱环可将含能化合物的密度提高0.06~0.08 g·cm-3, 爆速增加300 m·s-1[44].计算结果证实, 分子中引入不同的噁二唑环的是一种有效的降低感度并获得良好爆轰性能的方法[45]. Kuklja等[46]以4-氨基- 1, 2, 5-呋咱-3-偕胺肟及草酸二乙酯为原料, 以高位阻有机碱2, 4, 6-三甲基吡啶为催化剂, 脱水环化得化合物DATO (15), 再经硝化得LLM-200 (16). LLM-200理论密度达1.940 g·cm-3, 爆速达8780 m·s-1, 熔点180 ℃, Tdec=295 ℃, 特性落高H50=62 cm, 可见LLM-200是一种爆轰性能与RDX相当, 稳定性好的高能钝感耐热炸药.
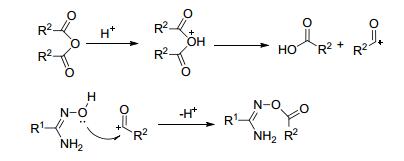
酸酐是很强的酰化剂, 易于和偕胺肟发生O-酰化反应.该反应可同时被酸催化和碱催化.在酸催化下, 酸酐与偕胺肟的酰化反应机理如图 13所示.
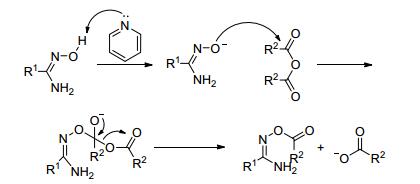
在碱催化下, 酸酐与偕胺肟的酰化反应机理如图 14所示(以吡啶为例).
Baykov等[47]报道了21种芳香型偕胺肟与环内酸酐的O-酰化反应, 在DMSO中室温反应1 h便可以高收率地得到酰化产物, 酰化产物在NaOH催化下反应即可发生脱水成环(Eq. 12).
|
|
(12) |
Vidavalur等[48]报道了以二氧化硅负载的高氯酸(HClO4-SiO2)作为催化剂, 在无溶剂条件下, 芳香型偕胺肟与7种酸酐一锅法制备1, 2, 4-噁二唑, 反应时间短, 收率高(Eq. 13).
|
|
(13) |
Klapötke等[38]报道了5, 5'-三氟甲基-3, 3'-联-1, 2, 4-噁二唑的合成.三氟乙酸酐与二氨基乙二肟在35 ℃下反应3 h即可得到产品(Eq. 14).
|
|
(14) |
联-1, 2, 4-噁二唑具有结构稳定、紧凑的特点, 三氟甲基是非能量基团, 但可以提高化合物的密度.该化合物的密度达2.01 g·cm-3.熔点为98 ℃, 分解温度为142 ℃.此外, 氟原子可以被硝基逐步取代[37, 38], 有望得到新型含能材料.
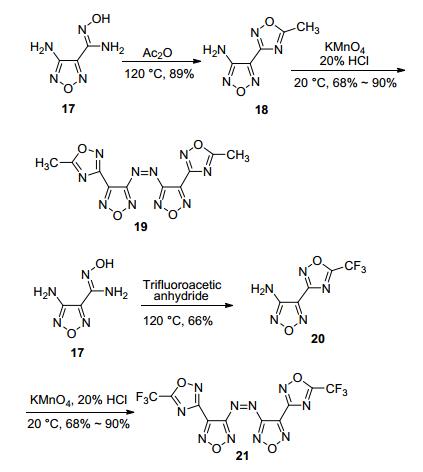
Qu等[20]以3-氨基-4-偕胺肟基呋咱(17) (图 15)为原料, 分别与乙酸酐、三氟醋酐反应, 经O-酰化环化得到1, 2, 4-噁二唑, 最后分别在KMnO4/HCl体系中反应得到偶氮产物(19, 21).偶氮结构可有效提高化合物的氮含量和生成焓, 并保持原有的平面结构[49~51].其中, 偶氮三氟甲基产物(21)密度达1.94 g·cm-3, 理论爆速为6602 m·s-1, 撞击感度大于80 J, 摩擦感度大于360 N.
酰胺的反应活性低, 高温下易分解[52], 因此偕胺肟与酰胺的反应研究不多. Shaposhnikov等[52]报道了4-氨基-1, 2, 5-呋咱-3-偕胺肟与酰胺的“一锅”法合成1, 2, 4-噁二唑, 收率为33%~50% (Eq. 15).
|
|
(15) |
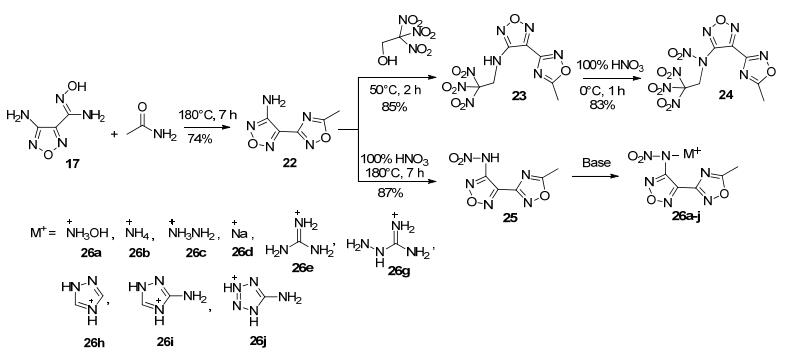
Yang等[24]采用这一方法合成4-氨基-3-(5-甲基- 1, 2, 4-噁二唑-3-基)呋咱(22), 随后利用呋咱上的活性氨基进行衍生反应: (1)经硝化得到硝氨基(25), 进而利用酸性氢成盐(26a~26j); (2)与三硝基乙醇反应, 经脱水缩合引入三硝基乙基高能高氧基团(23).这一系列化合物密度1.63~1.78 g·cm-3, 其中化合物3-(N, 2, 2, 2-四硝基乙氨基)-4-(5-甲基-1, 2, 4-噁二唑)呋咱(24)爆速达8602 m·s-1, 密度1.78 g·cm-3(图 16).
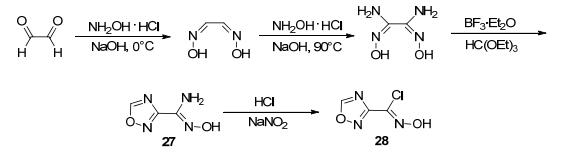
Andrianov等[53]发现在醚合三氟化硼的催化作用下, 原甲酸乙酯与二氨基乙二肟单边的偕胺肟反应, 生成1, 2, 4-噁二唑-3-偕胺肟(27), 27再与亚硝酸钠和盐酸反应得到1, 2, 4-噁二唑-3-氯肟(28) (图 17).
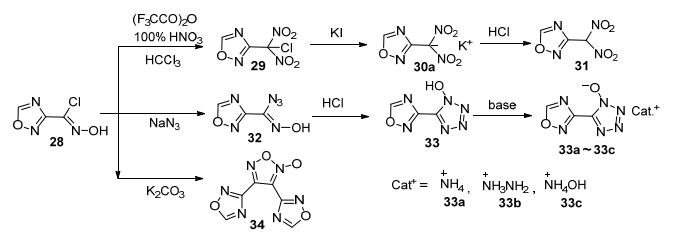
Cheng等[54]以1, 2, 4-噁二唑-3-氯肟(28)为起始反应物进行了后续研究, 通过硝化、叠氮和与环化等反应, 成功合成了3-二硝基甲基-1, 2, 4-噁二唑(31)、5-(1, 2, 4-噁二唑-3-基)-1-羟基四唑(33)(及其三种含能盐)和34 (图 18).其中, 3-二硝基甲基-1, 2, 4-噁二唑的钾盐(30a)理论密度达2.07 g·cm-3, 理论爆速8800 m·s-1.结果表明1, 2, 4-噁二唑-3-氯肟作为重要的含能中间体, 在合成1, 2, 4-噁二唑含能衍生物中具有广阔的应用前景.
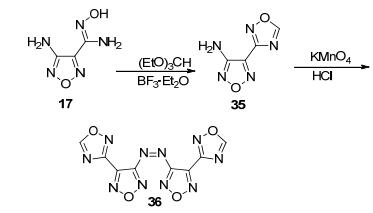
Qu等[20]以4-氨基-3-肟基-1, 2, 4-噁二唑和原甲酸酯为原料, 经环化、氧化偶联得到偶氮产物(36) (图 19).
Dimsdale[55]确定, 2, 6-二氯苄基偕胺肟与溴化氰在碳酸氢钾的催化下, 首先发生O-酰化反应而非N-酰化反应, 随后脱水环化得到1, 2, 4-噁二唑(Eq. 16).可能的反应机理见图 20.
|
|
(16) |
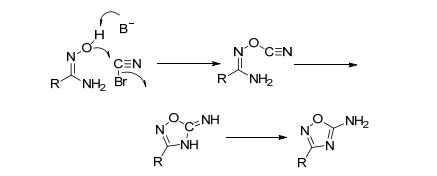
1, 2, 4-噁二唑的5位活性氨基可进一步进行硝化、氧化等反应得到多种结构的含能化合物. Shreeve等[56]以化合物37为原料与BrCN合环, 没有得到预期产物41, 反而获得了两种产物38和39.这是一种肟基α-C发生C—C键断裂-偶联构建联1, 2, 4-噁二唑的新反应.将化合物38分离, 再经硝化、成盐等得到3种含能离子盐.其中, 乙胺盐(40b)的IS=25 J, FS=40 J, 理论爆速9078 m·s-1, 是一种新型高能钝感含能离子盐(图 21).
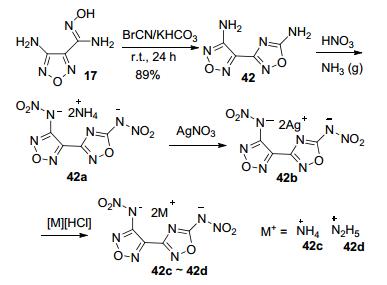
Shaposhnikov等[57]首次报道了3-氨基-4-(5-氨基- 1, 2, 4-噁二唑)呋咱(42)的合成. Shreeve等[58]以42为原料, 再经过硝化、成盐等反应得到其含能离子盐.其中42c密度为1.85 g·cm-3, 理论爆速达9046 m·s-1, 撞击感度为16 J, 摩擦感度为160 N (图 22).
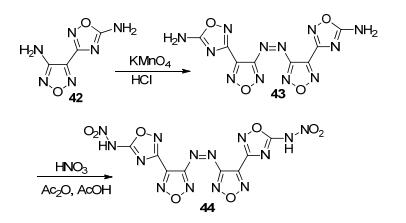
Qu等[20]以42为原料, 经氧化偶联得到43(由于呋咱侧链的氨基活性高于异呋咱侧链的氨基, 偶氮反应优先在呋咱侧链氨基发生), 随后硝化得到DDAzF (44).其中, 44密度高达2.12 g·cm-3, 理论爆速达10114 m· s-1, 高于RDX和HMX, 与CL-20相当, 分解温度为317 ℃, IS和FS均在可接受范围内, 是一个综合性能突出的高能材料, 其高密度可能归因于硝氨基参与多个分子氢键相互作用和其共轭平面结构(未得到单晶结构) (图 23).
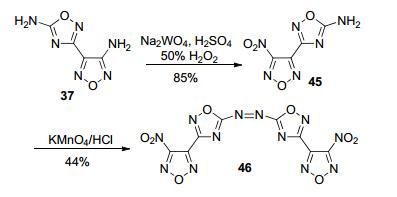
Lu等[59]则利用42的两个氨基的活性不同, 先将呋咱侧链的氨基氧化成硝基, 得到化合物45, 再用KMnO4/HCl处理, 得到偶氮化合物46, 总收率约为40.33%.该化合物的理论密度达1.92 g·cm-3, 理论爆速达9240 m·s-1, IS为18 J, FS为220 N, 熔点157 ℃, 分解点256 ℃, 综合性能优于HMX, 是一种有着潜在应用前景的高能钝感炸药(图 24).
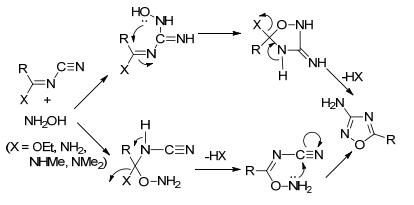
腈和偕胺肟的反应分两种反应机理进行: (ⅰ) O-酰化合环反应; (ⅱ)腈氧化物的1, 3-偶极环加成.
Yarovenko等[60]首次报道了有机腈与偕胺肟合成1, 2, 4-噁二唑.乙腈与苯甲酰肟反应需要在密封管中进行, 180 ℃反应12 h, 产率仅为21% (Eq. 17).该反应可能的机理为O-酰化环化, 见图 25.
|
|
(17) |
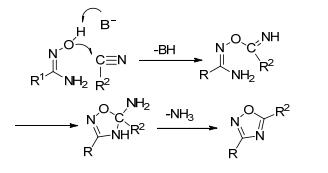
另一种反应机理为[3+2]偶极环加成, 这种反应方式可大大提升产率. Augustine等[61]利用PTSA-ZnCl2作催化剂, 偕胺肟与有机腈以高收率(77%~91%)得到25种1, 2, 4-噁二唑(Eq. 18).可能的原因是PTSA-ZnCl2活化偕胺肟离去一分子NH3形成氧化腈, 与腈发生[3+2]偶极环加成反应(图 26).
|
|
(18) |
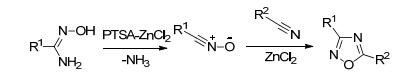
Shreeve等[58]利用17与3-氨基-4-氰基呋咱(47), 在TsOH/ZnCl2催化体系中环化得到双呋咱基1, 2, 4-噁二唑化合物48, 经硝化、成盐反应得到硝氨基化合物49及其含能离子盐(49a~79b), 其中49a的密度为1.71 g· cm-3, 爆速8603 m·s-1 (图 27).
1963年, Huffman等[62]首次报道了利用N-氰基亚胺和羟胺盐酸盐合成3-氨基-1, 2, 4-噁二唑(Eq. 19).
|
|
(19) |
Dimsdale等[55]分析了该反应可能的反应机理, 反应可能通过两种方式进行: (i)羟胺氮首先亲核加成氰基碳, 随后羟胺氧进攻亚氨酸酯的碳环化后脱HX; (ii)羟胺氧首先亲核加成亚氨酸酯的碳氰基碳, 随后羟胺氮进攻氰基碳后脱HX环化(图 28).
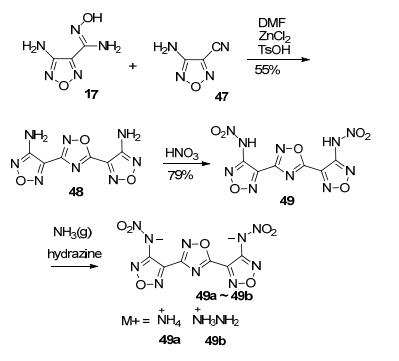
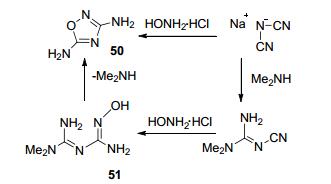
Huttunen等[63]以二氰基胺钠为原料, 在二甲基胺的催化作用下与盐酸羟胺反应得到重要的反应前体化合物1, 3-二氨基-1, 2, 4-噁二唑(50).可能的机理是:二氰基胺钠与二甲基胺发生单边亲核加成反应, 再与盐酸羟胺发生加成, 碱性条件下O进攻C发生合环反应, 再脱去一分子二甲基胺得到51(图 29).
这一反应中间体50在3, 5位均有氨基, 利用不同位点氨基的反应活性不同, 通过控制反应条件, 可得到多种性能优异的含能化合物.
Shreeve课题组[64]以50为原料, 经高锰酸钾/盐酸体系氧化, 1, 2, 4-噁二唑的3位氨基可偶联生成偶氮产物48.然后, 进一步采用不同的硝化体系得到不同的硝化产物[65].偶氮产物在100% HNO3中硝化, 可得到5, 5'-二硝氨基偶氮-1, 2, 4-噁二唑52, 进一步合成相应的含能离子盐.这一系列化合物爆速较高(8381~9243 m·s-1), 其中羟胺含能离子盐(49a)密度为1.864 g·cm-3, 爆速9243 m·s-1, FS为160 N, IS为10 J, 是一种具有潜在应用价值的高能离子盐; 偶氮产物在硝酸/醋酐体系中硝化, 可得到偶氮1, 2, 4-噁二唑酮化合物50, 中和得到3种盐衍生物(50a~50c)[64](图 30).
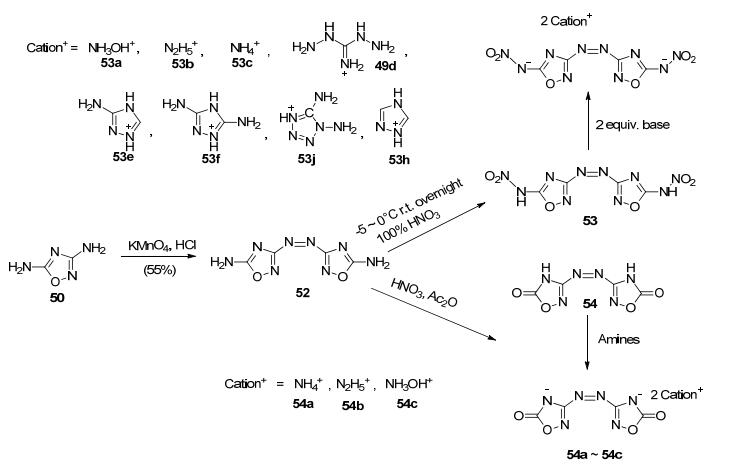
如采用Na2WO4/H2O2/H2SO4体系氧化50, 可以得到3-位硝化的产物3-硝基-5-氨基-1, 2, 4-噁二唑(55). Chen等[66]在二氯异氰尿酸钠(SDCI)/醋酸条件下处理55, 同时得到以一个氮相连的二硝基噁二唑的钠盐(56)和三氮烯相连的二硝基噁二唑的钠盐(57), 并获得了其它6种含能盐.其中, 双(3-硝基-1, 2, 4-噁二唑-5-基)酰胺胍盐密度为1.799 g·cm-3, 爆速8351 m·s-1, 分解温度(Tdec) 241 ℃, IS>40 J(图 31).
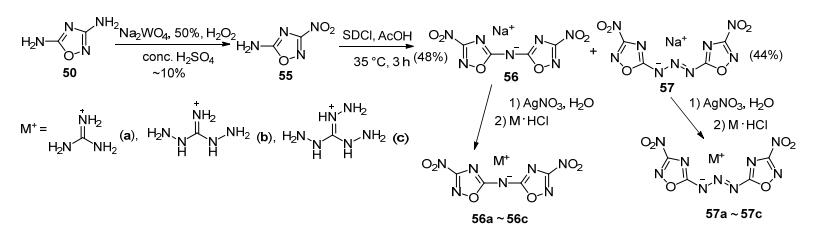
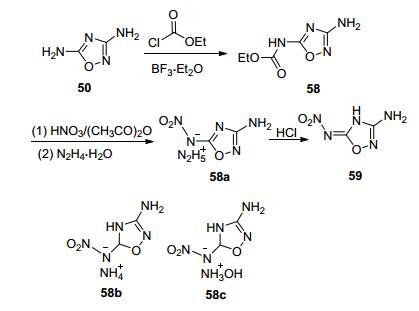
Shreeve等[67]以50为原料, 在醚合三氟化硼的催化下, 氯甲酸乙酯仅与一个氨基发生酰胺化反应, 硝化后碱性条件水解得到58a及其2种富氮盐(58b~58c).这一系列化合物爆速较高(8493~8897 m·s-1), 其中58a的爆速8897 m·s-1, IS=20 J, FS=240 N, 且原材料低廉, 步骤少, 综合性能与RDX相当, 是一种潜在的RDX替代物(图 32).
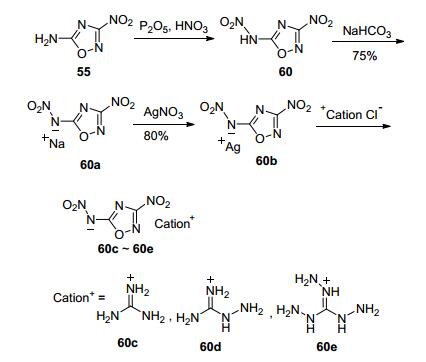
Chen等[68]在P2O5/HNO3条件下对55进行硝化后得到硝氨化产物(60), 碱性条件水解得到五种含能盐(60a~60e).其中60a的理论密度1.888 g·cm-3, IS>40 J, 理论爆速9354 m·s-1, 熔点146~148 ℃, 分解温度258 ℃.是一种综合性能优于RDX的含能离子盐(图 33).
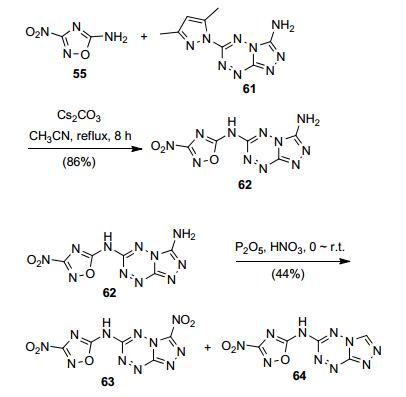
此外, Chen等[69]以化合物61为原料, 在Cs2CO3催化下, 55成功将四嗪上的氮杂五元环取代, 得到化合物62, 经P2O5/HNO3硝化得到化合物63和化合物TTNOA(64), 63的密度为1.84 g·cm-3(298 K), 理论爆速8976 m·s-1, IS>40 J, 综合性能优良(图 34).
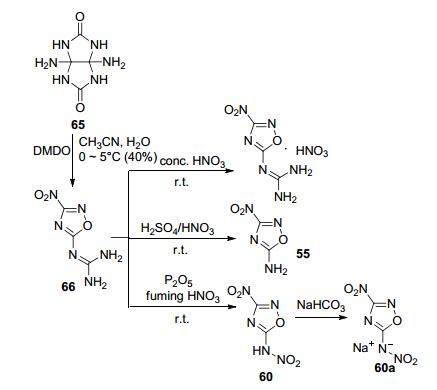
二氨基甘脲等氮杂环的重排也可得到1, 2, 4-噁二唑. Chen等[17]在接近中性条件下用二甲基过氧化酮处理二氨基甘脲, 重排后得到3-硝基-5-胍基-1, 2, 4-噁二唑(66).利用胍基良好的反应活性, 采用不同的硝化反应条件的到55, 60.其中, 60密度为1.883 g·cm-3, 爆速9095 m· s-1, 分解温度(Tdec)219 ℃, IS为15 J(图 35).
将1, 2, 4-噁二唑结构单元引入中含能化合物中, 期望利用1, 2, 4-噁二唑的稳定性和不敏感性, 同时提高化合物的具备高氧平衡值, 设计、合成新型的高能低感含能材料.
1, 2, 4-噁二唑基含能化合物性能对比可以看出, 此类化合物普遍具有感度低, 热稳定性好的特点; 共轭型化合物(如偶氮类)具有更好的综合性能.其中不乏一些性能优异的化合物, 如DDAzF的密度高达2.12 g· cm-3, 理论爆速达10114 m·s-1, 高于RDX和HMX, 与CL-20相当, 分解温度为317 ℃, IS和FS均在可接受范围内, 是一个综合性能突出的新型含能材料, 其高密度可能归因于硝氨基参与多个分子氢键相互作用和其共轭平面结构.
 下载:
导出CSV
下载:
导出CSV
| Substrate | FS/N | IS/J | Tdec/℃ | p/ (g·cm-3) | Vdet (m·s-1) | H50 / cm |
| RDX [70] | 120 | 7.4 | 210 | 1.86 | 8830 | 22 |
| HMX[71] | 120 | 7.0 | 260 | 1.91 | 9110 | — |
| DDAzF[20] | 80 | 6.0 | 317 | 2.12 | 10114 | — |
| LLM-200[46] | — | — | 295 | 1.94 | 8780 | 62 |
| 2h[7] | 160 | 4.0 | 156 | 1.95 | 8935 | — |
| 3[38] | 192 | 10 | 124 | 2.04 | 8367 | — |
| 4[39] | 80 | 10 | 151 | 2.02 | 8814 | — |
| 42[59] | 220 | 18 | 256 | 1.92 | 9240 | — |
| 57a[68] | — | >40 | 258 | 1.89 | 9319 | — |
| 60[17] | — | 15 | 219 | 1.88 | 9095 | — |
酰肼和溴化氰的反应活性高.在弱碱催化下, 即可以高收率获得环化产物.可能的反应机理见图 36.
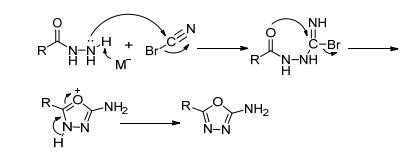
Klapötke等[72]利用二草酰肼与溴化氰缩合得到反应前体5, 5'-二氨基-1, 3, 4-噁二唑(62), 然后用100% HNO3硝化得到化合物63, 进一步成盐得到6种含能离子盐.其中铵盐(63a)的理论密度达1.95 g·cm-3, 理论爆速达9255 m·s-1, IS=10 J, FS=360 N, 综合性能优于RDX, 且反应步骤短, 收率高, 是一种有潜在应用价值的钝感高能离子盐. Zhang等[73]对化合物63进行了进一步的研究, 成功获得了单晶结构, 并进行了系统解析, 其晶体密度达1.99 g·cm-3 (298 K), 实测爆速9481 m· s-1, IS=5 J, FS=60 N, 感度略低于ε-CL-20, 反应路径短, 原料低廉, 是一种具有应用潜力的、爆轰性能突出的新型高能炸药(图 37).
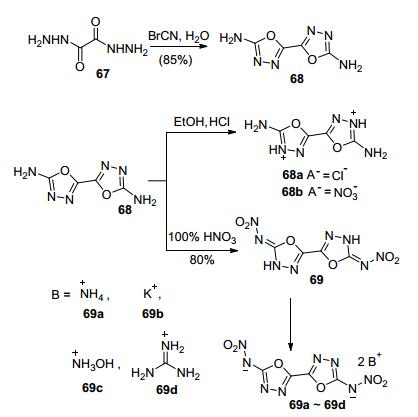
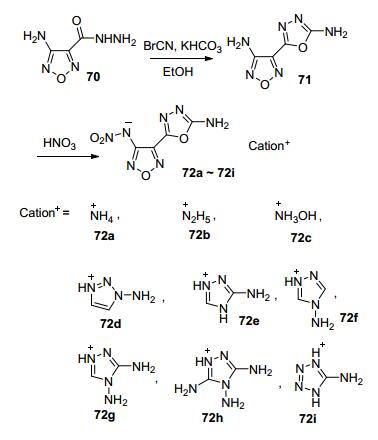
Shreeve等[74]利用溴化氰与4-氨基-1, 2, 5-噁二唑-3-乙酰肼缩合得到3-氨基-4-(2-氨基-1, 3, 4-噁二唑基)-1, 2, 5-噁二唑(71).由于与1, 3, 4-噁二唑环的氨基非常不活泼, 经硝化仅能得到单硝化产物, 进一步进行中和反应合成系列含能离子盐(72a~72i), 其理论密度1.61~1.81 g·cm-3, 理论爆速7493~8711 m·s-1(图 38).
二酰肼一般是由酰肼和羧酸衍生物的合成, 反应可在添加缚酸剂的条件下进行, 不同的取代基导致反应收率的不同.可能的反应机理如图 39(以吡啶为例).
酰氯[75, 76]和酸酐[77, 78]反应活性高, 在室温下即完成反应, 收率与取代基有关(Eq. 20).酯的反应活性较低, 通常需要在高温下回流[79]才能反应(Eq. 21).
|
|
(20) |
|
|
(21) |
通常, 二酰肼的脱水环化多使用的是五氧化二磷[80]、三氯氧磷[81]、亚硫酰氯[82]等酸性脱水试剂, 一般需要在长时间加热条件下完成环化, 后处理困难, 这种试剂不利于酸敏感型原料的环化.目前的研究主要侧重于改良实验条件, 使反应条件更温和, 产率更高, 副产物少且易于分离. Ishikawa等[83]在几乎中性的条件下使用2-氯-1, 3-二甲基咪唑氯化物(DMC)作为强力脱水剂, 在室温下用的DMC处理二酰基肼, 以优异的收率得到1, 3, 4-噁二唑(Eq. 22).
|
|
(22) |
James等[84]用二酰肼在六氯乙烷/乙腈/N, N-二异丙基乙胺Hunig碱的体系中进行处理, 三苯基膦作为温和有效的环化脱水剂迅速进行脱水环化, 在室温下即可得到产物噁二唑(Eq. 23).
|
|
(23) |
Liras等[85]以二酰基肼为原料, 以二氯甲烷作溶剂, 在吡啶/三氟甲磺酸酐体系中, 在温和的温度(-10 ℃至环境温度)下即可生成1, 3, 4-噁二唑(Eq. 24).
|
|
(24) |
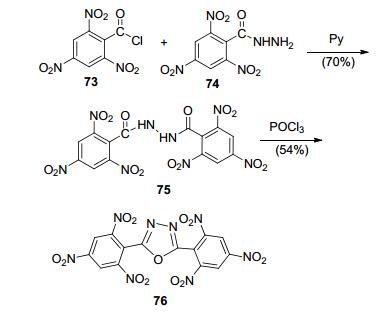
Sharnin[86]首次报道了2, 5-二三硝基苯基-1, 3, 4-噁二唑(76)的合成, 用三硝基苯酰肼和三硝基苯酰氯反应, 再在POCl3下脱水环化, 得化合物76 (图 40).
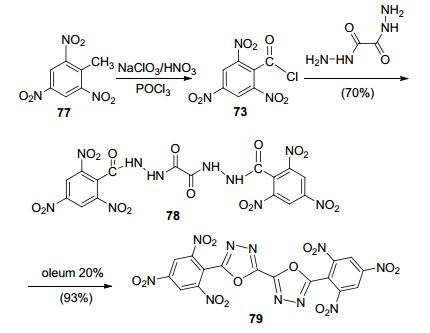
Klapötke课题组[21]三硝基甲苯(77)为原料, 经酰化得三硝基苯酰氯, 与草酰肼反应得到二酰肼, 随后在20%的发烟硫酸下脱水, 环化得到TKX-55 (79), 其有密度高(1.837 g·cm-3)、爆轰性能好(爆速8030 m·s-1, 爆压27.3 GPa)、热稳定性好(DSC初始分解温度335 ℃)、感度低(FS>360 N)的特点, 综合性能优异(图 41).
Klapötke等[87]报道了另一种合成联-1, 3, 4-噁二唑的方法:利用联四唑(80)与三氟酸酐反应得到5, 5'-三氟甲基-联-1, 3, 4-噁二唑(81), 173 K下理论密度为1.980 g· cm-3, 未报道其理论爆速(Eq. 25).
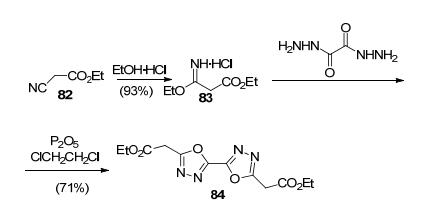
Yang等[88]以二氨基乙二肟和丙二酸二己酯为起始反应原料, 两步法合成3, 3'-联-1, 3, 4-噁二唑-5, 5'-二乙酯(84)(图 42).
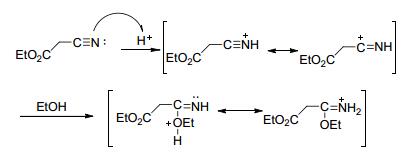
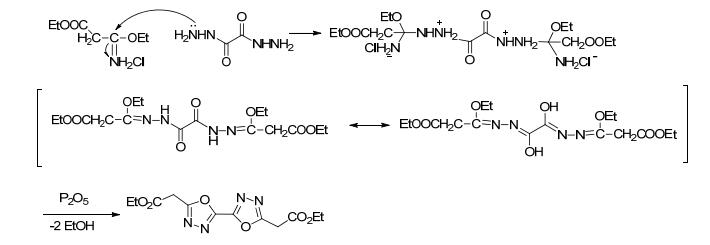
可能的反应机理为:在酸性条件下, 首先氰基N经亲核进攻、共振得到3-乙氧基-亚氨基丙酸乙酯盐酸盐(图 43), 再经亲核反应、共振、脱去两分子的乙醇后生成3, 3'-联1, 3, 4-噁二唑-5, 5'-二乙酯(图 44).
Yang等[88]以84为底物, 分别将其(1)经水解、硝化合成了具有多硝基结构的5, 5'-二(三硝基甲基)-2, 2'-联- 1, 3, 4-噁二唑(86); (2)经硝化、水解, 与不同的富氮碱反应生成含能离子盐88a~88k.这一系列化合物爆速为8770~7978 m·s-1, 其中羟胺盐(88f)的爆速为8770 m· s-1, 密度为1.85 g·cm-3, IS=20 J, FS=240 N, 综合性能与RDX相当(图 45).
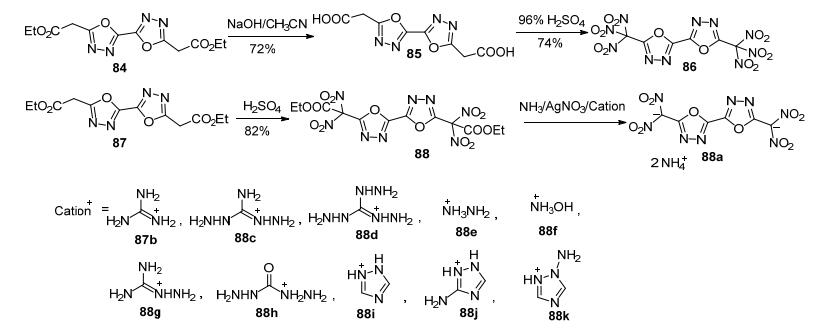
由于1, 3, 4-噁二唑的2, 5位很不活泼(2-硝基-5-氨基- 1, 3, 4-噁二唑及2, 5-二硝基-1, 3, 4-噁二唑均尚未见报道), 因此人们考虑利用酯基α-碳的活泼性引入高能基团. Shreeve等[74]利用1, 3, 4-噁二唑-2, 5-二乙酸酯(89)的α-碳引入硝基, 通过硝化得到其二硝基产物(91)、三硝基产物(92)及其6种含能盐(90a~90h).这也是目前报道的唯一1, 3, 4-噁二唑单环型含能化合物(图 46).
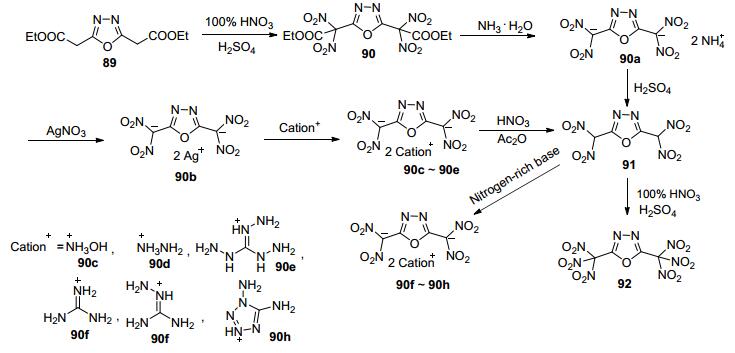
Shreeve等[89]以93为原料, 经硝化、碱性条件下脱酯生成4种含能离子盐; 再氧化成酮得到化合物96.这一系列化合物理论爆速为8042~8821 m·s-1, 其中羟胺盐的爆速为8561 m·s-1, 密度为1.87 g·cm-3, IS=33 J, FS=360 N, 是一种新型钝感含能离子盐(图 47).
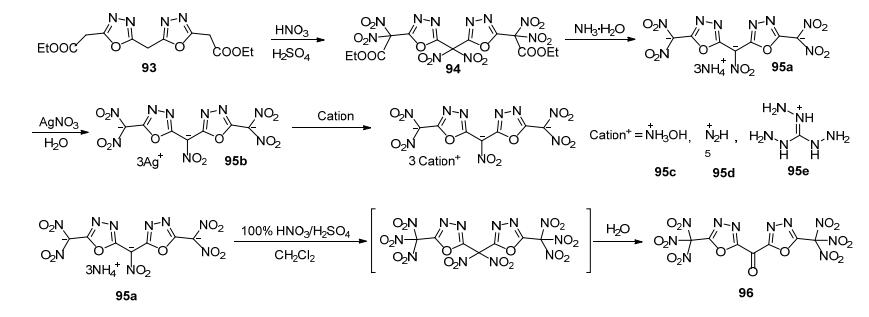
与1, 2, 4-噁二唑相比, 1, 3, 4-噁二唑生成焓更低, 且1, 3, 4-噁二唑的2, 5位很不活泼(如氨基很难氧化成硝基), 难以引入一些高能基团, 因此目前研究较少.但是1, 3, 4-噁二唑生成焓低, 含氧量高, Shreeve课题组[89]认为它可能对呋咱衍生物具有很好的稳定作用.
虽然目前报道的1, 3, 4-噁二唑基含能化合物并不丰富, 但可以看到, 1, 3, 4-噁二唑环基含能化合物普遍感度低, 有着可接受的密度和爆速, 并且已经出现一些有应用潜力的含能化合物, 如TKX-55等为代表的熔铸型炸药和以69为代表的高能炸药(表 2).
下载:
导出CSV
| Substrate | FS/N | IS/J | Tdec /℃ | p/(g·cm-3) | Vdet/(m·s-1) | H50/cm |
| RDX [70] | 120 | 7.4 | 210 | 1.86 | 8830 | 22 |
| HMX[71] | 120 | 7.0 | 260 | 1.91 | 9110 | — |
| TKX-55[21] | >360 | 5 | 335 | 1.837 | 8030 | |
| 69[72, 73] | 60[73], 72[72] | 1[72], 5[72] | 200[72], 210 [73] | 1.99[73] | 9481[73] | |
| 69a[72] | 360 | 10 | 197 | 1.95 | 9255 | |
| 88f[88] | 240 | 20 | 160.2 | 1.85 | 8770 | |
| 90c[89] | 360 | 20 | 146 | 1.89 | 9266 | |
| 95c[90] | 360 | 33 | 180 | 1.87 | 8561 |
综上所述, 异呋咱类含能化合物的设计及合成已经取得了一定的进展.事实证明, 异呋咱环是优良的含能化合物结构单元, 可有效的降低含能化合物感度.部分异呋咱类含能化合物和含能离子盐表现出结构致密, 能量水平高且感度低的特点, 特别是部分平面型异呋咱含能化合物综合性能已优于HMX, 具有潜在应用价值.
氮杂环骨架的构建是含能材料合成的灵魂和核心.迄今为止, 相对于1, 2, 5-噁二唑及1, 2, 4-噁二唑为母体结构单元的含能化合物, 1, 3, 4-噁二唑类含能化合物报道相对较少, 这主要是由于1, 3, 4-噁二唑的环化反应收率受取代基影响较大, 其次, 其2, 5位反应活性较差, 进行后续反应(如氨基偶氮化、氧化等)较为困难, 制约了1, 3, 4-噁二唑类含能化合物的发展, 如何高效构建1, 3, 4-噁二唑母体单元及在其邻位引入其它高氮杂环构建母体骨架是今后1, 3, 4-噁二唑类含能化合物研究的重点和难点.异呋咱类含能化合物研究才刚起步, 意味着该领域发展空间很大.随着含能化合物合成技术的不断进步, 有望涌现有工业化价值的新型异呋咱类不敏感含能材料.
Zhang, J. H.; Zhang, Q. H.; Thao, T. V.; Parrish, D. A.; Shreeve, J. M. J. Am. Chem. Soc. 2015, 137, 1697. doi: 10.1021/ja5126275
Zhang, J. H.; Shreeve, J. M. J. Phys. Chem. C 2015, 119, 12887.
Zhang, J. H.; Shreeve, J. M. J. Am. Chem. Soc. 2014, 136, 4437. doi: 10.1021/ja501176q
Klapötke, T. M.; Krumm, B.; Scherr, M.; Haiges, R.; Christe, K. O. Angew. Chem., Int. Ed. 2007, 46, 8686. doi: 10.1002/anie.v46:45
Joo, Y. H.; Twamley, B.; Garg, S.; Shreeve, J. M. Angew. Chem., Int. Ed. 2008, 47, 6263. doi: 10.1002/anie.v47:33
张俊林, 肖川, 翟连杰, 王锡杰, 毕福强, 王伯周, 有机化学, 2016, 36, 1197.Zhang, J. R.; Bi, F. Q.; Lian, P.; Zhang, J. L.; Wang, B. Z. Chin. J. Org. Chem. 2016, 36, 1197 (in Chinese).
Klapötke, T. M.; Mayr, N.; Stierstorfer, J.; Weyrauther, M. Chem. Eur. J. 2014, 20, 1410. doi: 10.1002/chem.v20.5
Thottempudi, V.; Yin, P.; Zhang, J. H.; Parrish, D. A.; Shreeve, J. M. Chem. Eur. J. 2014, 20, 542. doi: 10.1002/chem.v20.2
Veauthier, J. M.; Chavez, D. E.; Tappan, B. C.; Parrish, D. A. J. Energy Mater. 2010, 28, 229. doi: 10.1080/07370651003601769
何金选, 卢艳华, 雷晴, 曹一林, 火炸药学报, 2011, 34, 9.He, J. X.; Lu, Y. H.; Lei, Q.; Cao Y. L. Chin. J. Explos. Propellants 2011, 34, 9 (in Chinese).
Sheremetev, A. B.; Ivanova, E. A.; Spiridonova, N. P.; Melnikova S. F.; Tselinsky I. V.; Suponitsky K. Y.; Antipin, M. Y. J. Heterocycl. Chem.
Lim, C. H.; Kim, T. K.; Kim, K. H.; Chung, K. H.; Kim, J. S. Bull. Korean Chem. Soc. 2010, 31, 1400. doi: 10.5012/bkcs.2010.31.5.1400
张家荣, 毕福强, 廉鹏, 张俊林, 王伯周, 有机化学, 2017, 37, 2736. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346122.shtmlZhang, J. R.; Bi, F. Q.; Lian, P.; Zhang, J. L.; Wang, B. Z. Chin. J. Org. Chem. 2017, 37, 2736 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346122.shtml
Sheremetev, A. B.; Kharitonova, O. V.; Mantseva, E. V.; Kulagina, V. O.; Shatunova, E. V.; Aleksandrova, N. S.; Mel'nikova, T. M.; Ivanova, E. A.; Dmitriev, D. E.; Eman, V.; Yundin, I. L.; Kuz' min, V. S.; Strelenko, Y. A.; Novikova, T. S.; Lebedev, O. V.; Khmel'nitskii, L. I. Russ J. Org. Chem. 1999, 35, 1525.
葛忠学, 来蔚鹏, 廉鹏, 王伯周, 薛永强, 火炸药学报, 2007, 30, 5.Ge, Z. X.; Lai, W. P.; Lian, P.; Wang, B. Z.; Xue, Y. Q. Chin. J. Explos. Propellants 2007, 30, 5 (in Chinese).
Sun, Q.; Shen, C.; Li, X.; Lin, Q. H.; Lu, M. J. Mater. Chem. A 2017, 5, 11063. doi: 10.1039/C7TA02209C
Fu, Z. D.; Su, R.; Wang, Y.; Wang, Y. F.; Zeng, W.; Xiao, N.; Wu, Y. K.; Zhou, Z. M.; Chen, J.; Chen, F. X. Chem.-Eur. J. 2012, 18, 1886. doi: 10.1002/chem.201103159
Korkin, A. A.; Bartlett, R. J. J. Am. Chem. Soc. 1996, 118, 12244. doi: 10.1021/ja962744b
Kamlet, M. J.; Jacobs, S. J. J. Chem. Phys. 1968, 48, 23. doi: 10.1063/1.1667908
Qu, Y. Y.; Zeng, Q.; Wang, J.; Ma, Q.; Li, H. Z.; Li, H. N.; Yang, G. C. Chem.-Eur. J. 2016, 22, 12527. doi: 10.1002/chem.v22.35
Klapötke, T. M.; Witkowski, T. G. ChemPlusChem 2016, 81, 357. doi: 10.1002/cplu.201600078
Johnson, E. C.; Sabatini, J. J.; Chavez, D. E.; Sausa, R. C.; Byrd, E. F. C.; Wingard, L. A.; Guzmàn, P. E. Org. Process Res. Dev. 2018, 22, 736. doi: 10.1021/acs.oprd.8b00076
Chavez, D.; Klapötke, T. M.; Parrish, D.; Piercey, D. G.; Stierstorfer, J. Propellants Explos. Pyrotech. 2014, 39, 641. doi: 10.1002/prep.201300135
Yu, Q.; Cheng, G. B.; Ju, X. H.; Lu, C. X.; Lin, Q. H.; Yang. H. W. New J. Chem. 2017, 41, 1202. doi: 10.1039/C6NJ03333D
Tamura, M.; Ise, Y.; Okajima Y.; Nishiwaki, N.; Ariga, M. Synthesis 2006, 3453.
Tsiulin, P. A.; Sosnina, V. V.; Krasovskaya, G. G.; Danilova, A. S.; Baikov, S. V.; Kofanov, E. R. Russ. J. Org. Chem. 2011, 47, 1874. doi: 10.1134/S1070428011120153
Eloy, F.; Lenaers, R. Chem. Rev. 1962, 62, 155. doi: 10.1021/cr60216a003
Tiemann, F.; Kruger, P. Chem. Ber. 1884, 17, 1658.
Ooi, N. S.; Wilson, D. A. J. Chem. Soc., Perkin Trans. 2 1980, 1792.
Chiou, S. S.; Shine, H. H. J. Heterocycl. Chem. 1989, 26, 125. doi: 10.1002/jhet.v26:1
Gangloff, R. A.; Litvak, J; Shelton, E. J.; Sperandio, D.; Wang, V. R.; Rice, K. D. Tetrahedron Lett. 2001, 42, 1441. doi: 10.1016/S0040-4039(00)02288-7
Lukin, K.; Kishore, V. J. Heterocycl. Chem. 2014, 51, 256. doi: 10.1002/jhet.1689
Otaka, H.; Ikeda, J.; Tanaka, D.; Tobe, M. Tetrahedron Lett. 2014, 55, 979. doi: 10.1016/j.tetlet.2013.12.016
Kaboudin, B.; Navaee, M. Heterocycles 2003, 60, 2287. doi: 10.3987/COM-03-9769
Kiseleva, V. V.; Gakh, A. A.; Fainzil'berg, A. A. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1991, 100, 1888.
Huang, X. C.; Guo, T.; Wang, Z. J.; Liu, M.; Qiu, S. J.; Yao, B. J.; Li, H. L.; Jiang, J.; Tang, W.; Lv, Y. D.; Zhang, Y.; Chen, Z. Q.; Shi, Q.; Zheng, X. D. CN 106632124, 2017.
黄晓川, 王子俊, 郭涛, 秦明娜, 刘敏, 邱少君, 含能材料, 2017, 25, 603. doi: 10.11943/j.issn.1006-9941.2017.07.012Huang, X. C.; Wang, Z. J.; Guo, T.; Qin, M. N.; Liu, M.; Qiu, S. Chin. J. Energ. Mater. 2017, 25, 603 (in Chinese). doi: 10.11943/j.issn.1006-9941.2017.07.012
Kettner, M. A.; Karaghiosoff, K; Klapötke, T. M.; Sućeska, M.; Wunder, S. Chem.-Eur. J. 2014, 20, 7622. doi: 10.1002/chem.201402291
Kettner, M. A.; Klapötke, T. M. Chem. Commun. 2014, 50, 2268. doi: 10.1039/C3CC49879D
Gregory, G. I.; Warburton, W. K.; Seale, P. W. DE 2224338, 1972.
Pagoria, P. F.; Zhang, M. X.; Zuckerman, N. B.; DeHope, A. J.; Parrish, D. A. Chem. Heterocycl. Compd. 2017, 53, 760. doi: 10.1007/s10593-017-2122-9
Dolbier, W. R.; Burkholder, C. R.; Médebielle, M. J. Fluorine Chem. 1999, 95, 127. doi: 10.1016/S0022-1139(99)00007-X
Neves Filho, R. A. W.; Silva-Alves, D. C. B.; Anjos, J. V. D.; Srivastava, R. M. Synth. Commum. 2013, 43, 2596. doi: 10.1080/00397911.2012.724757
Zhang C. J. Mol. Struc. Theochem 2006, 795, 77
Dippold, A. A.; Izsák, D.; Klapötke, T. M. Chem. Eur. J. 2013, 19, 12042. doi: 10.1002/chem.201301339
Tsyshevsky, R.; Pagoria, P.; Zhang, M. X.; Racoveanu, A.; Parrish, D. A.; Smirnov, A. S.; Kuklja. M. M. J. Phys. Chem. C 2017, 121, 23853. doi: 10.1021/acs.jpcc.7b07584
Tarasenko, M.; Duderin, N.; Sharonova, T.; Baykov, S.; Shetnev, A.; Smirnov. V. A. Tetrahedron Lett. 2017, 58, 3672. doi: 10.1016/j.tetlet.2017.08.020
Tadikonda, R.; Nakka, M.; Gajula, M. B.; Rayavarapu, S.; Gollamudi, P. R.; Vidavalur, S. Synth. Commum. 2014, 44, 1978. doi: 10.1080/00397911.2014.883633
Klapçtke, T. M.; Piercey, D. G. Inorg. Chem. 2011, 50, 2732. doi: 10.1021/ic200071q
Li, Y. C.; Qi, C.; Li, S. H.; Zhang, H. J.; Sun, C. H.; Yu, Y. Z.; Pang, S. P. J. Am. Chem. Soc. 2010, 132, 12172. doi: 10.1021/ja103525v
Yin, P.; Parrish, D. A.; Shreeve, J. M. Chem.-Eur. J. 2014, 20, 6707. doi: 10.1002/chem.v20.22
Shaposhnikov, S. D.; Korobov, N. V.; Sergievskii, A. V.; Pirogov, S. V.; Mel'nikova, S. F.; Tselinskii, I. V. Russ. J. Org. Chem. 2002, 38, 1351. doi: 10.1023/A:1021668216426
Andrianov, V. G.; Semenikhina, V. G.; Eremeev, A. V. J. Heterocycl. Compd. 1994, 30, 4.
Yan. C; Wang, K. C.; Liu, T. L.; Yang, H. W.; Cheng, G. B.; Zhang, Q. H. Dalton Trans. 2017, 46, 14210. doi: 10.1039/C7DT03320F
Dimsdale, M. J. J. Heterocycl. Chem. 1981, 18, 37. doi: 10.1002/jhet.v18:1
Zhao, G.; He, C. L.; Yin, P.; Imler, G. H.; Parrish, D. A.; Shreeve, J. M. J. Am. Chem. Soc. 2018, 140, 3560. doi: 10.1021/jacs.8b01260
Shaposhnikov, S. D.; Korobov, N. V.; Sergievskii, A. V.; Pirogov, S. V.; Mel nikova, S. F.; Tselinskii, I. V. Russ. J. Org. Chem. 2001, 38, 1351.
Wei, H.; He, C. L.; Zhang, J. H.; Shreeve, J. M. Angew. Chem., Int. Ed. 2015, 54, 9367. doi: 10.1002/anie.201503532
Wang, Q.; Shao, Y. L.; Lu, M. Cryst. Growth Des. 2018, 18, 6150. doi: 10.1021/acs.cgd.8b01016
Yarovenko, V. N.; Taralashvili, V. K.; Zavarzin, I. V.; Krayushkin, M. M. Tetrahedron 1990, 46, 3941. doi: 10.1016/S0040-4020(01)90529-0
Augustine, J. K.; Akabote, V.; Hegde, S. G.; Alagarsamy, P. J. Org. Chem. 2009, 74, 5640. doi: 10.1021/jo900818h
Huffman, K. R.; Schaefer, F. C. Org. Chem. 1963, 28, 1816. doi: 10.1021/jo01042a018
Huttunen, K. M.; Leppänen, J.; Kemppainen E.; Palonen, P.; Rautio, J.; Järvinen, T.; Vepsäläinen, J. Synthesis 2008, 3619.
Thottempudi, V.; Zhang, J. H.; Hea, C. L.; Shreeve, J. M. RSC Adv. 2014, 4, 50361. doi: 10.1039/C4RA10821C
袁余斌, 聂进, 王烁今, 张正波, 有机化学, 2005, 25, 394. doi: 10.3321/j.issn:0253-2786.2005.04.006Yuan, Y. B.; Nie, J.; Wang, S. J.; Zhang, Z. B. Chin. J. Org. Chem. 2005, 25, 394 (in Chinese). doi: 10.3321/j.issn:0253-2786.2005.04.006
Pang, F. Q.; Wang, G. L.; Lu, T.; Fan, G. J.; Chen, F. X. New J. Chem. 2018, 42, 4036. doi: 10.1039/C7NJ03548A
Tang, Y. X.; Gao, H. X.; Mitchell, L. A.; Parrish, D. A.; Shreeve, J. M. Angew. Chem., Int. Ed. 2016, 55, 1147. doi: 10.1002/anie.201509985
Yong, T. G.; Lin, M. Z.; Fu, Q. P.; Xiu, J. Q.; Jing, L. H.; Chen, F. X. Chin. Chem. Lett. 2016, 27, 433. doi: 10.1016/j.cclet.2015.12.008
Wang, G. L.; Lu, T.; Fan, G. J.; Li, C. Q.; Yin, H. Q.; Chen, F. X. Chem-Asian J. 2018, 13, 3718. doi: 10.1002/asia.v13.23
Vroom, A. H.; Winkler, C. A. Can. J. Res. 1950, 28B, 701. doi: 10.1139/cjr50b-085
Maycock, J. N.; Verneker, V. R.; Lochte, W. Phys. Status Solidi B 1969, 35, 849. doi: 10.1002/(ISSN)1521-3951
Hermann, T. S.; Karaghiosoff, K, ; Klapötke, K. M.; Stierstorfer, J. Chem.-Eur. J. 2017, 23, 12087. doi: 10.1002/chem.201702191
Zhang, W. Q.; Zhang, J. H.; Deng, M. C.; Qi, X. J.; Nie, F.; Zhang, Q. H. Nat. Commun. 2017, 18, 1..
Tang, Y. X.; He, C. L.; Mitchell, L. A.; Parrish, D. A.; Shreeve, J. M. J. Mater. Chem. A 2015, 3, 23143. doi: 10.1039/C5TA06898C
Fang, T.; Tan, Q. T.; Ding, Z. W.; Liu, B. X.; Xu, B. Org. Lett. 2014, 16, 2342. doi: 10.1021/ol5006449
Si, Z. J.; Li, J.; Li, B.; Zhao, F. F.; Liu, S. Y.; Li, W. L. Inorg. Chem. 2007, 46, 6155. doi: 10.1021/ic061645o
Dufau, L.; Ressurreição, A. S. M.; Fanelli, R.; Kihal, N.; Vidu, A.; Milcent, T.; Soulier, J. L.; Rodrigo, J.; Desvergne, A.; Leblanc, K.; Bernadat, G.; Crousse, B.; Ravaux, M. R.; Ongeri, S. J. Med. Chem. 2012, 55, 6762. doi: 10.1021/jm300181j
Balicki, R.; Nantka-Namirski, P. Acta Pol. Pharm. 1988, 45, 1.
Saudi, M. N. S.; El-Semary, M. M. A.; Elbayaa, R. Y.; Jaeda, M. I.; Eissa, M. M.; Amer, E. I.; Baddour, N. M. Med. Chem. Res. 2012, 21, 257. doi: 10.1007/s00044-010-9532-x
Carlsen, P. H. J.; Jørgensen, K. B. J. Heterocycl. Chem. 1994, 31, 805. doi: 10.1002/jhet.v31:4
Zhang, P.; Qu, S. N.; Wang, H. T.; Bai, B. L.; Li, M. Liq. Cryst. 2008, 35, 389. doi: 10.1080/02678290801902564
Al-Talib, M.; Tastoush, H.; Odeh, N. Synth. Commun. 1990, 20, 1811. doi: 10.1080/00397919008053105
Isobe, T.; Ishikawa, T. J. Org. Chem. 1999, 64, 6989. doi: 10.1021/jo9909756
James, C. A.; Poirier, B.; Grisé, C.; Martel, A.; Ruediger, E. H. Tetrahedron Lett. 2006, 47, 511. doi: 10.1016/j.tetlet.2005.11.057
Liras, S.; Allen, M. P.; Segelstein, B. E. Synth. Commun. 2000, 30, 437. doi: 10.1080/00397910008087340
Sharnin, G. P. Khim. Geterotsikl. 1977, 6, 741.
Kettner, M. A.; Klapötke, T. M.; Witkowski, T. G.; Hundling, F. V. Chem.-Eur. J. 2015, 21, 4238. doi: 10.1002/chem.v21.11
Tian, J. W.; Xiong, H. L.; Lin, Q. H.; Cheng, G. B.; Yang, H. W. New J. Chem. 2017, 41, 1918. doi: 10.1039/C6NJ03608B
Yu, Q.; Imler, G. M.; Parrish, D. A.; Shreeve, J. M. Chem.-Eur. J. 2017, 23, 17682. doi: 10.1002/chem.201704939

图 1 呋咱与异呋咱的结构及生成焓[7]
Figure 1 Structures, and enthalpies of formation of furazan and isofurazan

图 2 异呋咱的引入对爆轰性能的影响
Figure 2 Effect of introduction for isofurazan on detonation performances

图 3 酰氯与偕胺肟O-酰化反应机理
Figure 3 Proposed O-acylation reaction mechanism of acylchloride and amidoximes

图 4 O-酰化中间体加热环化机理
Figure 4 Proposed heating cyclization mechanism of O-acylation intermediate

图 5 O-酰化中间体碱催化环化机理
Figure 5 Proposed cyclization mechanism of O-acylation intermediate via base catalysis

图 11 偕胺肟与酯的脱水环化反应机理
Figure 11 Proposed dehydration and cyclization mechanism of amidoximes and ester

图 13 偕胺肟与酸酐的O-酰化反应机理(酸催化)
Figure 13 roposed O-acylation mechanism of amidoximes and anhydride via acid catalysis

图 14 偕胺肟与酸酐的O-酰化反应机理(碱催化)
Figure 14 Proposed O-acylation mechanism of amidoximes and anhydride via base catalysis

图 18 化合物33, 34及其含能离子盐的合成
Figure 18 Synthesis of compounds 33, 34 and its energetic ion salts

图 20 偕胺肟与溴化氰缩合环化反应机理
Figure 20 Proposed reaction mechanism of amidoximes and cyanogen bromide

图 21 化合物38, 39及其含能离子盐的合成
Figure 21 Synthesis of compounds 38, 39 and its energetic ion salts

图 25 偕胺肟与有机腈合环反应机理
Figure 25 Proposed reaction mechanism of amidoxime and organic nitrile

图 26 偕胺肟与有机腈合环反应机理
Figure 26 Proposed reaction mechanism of amidoxime and organic nitrile via PTSA-ZnCl2

图 27 化合物48, 49及其含能离子盐的合成
Figure 27 Synthesis of compounds 48, 49 and its energetic ion salts

图 28 N-氰基亚胺与羟胺环化反应机理
Figure 28 Proposed cyclization reaction mechanism of N-cya- noimine and hydroxylamine

图 30 化合物53和54及其含能离子盐的合成
Figure 30 Synthesis of compounds 53 and 54 and its energetic ion salts

图 31 化合物56和57及其含能离子盐的合成
Figure 31 Synthesis of compounds 56 and 57 and its energetic ion salts

图 36 酰肼与溴化氰环化反应机理
Figure 36 Proposed cyclization reaction mechanism of acylhydrazide with cyanogen bromide

图 37 化合物68和69及含能离子盐的合成
Figure 37 Synthesis of compounds 68 and 69 and its energetic ion salts

图 45 化合物86及含能离子盐88a~88k的合成
Figure 45 Synthesis of compound 86 and energetic ion salts 88a~88k

图 46 化合物91和92和含能离子盐的合成
Figure 46 Synthesis of compounds 91 and 92 and its energetic ion salts

图 47 化合物94和96及含能离子盐的合成
Figure 47 Synthesis of compounds 94 and 96 and energetic ion salt
表 1 1, 2, 4-噁二唑含能化合物与RDX及HMX性能对比
Table 1. Properties of some 1, 2, 4-oxadiazole energetic compounds compared with RDX and HMX
| Substrate | FS/N | IS/J | Tdec/℃ | p/ (g·cm-3) | Vdet (m·s-1) | H50 / cm |
| RDX [70] | 120 | 7.4 | 210 | 1.86 | 8830 | 22 |
| HMX[71] | 120 | 7.0 | 260 | 1.91 | 9110 | — |
| DDAzF[20] | 80 | 6.0 | 317 | 2.12 | 10114 | — |
| LLM-200[46] | — | — | 295 | 1.94 | 8780 | 62 |
| 2h[7] | 160 | 4.0 | 156 | 1.95 | 8935 | — |
| 3[38] | 192 | 10 | 124 | 2.04 | 8367 | — |
| 4[39] | 80 | 10 | 151 | 2.02 | 8814 | — |
| 42[59] | 220 | 18 | 256 | 1.92 | 9240 | — |
| 57a[68] | — | >40 | 258 | 1.89 | 9319 | — |
| 60[17] | — | 15 | 219 | 1.88 | 9095 | — |
 下载: 导出CSV
下载: 导出CSV
表 2 1, 3, 4-噁二唑化合物与RDX及HMX性能对比
Table 2. Properties of some 1, 3, 4-oxadiazole compounds compared with RDX and HMX
| Substrate | FS/N | IS/J | Tdec /℃ | p/(g·cm-3) | Vdet/(m·s-1) | H50/cm |
| RDX [70] | 120 | 7.4 | 210 | 1.86 | 8830 | 22 |
| HMX[71] | 120 | 7.0 | 260 | 1.91 | 9110 | — |
| TKX-55[21] | >360 | 5 | 335 | 1.837 | 8030 | |
| 69[72, 73] | 60[73], 72[72] | 1[72], 5[72] | 200[72], 210 [73] | 1.99[73] | 9481[73] | |
| 69a[72] | 360 | 10 | 197 | 1.95 | 9255 | |
| 88f[88] | 240 | 20 | 160.2 | 1.85 | 8770 | |
| 90c[89] | 360 | 20 | 146 | 1.89 | 9266 | |
| 95c[90] | 360 | 33 | 180 | 1.87 | 8561 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

















































 下载:
下载: