
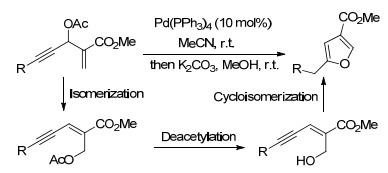
图式 1.
5-取代呋喃-3-甲酸酯的合成
Scheme 1.
Synthesis of 5-substituted furan-3-carboxylates

多取代呋喃广泛存在于天然产物和药物中[1], 同时也是重要的有机合成反应砌块[2]和构建高分子材料的基础骨架[3].因此, 发展新型高效的多取代呋喃合成方法一直是有机合成的热点之一.对近5年来的合成方法新进展进行了综述.
Ramón课题组[4]以芳炔为原料, 首先在CuO负载于Fe3O4得到的非均相催化剂和t-BuOK作用下, 通过无溶剂反应得到1, 3-二炔中间体(Eq. 1).经回收催化剂再利用之后, 向反应体系中加入KOH/H2O/DMSO, 在80 ℃下反应3 d, 将1, 3-二炔中间体转化为2, 5-二芳基取代的呋喃.原料芳炔的苯环上含有4-甲基或4-三氟甲基取代基时, 反应收率高于90%.但是苯环上含有甲氧基取代基, 反应收率明显降低(50%).脂肪炔在同样的条件下则不能完成反应.
|
|
(1) |
Reddy等[5]研究发现, 以炔醛和丙烯酸甲酯为底物通过Morita-Baylis-Hillman反应得到的产物, 再和乙酸之间发生酯化反应得到一种有价值的中间体.该中间体在Pd(PPh3)4催化下, 在温和的室温条件下通过AcO基团的1, 3-迁移异构化、脱酰基和异构化环合三步串联反应, 构建5-取代呋喃-3-甲酸甲酯类化合物(Scheme 1).芳炔上的吸电子或供电子基团对反应活性几乎没有影响, 均可以实现高于80%的收率.当R为烯基或烷基时, 也可以获得64%~72%的产物.
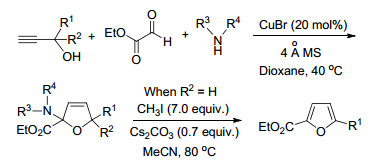
麻生明等[6]通过铜催化的炔丙基叔醇、乙醛酸乙酯和仲胺的三组分反应, 在温和条件下得到2, 2, 5, 5-四取代的二氢呋喃(Scheme 2).以炔丙基仲醇(R2=H)为底物时, 该中间体在CH3I-Cs2CO3-MeCN体系中80 ℃加热, 通过生成季铵盐和消除反应实现芳构化, 可以转化为2, 5-二取代呋喃.该合成二取代呋喃方法适用于R1为烷基、芳基(含呋喃环或萘环)或烯基的各种二级炔丙醇底物, 产物的分离收率达到50%~65%.
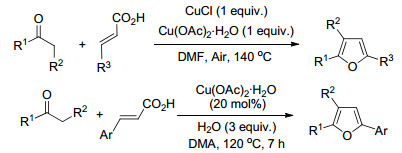
张玉红等[7]从含α-亚甲基的酮和α, β-不饱和酸两种易得原料出发, 以N, N-二甲基甲酰胺(DMF)为溶剂, 在CuCl-Cu(OAc)2·H2O促进下通过脱羧环化异构化反应, 区域选择性的合成了2, 3, 5-三取代呋喃化合物(Scheme 3).该合成策略对于大多数的底物通用性较好.但当R3为含有供电子的烷氧基团, 或R1为t-Bu、2-萘基或2-噻吩基等大位阻取代基团时, 产物的收率会明显下降(低于40%). Hajra等[8]以同样类型的底物, 在水合乙酸铜催化下, 通过脱羧环合反应得到2, 3, 5-三取代的呋喃.他们的研究表明, 向反应体系中加入水对该条件下的反应转化起很关键的作用.上述合成方法不仅合成原料易得、催化剂的成本低廉, 而且操作简便, 不需要在特殊的惰性气体保护下完成.
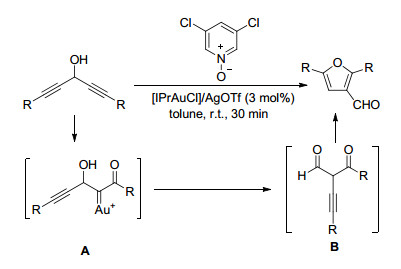
Hashmi等[9]以1, 4-二炔-3-醇为原料, 通过Au催化活化得到Au-卡宾-α-酮中间体A, 再经过1, 2-炔基迁移后异构化为1, 3-二酮的中间体B, 最后发生环化后得到2, 5-二取代呋喃-3-甲醛(Scheme 4).该合成策略条件温和(室温), 反应效率高(30 min), 对于R为芳基的各种底物均有很好的普适性(收率80%~95%); 当R=n-Bu时, 也可以达到63%的收率.
Chuang等[10]研究了1, 3-二炔二甲酸酯、三取代膦和芳醛之间的三组分反应, 得到含有活性膦叶立德(P- Ylide)取代基的三取代呋喃(Eq. 2).目标产物中的叶立德基团可以氧化得到α-酮酸酯官能团, 也可以转化为烯键.该反应仅局限于苯环上含吸电子基团的芳醛和三芳基膦的底物.亲核性更强的烷基膦以及无吸电子取代基的苯甲醛和吡啶醛则不能得到预期产物.对三芳基膦底物的进一步考察发现, 芳环上含有供电子基团时有利于活性的提高, 而含有吸电子基团或是杂环代替苯环的底物则活性相对较差.
|
|
(2) |
Hajra等[11]报道了一种以α-亚甲基酮和β-硝基苯乙烯为原料, 通过Cu(Ⅰ)催化的分子间环化反应, 以中等收率区域选择性的合成2, 3, 5-三取代呋喃的方法(Eq. 3).该反应中的酮既可以是芳香(杂环)酮, 也可以是甲基酮或环酮.原料β-硝基苯乙烯中苯环上取代基的电子效应对反应活性无明显影响.
|
|
(3) |
雷爱文等[12]选择了I2-CHP(过氧化氢异丙苯)催化氧化体系调节自由基反应的活性, 完成了β-酮酸酯和炔的氧化偶联和环化异构化的串联反应, 以中等的收率(46%~60%)得到三取代的呋喃-3-甲酸酯类化合物(Eq. 4).对底物的通用性考察表明:脂肪炔和芳炔具有相似的反应活性, 芳炔苯环上的取代基电子效应对反应收率也无明显影响, 但是以1, 3-二酮代替β-酮酸酯的反应未能得到相应的目标产物.
|
|
(4) |
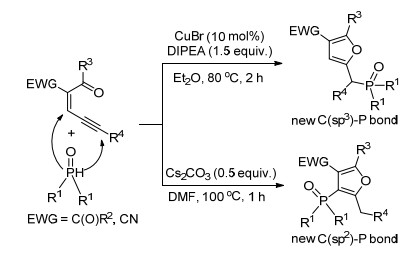
江焕峰课题组[13]以含有吸电子基团的共轭炔烯基酮和二取代磷酸酯为原料, 实现了膦基化的多取代呋喃构建(Scheme 5).在Cu(Ⅰ)催化下, 该两种底物转化为含C(sp3)—P键的2, 3, 5-三取代呋喃化合物.如果在Cs2CO3促进下, 则构建的是2, 3, 4, 5-四取代的呋喃环.
Hajra等[14]报道了Cu(Ⅱ)催化下, 在空气介质中通过芳基酮和芳基乙烯的环化反应构建三取代或四取代呋喃的方法(Eq. 5).该方法经历了铜催化的自由基反应历程, 底物适应性范围较广, 收率高.以单芳基乙烯为原料得到的是2, 3, 5-三取代呋喃; 若以二芳基乙烯为底物, 则构建的是四取代的呋喃化合物.
|
|
(5) |
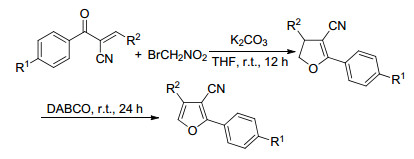
Chen和He等[15]以α, β-不饱和烯酮和溴代硝基甲烷为原料, 合成了一系列的多取代呋喃(Scheme 6).该反应在四氢呋喃(THF)中进行, 向反应体系中加入1 equiv.的K2CO3, 室温搅拌12 h, 主要产物是二氢呋喃.如果再向反应液中加入2 equiv.的DABCO, 继续室温搅拌24 h, 进一步脱氢芳构化得到三取代的呋喃.
我们课题组[16]以1, 1-二溴代芳基乙烯和乙酰乙酸乙酯为原料, 在Ag2CO3-Cs2CO3-KOAc体系的促进下, 通过氧化偶联-环合反应合成了一系列的2, 3, 5-三取代呋喃(Eq. 6).反应所需的底物1, 1-二溴代芳烯烃可通过芳醛的Corey-Fuchs反应定量的获得.底物1, 1-二溴代芳烯在相应的反应条件下首先发生消除生成炔中间体.该中间体在Ag促进下和乙酰乙酸乙酯通过C(sp3)—H和C(sp)—H的氧化偶联发生炔基化、再经历环化过程最终得到目标产物.
|
|
(6) |
Liang等[17]报道了一种环戊烷并多取代呋喃的合成方法.该方法以2-炔基-2-烯-1-酮和1, 1-二芳基乙烯为原料, 通过金催化在室温下反应1 h, 得到了该类特殊结构的多取代呋喃(Eq. 7).吸电子和供电子取代基对该合成方法的收率没有明显的影响.
|
|
(7) |
Song和You等[18]以端基炔和1, 3-二羰基化合物为原料, 在Au催化下通过C(sp3)—H和C(sp)—H的氧化偶联-环化串联反应, 得到了一系列的3-炔基多取代呋喃类化合物(Eq. 8).该反应条件温和, 产物的区域选择性高, 底物的普适性强, 1, 3-二羰基底物的结构对反应活性没有显著的影响.芳炔底物的活性则明显高于烷基炔和含有杂环的炔类.
|
|
(8) |
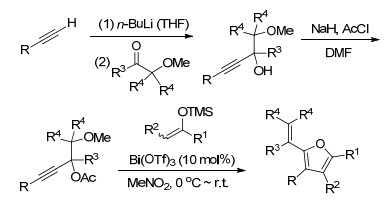
Bach等[19]以甲氧基取代的炔丙醇乙酸酯和三甲硅基烯醇醚为原料, 在Bi(OTf)3催化下合成2-烯基四取代呋喃(Scheme 7).该反应的特点是条件温和, 效率高, 在0~25 ℃条件下5 min即可完成转化.反应所需的底物炔丙醇乙酸酯可以由端基炔烃为原料, 经过两步反应制备.
徐政虎等[20]以Cu(OAc)2-Pd(OAc)2混合“转移”催化, 以CH3CN-DMSO为混合溶剂, 在较低的温度和较短的反应时间将环丙烯衍生物转化为2, 3, 4, 5-四取代呋喃类化合物(Eq. 9).
|
|
(9) |
卓克磊课题组[21]以α, β-不饱和酮和1, 3-二羰基化合物为底物, 在MnO2的促进下通过自由基加成和C—O偶联的闭环反应构建了含3, 4-二酰基的四取代呋喃(Eq. 10).反应的通用性考察表明: α, β-不饱和酮底物中的取代基电子效应对反应转化无显著影响. 1, 3-二羰基化合物底物则既可以是1, 3-二酮, 也可以是β-酮酸酯类.
|
|
(10) |
Reddy等[22]报道了一种在有机碱(DABCO和DBU)促进下, 通过MBH酯和羰基α-位亚甲基之间的取代反应和环化异构化构建四取代呋喃环的方法(Eq. 11).该合成方法的反应条件温和(室温下完成), 无需金属催化, 且原料易得(MBH酯可以通过Morita-Baylis-Hillman反应制备).
|
|
(11) |
Namboothiri等[23]以α-硝基苯乙酮亲核进攻MBH-乙酸酯, 在室温条件下以中等收率(45%~59%)得到含硝基的具有区域选择性的四取代呋喃(Eq. 12), 芳环上的供电子或吸电子取代基对反应收率没有明显的影响, 邻位取代基的位阻效应也不明显.该方法以DABCO为碱, 反应过程涉及分子内Michael加成和分子内Mannich反应等关键步骤.
|
|
(12) |
贺峥杰等[24]以三丁基膦促进的取代丙炔酸酯、芳香醛和酰氯四组分环化反应, 以中等收率生成四取代的呋喃(Eq. 13).脂肪和芳香酰氯都可以参与反应.大多数的芳香醛在优化条件下可以得到预期产物, 但是脂肪醛在同样的条件下不能完成反应.直接以丙炔酸酯为原料时, 则会得到2, 3, 5-三取代的呋喃.该合成方法首先经过原位生成磷叶立德活性中间体, 接着发生分子内Wittig反应, 再脱去三丁基氧膦得到目标产物.该反应使用简单易得的原料, 在温和的室温条件下完成, 为多取代呋喃的合成提供了简便的新方法.
|
|
(13) |
王存德等[25]报道了一例DABCO促进下, 由1-腈基环丙烷-1-甲酸酯和醛之间的开环-环加成反应合成四取代呋喃的方法, 最高收率可达到98% (Eq. 14).该反应适用于苯环上含各种供电子和吸电子基团的反应底物, 多数情况下均能取得满意的收率, 只有甲氧基的存在会导致反应收率稍微下降.该合成方法无需添加溶剂, 符合绿色合成的理念.反应的历程包含了环丙烷开环、分子间亲核加成、分子内O-亲核加成和芳构化等多个关键步骤.
|
|
(14) |
Selander等[26]开发了一种In(Ⅲ)催化的环戊烷并呋喃化合物合成方法(Eq. 15).该反应采用环境友好的Lewis酸催化体系, 以α-炔基不饱和酮、醛和仲胺为原料发生三组分反应, 具有很好的原子经济性.该方法的底物普适性也较好, 大多数的反应底物在优化后的条件下均能获得理想的反应收率和立体选择性.
|
|
(15) |
Sahoo等[27]以酚和非活化的内炔为原料, 通过Pd-催化环化的加成偶联反应, 构建了多取代的苯并呋喃类化合物(Eq. 16).该方法的反应底物普适性宽泛.酚环上取代基的电子效应对反应活性无明显的影响, 但是酚羟基邻位有取代基时, 空间位阻效应则会导致反应收率的显著降低.内炔的两个取代基既可以是芳基, 也可以是烷基.芳基-烷基混合内炔参与反应时, 产物的区域选择性很高, 主要得到的是芳基取代基在呋喃环α-位的产物.
|
|
(16) |
Maiti等[28]在相似的Pd-催化条件下, 利用酚和端基烯烃之间的偶联环化反应合成α-取代的苯并呋喃, 反应的收率从中等到较高(Eq. 17).两种底物苯环上的邻位取代基的位阻效应不显著.酚环上吸电子基团的存在不会降低反应的收率.因此, 该合成策略提供了一种用其他方法很难得到的苯环上含吸电子基团苯并呋喃的合成途径.
|
|
(17) |
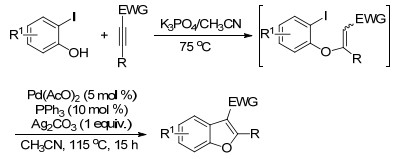
张卫东等[29]以邻碘代酚和连有吸电子基团的炔为原料, 通过酚羟基对叁键的亲核加成和Pd催化的分子内Heck反应两步串联的“一锅法”反应, 高收率地构建2, 3-二取代苯并呋喃环(Scheme 8).芳环和炔两种底物中的各种取代基团对反应效率均没有明显的影响.该合成策略还被成功应用于天然产物Daphnodorin B的全合成.
Larock等[30]以邻碘代酚、端基芳炔和芳基碘为原料, 通过微波促进下和Pd催化的三种组分“一锅”反应, 用较短的反应时间高收率地转化为2, 3-取代的苯并呋喃化合物(Eq. 18).反应效率几乎不受酚环上取代基电子效应的影响, 但是酚羟基邻位有较大体积取代基如烯丙基等存在时, 位阻会导致收率下降.此外, 部分溶解性较差的底物会影响第一步Sonogashira偶联反应的发生, 从而导致两步反应的总收率下降.底物扩展研究表明:当芳炔底物中有强吸电子基团如腈基存在时, 环合反应不能发生.脂肪族端基炔在同样的条件下也无法得到相应的呋喃产物.
|
|
(18) |
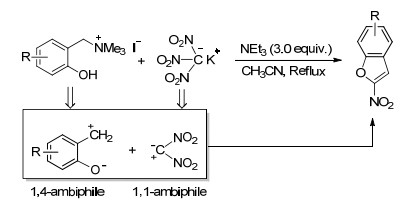
Osyanin等[31]报道了一例合成2-硝基取代的苯并呋喃衍生物的新方法.该反应以三硝基甲烷化钾作为1, 1-亲电-亲核两性等价体, 和1, 4-两性等价体环合, 反应收率中等(Scheme 9).该类化合物不仅具有重要的生理活性, 还是非常有价值的合成中间体.苯环上的各种供电子和吸电子基团以及邻位取代基均不会影响反应活性.
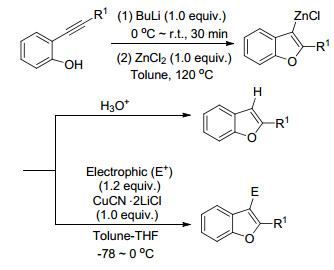
Tsuji和Nakamura等[32]以邻炔基酚为原料, 通过BuLi-ZnCl2促进的分子内环化反应得到苯并呋喃-β-氯化锌中间体.该中间体质子化后直接得到三取代的呋喃, 若在低温下接受亲电试剂进攻, 则可以转化为四取代的呋喃(Scheme 10).该方法适用于大多数的反应底物, 转化率最高可接近100%.
近年来, 有机化学家在呋喃衍生物的合成方面做了大量的研究, 发现了很多新颖的合成方法.由于呋喃化合物在天然产物、药物和合成领域有着重要的作用, 开发新的合成方法, 尤其是从廉价易得的原料出发, 使用简便的操作和温和的反应条件, 底物通用性和官能团兼容性宽泛的新方法, 仍将是化学家们持续关注的热点.通过原子经济性高的多组分反应和串联反应构建含有各种官能团的多取代呋喃, 是有机化学和绿色化学领域面临的新挑战.
(a) Hou, X. L.; Yang, Z.; Wong, H. N. C. Prog. Heterocycl. Chem. 2003, 15, 167.
(b) Keay, B. A.; Dibble, P. W. In Comprehensive Heterocyclic Chemistry Ⅱ, Vol. 2, Eds.: Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Elsevier, Oxford, 1997, p. 395.
(c) Suhre, M. H.; Reif, M.; Kirsch, S. F. Org. Lett. 2005, 7, 3925.
(d) Rao, A. U.; Xiao, D.; Huang, X.; Zhou, W.; Fossetta, J.; Lundell, D.; Tian, F.; Trivedi, P.; Aslanian, R.; Palani, A. Bioorg. Med. Chem. Lett. 2012, 22, 1068.
(e) Kumari, N.; Mishra, C. B.; Prakash, A.; Kumar, N.; Mongre, R.; Luthra, P. M. Neurosci. Lett. 2014, 558, 203.
(f) Hasegawa, F.; Niidome, K.; Migihashi, C.; Murata, M.; Negoro, T.; Matsumoto, T.; Kato, K.; Fujii, A. Bioorg. Med. Chem. Lett. 2014, 24, 4266.
(a) Kalaitzakis, D.; Triantafyllakis, M.; Alexopoulou, I.; Sofiadis, M.; Vassilikogiannakis, G. Angew. Chem., Int. Ed. 2014, 53, 13201.
(b) Wang, Z.-L.; Li, H.-L.; Ge, L.-S.; An, X.-L.; Zhang, Z.-G.; Luo, X.; Fossey, J. S.; Deng, W.-P. J. Org. Chem. 2014, 79, 1156.
(a) Zeng, C.; Seino, H.; Ren, J.; Hatanaka, K.; Yoshie, N. Macromolecules 2013, 46, 1794.
(b) Zeng, C.; Seino, H.; Ren, J.; Hatanaka, K.; Yoshie, N. Polymer 2013, 54, 5351.
(c) Gidron, O.; Shimon, L. J. W.; Leitus, G.; Bendikov, M. Org. Lett. 2012, 14, 502.
(d) Gidron, O.; Dadvand, A.; Sheynin, Y.; Bendikov, M.; Perepichka, D. F. Chem. Commun. 2011, 47, 1976.
(e) Bunz, U. H. F. Angew. Chem., Int. Ed. 2010, 49, 5037.
(f) Gidron, O.; Diskin-Posner, Y.; Bendikov, M. J. Am. Chem. Soc. 2010, 132, 2148.
Pérez, J. M.; Cano, R.; Yus, M.; Ramón, D. J. Synthesis 2013, 45, 1373. doi: 10.1055/s-00000084
Reddy, C. R.; Krishna, G.; Reddy, M. D. Org. Biomol. Chem. 2014, 12, 1664. doi: 10.1039/c3ob42396d
Fan, W.; Ma, S. M. Eur. J. Org. Chem. 2015, 3531.
Yang, Y. Z.; Yao, J. Z.; Zhang, Y. H. Org. Lett. 2013, 15, 3206. doi: 10.1021/ol400912v
Ghosh, M.; Mishra, S.; Monir, K.; Hajra, A. Org. Biomol. Chem. 2015, 13, 309. doi: 10.1039/C4OB01320D
Wang, T.; Shi, S.; Hansmann, M. M.; Rettenmeier, E.; Rudolph, M.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2014, 53, 3715. doi: 10.1002/anie.v53.14
Deng, J. C.; Chuang, S. C. Org. Lett. 2014, 16, 5792. doi: 10.1021/ol502879c
Ghosh, M.; Mishra, S.; Hajra, A. J. Org. Chem. 2015, 80, 5364. doi: 10.1021/acs.joc.5b00704
Tang, S.; Liu, K.; Long, Y.; Qi, X. T.; Lan, Y.; Lei, A. W. Chem Commun. 2015, 8769.
Yu, Y.; Yi, S. G.; Zhu, C. L.; Hu, W. G.; Gao, B. J.; Chen, Y.; Wu, W. Q.; Jiang, H. F. Org. Lett. 2016, 18, 400. doi: 10.1021/acs.orglett.5b03415
Dey, A.; Ali, M. A.; Jana, S.; Hajra, A. J. Org. Chem. 2017, 82, 4812. doi: 10.1021/acs.joc.7b00476
Chen, R. S.; Fan, X.; Xu, Z. Z.; He, Z. J. Tetrahedron Lett. 2017, 58, 3722. doi: 10.1016/j.tetlet.2017.08.027
Zhang, W. S.; Xu, W. J. Chem. Heterocycl. Compd. 2017, 53, 615. doi: 10.1007/s10593-017-2100-2
He, T.; Gao, P.; Qiu, Y. F.; Yan, X. B.; Liu, X. Y.; Liang, Y. M. RSC Adv. 2013, 13, 19913. https://www.researchgate.net/publication/272320384_Gold-catalyzed_intermolecular_reaction_of_2-1-alkynyl-2-alken-1-ones_with_diarylethenes_to_construct_polysubstituted_cyclopentacfurans_through_a_cascade_heterocyclization32_cycloaddition_sequence
Ma, Y. H.; Zhang, S.; Yang, S. P.; Song, F. J.; You, J. S. Angew. Chem., Int. Ed. 2014, 53, 7870. doi: 10.1002/anie.201402475
Nitsch, D.; Bach, T. J. Org. Chem. 2014, 79, 6372. doi: 10.1021/jo5009993
Song, C. L.; Wang, J. W.; Xu, Z. H. Org. Biomol. Chem. 2014, 12, 5802. doi: 10.1039/C4OB00987H
Yue, Y. Y.; Zhang, Y. L.; Song, W. W.; Zhang, X.; Liu, J. M.; Zhuo, K. L. Adv. Synth. Catal. 2014, 356, 2459. doi: 10.1002/adsc.v356.11/12
Reddy, C. R.; Mohammed, S. Z.; Kumaraswamy, P. Org. Biomol. Chem. 2015, 13, 8310. doi: 10.1039/C5OB00989H
Mane, V.; Kumar, T.; Pradhan, S.; Katiyar, S.; Namboothiri, I. N. N. RSC Adv. 2015, 5, 69990. doi: 10.1039/C5RA11471C
邹雯, 贺峥嵘, 贺峥杰, 有机化学, 2015, 35, 1739. http://sioc-journal.cn/Jwk_yjhx/EN/Y2015/V35/I8/1739Zou, W.; He, Z. R.; He, Z. J. Chin. J. Org. Chem. 2015, 35, 1739(in Chinese). http://sioc-journal.cn/Jwk_yjhx/EN/Y2015/V35/I8/1739
Liu, J. M.; Ye, W. J.; Qing, X. S.; Wang, C. D. J. Org. Chem. 2016, 81, 7970. doi: 10.1021/acs.joc.6b01259
Pathipati, S. R.; Werf, A.; Eriksson, L.; Selander, N. Angew. Chem., Int. Ed. 2016, 55, 11863. doi: 10.1002/anie.201606108
Kuram, M. R.; Bhanuchandra, M.; Sahoo, A. K. Angew. Chem., Int. Ed. 2013, 52, 4607. doi: 10.1002/anie.201210217
Sharma, U.; Naveen, T.; Maji, A.; Manna, S.; Maiti, D. Angew. Chem., Int. Ed. 2013, 52, 12669. doi: 10.1002/anie.201305326
Yuan, H.; Bi, K. J.; Li, B.; Yue, R. C.; Ye, J.; Shen, Y. H.; Shan, L.; Jin, H. Z.; Sun, Q. Y.; Zhang, W. D. Org. Lett. 2013, 15, 4742. doi: 10.1021/ol4021095
Markina, N. A.; Chen, Y.; Larock, R. C. Tetrahedron 2013, 69, 2701. doi: 10.1016/j.tet.2013.02.003
Osyanin, V. A.; Osipov, D. V.; Demidov, M. R.; Klimochkin, Y. R. N. J. Org. Chem. 2014, 79, 1192. doi: 10.1021/jo402543s
Tsuji, H.; Ilies, L.; Nakamura, E. Synlett 2014, 25, 2099. doi: 10.1055/s-00000083

图式 2 以炔、醛和胺为原料Cu(Ⅰ)-催化合成2, 5-二取代呋喃
Scheme 2 Copper(Ⅰ)-catalyzed synthesis of 2, 5-disubstituted furans from alkynes, aldehydes and amines

图式 3 以烷基酮和a, β-不饱和酸为原料合成2, 3, 5-三取代呋喃
Scheme 3 Synthesis of 2, 3, 5-trisubstituted furans from alkyl ketones and a, β-unsaturated carboxylic acids

图式 6 以α, β-不饱和烯酮和溴代硝基甲烷为原料合成多取代呋喃
Scheme 6 Synthesis of polysubstituted furansfrom bromonitromethane and oxodienes

图式 8 3-取代-3-功能化苯并呋喃的合成
Scheme 8 Synthesis of 2-substituted-3-functionalized benzofurans

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


















 下载:
下载:





 下载:
下载: