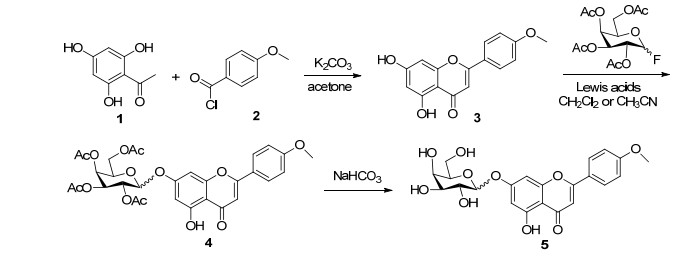
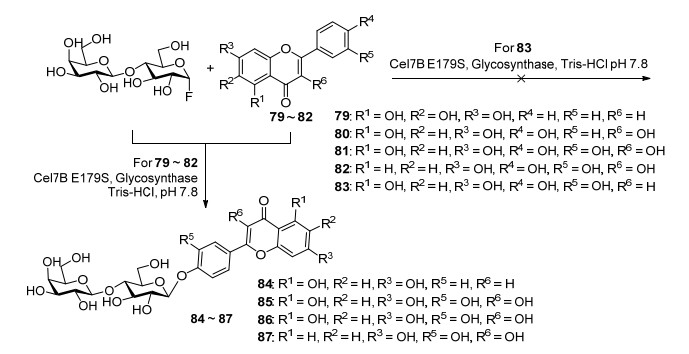
图式 1.
金合欢素7-O-β-D-吡喃半乳糖苷的全合成路线
Scheme 1.
Total synthetic route of acacetin-7-O-β-D-galactopyranoside

黄酮苷(flavonid glycoside)是黄酮苷元与糖基通过苷键连接形成的化合物.其中, 通过碳-氧键连接的称为黄酮氧苷, 碳-碳键连接的称为黄酮碳苷.黄酮苷类广泛存在于自然界植物, 具有抗炎[1, 2]、抗氧化[3~5]、抗肿瘤[6, 7]、抗菌[8, 9]、降血糖[10, 11]等药理活性.研究表明, 糖链是重要的生物分子和信息载体, 参与细胞生物几乎所有的生命过程, 特别是在细胞分化、发育、免疫、老化、癌变、信息传递等生命基础活动和重大疾病过程中起着特异性的识别、介导与调控作用[12~14].除此之外, 糖基化可改变药物的生物活性, 增加水溶性, 降低毒副作用, 提高特异靶向性等特点, 具有广阔的发展前景[15~17].目前黄酮苷主要是从植物中提取分离, 但是许多具有良好活性的黄酮苷在植物中的含量较低[18, 19].如nervilifordins F和G是自然分布于两广少数民族地区的特产名贵药材青天葵的黄酮氧苷, 具有良好的抗炎活性, 但药材资源稀缺, 且从青天葵的干燥地上部分提取的产率仅为1.2%和0.6%[20], 极大限制了对其研究及潜在药用价值的开发利用.正因为黄酮苷具有这些独特的性质, 其黄酮苷的合成研究日益受到重视. 2011年Kondo等[21]综述报道了2000年至2010年关于黄酮及黄酮苷的合成; 2013年Wang等[22]对天然黄酮及黄酮苷的首次合成进行了综述; 2014年梅青刚等[23]对黄酮醇的合成进行了概述; 2014年Yu等[24]对黄酮氧苷的合成作了综合报道; 2017年Iranshahi等[25]对一锅法合成黄酮作了综合概述; 2018年Suzuki等[26]对芳香糖苷的合成作了一个全面综述, 包括黄酮碳苷的合成.国内外对黄酮苷的合成研究虽然较多, 对于那些常用的糖苷化反应的合成产率仍较低, 特别是区域选择性糖苷化, 尤其是涉及到多个糖苷合成时, 产率更低.随着生物技术的发展, 酶催化生物合成的黄酮苷也越来越广泛, 2014年Xiao等[27]和2016年Hofer[28]分别综述报道了酶催化和微生物代谢工程合成黄酮苷. 2014年来关于黄酮的合成只针对某一面的综述, 因此本文对近几年黄酮苷的合成研究作一较系统全面的综述.
黄酮苷的合成主要包括化学合成和酶催化生物两种类型, 化学合成法又分为全合成和半合成.
在黄酮苷的化学合成中, 苷元与糖链的连接是最关键的步骤.全合成反应需要的步骤相对比较冗长, 但是可以根据目标分子的结构特点而灵活设计合成路线, 可以构建自然界中不存在的及大量合成自然界中含量极低的苷元.
目前黄酮苷元的全合成有经典的两种方法, 即β-丙二酮酸化关环法(Baker-Venkataraman, BK-VK法)和查尔酮氧化关环法(Algar-Flynn-Oyamada, AFO法).
Baker-Venkataraman (BK-VK)反应是合成黄酮的最常用的方法, 也是最简便的方法, 该反应经过了酯化和分子内重排两步反应, 反应条件较为温和.
由于传统BK-VK法通常需要剧烈的反应条件, 并且产率较低.近年来, 人们对所用碱催化剂、相转移催化法以及硅烷基羟基保护等方面进行不断改良, 同时设法使酯化和克莱森重排两步反应同步进行.
文献报道[29], 在大量合成5-甲氧基黄酮时, 传统BK-VK法存在产率低和产物分难离的问题.因此, 使用相转移催化剂使酯化和重排两步反应连续同步进行, 从而避免了反应物苯甲酰氯与克莱森重排目标产物分离困难的情况, 且提高了产率.传统BK-VK法由于剧烈的反应条件和长时间反应使敏感性的甲氧基水解, 导致传统BK-VK法合成5-甲氧基黄酮产率低.然而, 在碱性较弱K2CO3的温和条件下使用相转移催化剂叔丁基醇钾, 对BK-VK法这样的亲核反应是有利的, 从而使得回收率得到大大的提高[30].
在对影响BK-VK反应酯化和重排二步反应的因素如溶剂、碱催化剂和闭环试剂等的研究中, 发现以丙酮为溶剂、K2CO3为碱、HOAc-NaOAc闭环试剂反应体系最有效, 且具有反应条件温和、操作简便、收率提高、溶剂成本降低、易于回收、反应时间缩短等优点[31].
在合成金合欢素7-O-β-D-吡喃半乳糖苷(5)的研究中, 直接以廉价易得的2, 4, 6-三羟基苯乙酮(1)和对甲氧基苯甲酰氯(2)为原料, 在K2CO3催化下, 通过BK-VK反应一步合成金合欢素(3).考察了在不同溶剂、不同路易斯酸催化剂条件下, 不同构型的乙酰基保护的氟代半乳糖进行糖苷化反应对糖苷化产物构型影响, 结果发现, 各种溶剂、路易斯酸、糖基供体得到的主要是β构型的糖苷化产物, 其中以二氯甲烷为溶剂, BF3·OEt2为催化剂, 糖基供体为β构型时, 糖苷产物α、β构型的比例为3:97, 总收率高达46% (Scheme 1)[32].该反应步骤较短, 同时糖苷化产物主要为β构型, 与绝大多数天然糖苷类产物为β构型相一致, 对于天然糖苷化合物的合成具有重要参考意义.
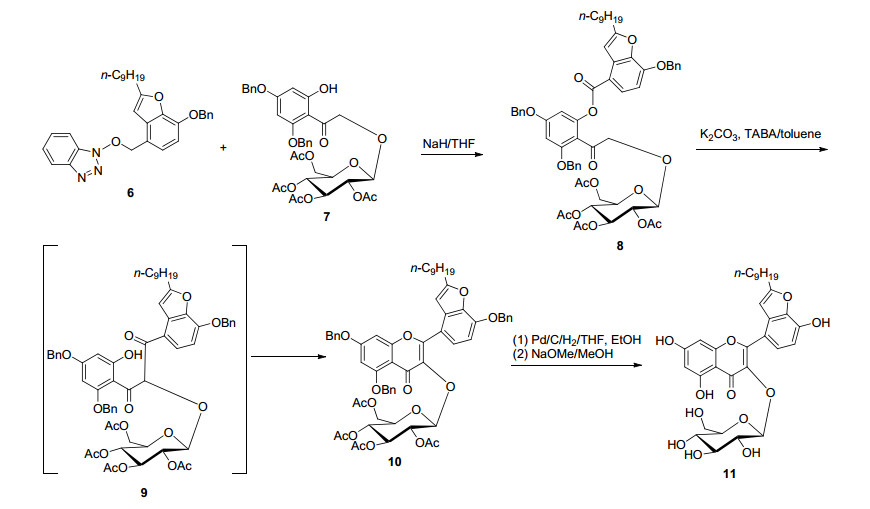
2015年首次报道了通过全合成法获得具有良好抗病毒活性的天然产物鱼腥草素B (11)[33].首先以苄基保护的白杨素通过逆醛醇反应降解得到苯乙酮衍生物, 接着通过Rubottom氧化反应得到羟基化产物, 然后在BF3·OEt2催化下, 与乙酰基保护的溴代半乳糖进行糖基化反应得到半乳糖基化的羟基苯乙酮(7)关键结构单元; 以3, 4-二羟基苯甲醛与炔烃合成苯并呋喃结构单元, 再与苯并三唑衍生成活性的芳基酯(6).最后, 在NaH存在下, 半乳糖基化的羟基苯乙酮与活性的芳基酯(6)进行酯化反应得到糖基芳基酯(8), 然后在K2CO3催化剂、四丁基溴化铵(TBAB)相转移催化剂催化下, 通过BK- VK重排、环化脱水构建黄酮碳骨架, 最后脱苄基和乙酰基得到鱼腥草素B (11), 总收率为11.0% (Scheme 2).在上述反应过程中, 首先通过合成目标化合物结构片段, 最后通过BK-VK重排、环化脱水关键步骤构建黄酮碳骨架, 这在合成苯并含O、N等杂环黄酮时有借鉴意义.
查尔酮是合成黄酮的关键中间体.查尔酮氧化关环法又称Algar-Flynn-Oyamada (AFO)法, Algar-Flynn- Oyamada法最初由爱尔兰的Algar、Flynn[34]和日本的Oyamada[35, 36]于1934年同时发现, 后来广泛应用于黄酮醇的合成[37~39].
多甲氧基黄酮具有显著抗癌、抗炎等活性, 但其水溶性差, 对生物受体的亲和能力不强, 而进行糖基化后可大大改善上述缺点, 近年来对其化学全合成及其糖苷化的研究日益受到重视[40].文献报道, 以相应甲氧基的苯乙酮和苯甲醛衍生物为原料, 通过AFO反应合成了一系列多氧甲基黄酮醇, 并以其中的3', 4', 7-三甲氧基黄酮醇为苷元与不同的乙酰基保护的溴代糖进行糖苷缩合反应, 得到一系列以常见的单糖为糖基的3', 4', 7-三甲氧基黄酮苷[41].
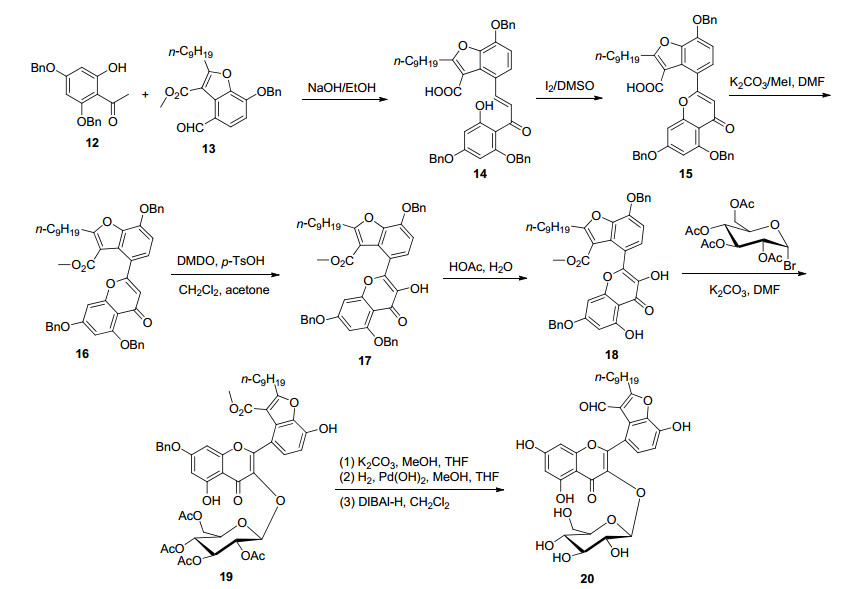
继2015年报道全合成鱼腥草素B之后, 2018年报道了全合成了鱼腥草素的重要成员之一鱼腥草素A (20)[42], 类似上例合成鱼腥草素B一样, 首先合成目标化合物结构片段, 再构建黄酮骨架.在本例中, 首先将3, 4-二羟基苯甲醛合成苯并呋喃醛(13)结构单元, 然后在NaOH碱性条件下, 苯并呋喃醛(13)与廉价易得的芳基酮(12)羟醛缩合反应生成查尔酮中间体(14), 然后I2氧化关环、过氧丙酮(DMDO)氧化羟基化、HOAc/H2O选择性去5-苄基后得到黄酮醇(18), 再在K2CO3催化下, 与乙酰基保护的溴代糖糖基供体进行缩合反应, 最后去乙酰基、去苄基得到目标产物鱼腥草素A (20), 总收率为8.0% (Scheme 3).值得注意的是, 本例与上例合成鱼腥草素B一样, 反应步骤繁多, 总收率不高.不同的是上例合成鱼腥草素B是首先合成糖基化的羟基苯乙酮关键结构单元, 再通过BK-VK反应构建黄酮骨架, 而本例是首先通过AFO反应合成黄酮醇, 最后再进行糖基化.
BK-VK反应和AFO反应均具有原料廉价易得, 溶剂成本降低, 反应步骤易于操作等优点.但是合成黄酮醇时, 需要经过黄酮中间体, 反应步骤多, 产率一般也不高.查尔酮是合成各类黄酮的重要中间体, 在不同的氧化条件下, 可得到不同类型的黄酮化合物. AFO反应是目前人们普遍应用于合成黄酮醇的一种方法, 但该反应受底物取代基和反应温度的影响较大, 副产物较多, 分离纯化较困难.人们可根据目标化合物情况, 灵活选择这两种方法.
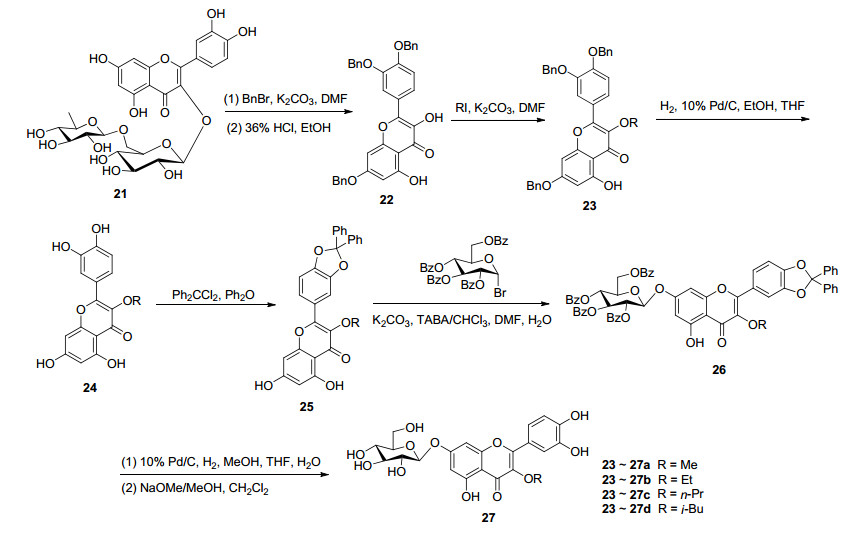
以黄酮苷为原料, 一般经过乙酰化、苯甲酰化或苄基化、酸水解等反应, 得到保护基保护的黄酮苷元, 再与糖基供体进行糖苷化反应.如以芦丁(21)为原料, 经过苄基化、酸水解得到苄基保护的槲皮素, 再与乙酰基保护的溴代糖进行糖苷化反应, 然后经过去乙酰基和去苄基等步骤获得系列槲皮素3-糖苷、3, 4'-O-二糖苷和3, 7-O-二糖苷[43].由于共轭效应而使得槲皮素的7、4'与3位羟基活性更高, 因此, 在合成槲皮素7-O-糖苷时, 3位和4'位羟基一般需选择性保护起来.文献报道, 芦丁(21)经过选择性苄基化、酸催化水解、选择性3-羟基烷基化、氢化去苄基后, 以二苯基甲叉选择性保护3', 4'位羟基, 再选择性7位羟基糖基化, 得到槲皮素7-O-糖基化的关键中间体(26a~26d), 再经过去二苯基甲叉保护基、苯乙酰基等步骤合成了一系列槲皮素7-O-β-D-吡喃葡萄糖苷(27a~27d)[44, 45](Scheme 4).
柚皮苷和橙皮苷是天然二氢黄酮苷, 广泛分布于柑桔、柚子等芸香科属植物中.文献报道, 柚皮苷和橙皮苷经过糖苷水解、碘/吡啶脱氢、甲基化或苄基保护、催化氢化脱苄基、糖基化等反应步骤, 得到了一系列多甲氧基黄酮苷[40, 46].
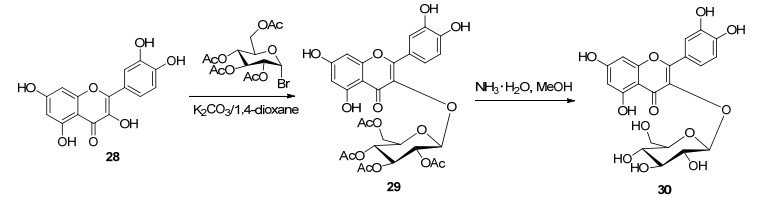
以天然的黄酮醇为原料, 利用苯环上酚羟基位置不同而导致反应活性大小不同的性质, 进行选择性取代得到多种取代的黄酮苷.其中黄酮醇以槲皮素、山奈酚最为常见. Park等[47]报道在K2CO3/1, 4-二氧六环体系中, 槲皮素(28)直接与乙酰基保护的溴代糖进行糖苷化反应, 然后在NH3/MeOH碱性条件下去乙酰基得到槲皮素3-O-β-D-葡糖苷(30), 收率为60% (Scheme 5). Yang等[48, 49]以山萘酚为原料, 合成了系列山萘酚3-O-糖苷和山萘酚3, 7-二糖苷化合物.山奈酚衍生物是淫羊藿苷具有良好抗肿瘤活性的黄酮苷元, 文献报道[50], 以山奈酚为原料, 经过11个线性步骤半合成了淫羊藿苷, 总收率为7%.芹菜素7-O-β-D-吡喃葡萄糖-4'-O-α-L-吡喃糖苷具有抑制乙型肝炎病毒复制、抗中风等活性的天然产物, 有研究报道直接以柚皮素为原料与苯甲酰基保护的溴代糖糖苷化, 2, 3位氧化脱氢, 再与苯甲酰基保护的溴代糖糖苷化反应, 碱水解苯甲酰基保护基四步反应, 合成了目标产物, 并经过同样的方法获得芹菜素7, 4'-二-O- β-D-吡喃葡萄糖苷, 总收率为45%[51].该法具有反应步骤较短、友好、廉价等优点.
糖苷链的合成一般是在糖基供体和糖基受体在催化剂作用下进行的.黄酮氧苷合成的方法目前已有二十多种, 目前常用的有三种: Koening-Knorr法、糖基三氯乙酰亚胺酯法、相转移催化法, 而金(Au (Ⅰ))催化的糖邻炔基苯甲酸酯法可高效合成黄酮5-O糖苷.目前最常用的糖基给体有糖基卤化物、糖基三氯乙酰亚胺酯、邻炔基苯甲酸糖基酯和硫苷等.
经典的Koenig-Knorr法始于1901年[52], 经过百年来不断发展已成为一个常用的合成糖苷和寡糖的方法, 在早期合成黄酮苷时被广泛应用[53~55].常以本乙酰基和乙酰基保护的卤代糖作糖基供体, 苯、甲苯、吡啶以及喹啉等作溶剂, 重金属盐(汞盐或银盐)作催化剂, 无水硫酸钙、4Å分子筛作除水剂. Koenig-Knorr法在糖苷合成中应用比较早, 条件简便并且比较成熟, 但是由于产率低、区域选择性差, 糖基给体不稳定, 无水条件苛刻, 并且使用的催化剂汞盐或银盐昂贵且有毒, 对环境不友好, 后处理步骤相对比较烦琐等缺点, 近年来该方法使用逐渐减少.
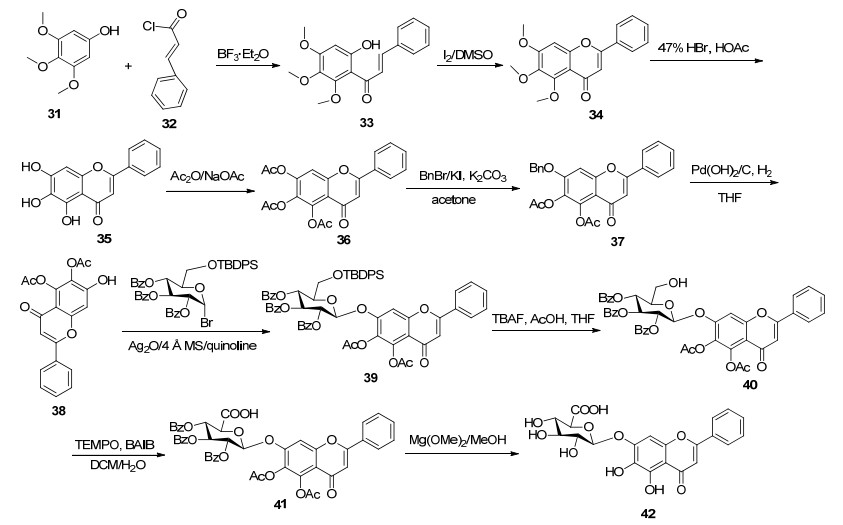
黄芩苷是传统中草药黄芩的主要成分之一, 具有抗氧化、抗病毒、抗HIV、保肝等生物活性. Mezey-Vandor等[56]以黄芩素为苷元, 直接以溴代的葡萄醛酸为糖基供体进行糖基化反应, 获得所需的目标产物, 产率较低(40%).在碳水化合物合成反应中, 其中具有挑战性的主要问题之一是糖苷化反应产率较低, 而葡萄糖醛酸糖基供体比一般的糖基供体的反应活性更低, 导致糖苷化反应产率进一步降低.并且, 在Koenig-Knorr反应条件下, 葡萄醛酸甲酯糖基供体不稳定, 进行消除反应产生2-酰氧基乙二醇副产物[57].因此, Li等[58, 59]对糖基供体进行改进, 首先以硅烷基、苯乙酰基保护的溴代葡萄糖为糖基供体进行糖基化反应, 获得所需的糖基化产物(糖苷化产率92%), 然后再选择性去6位保护基, 通过Widlanski氧化(TEMPO/BAIB)形成羧酸获得葡糖糖醛酸糖苷化的目标产物, 葡萄醛酸化产率59%.在反应过程中, 首先以3, 4, 5-三甲氧基苯酚(31)和肉桂酰氯(32)为原料, 经过酰化、查尔酮氧化闭环、去甲基等反应得到黄芩素(39), 接着, 再经过区域选择性5, 6, 7-三羟基乙酰化、7-乙酰基苄基化、去7-苄基等步骤得到5, 6-二乙酰基黄芩素(38).然而, 与N-苯基三氟乙酰亚胺糖基酯和金催化的邻炔基苯甲酸糖基酯为糖基供体进行糖苷化反应, 结果却得不到糖基化产物, 因此, 通过经典的Koenigs-Knorr法, 以喹啉作溶剂、Ag2O作催化剂、4Å分子筛作除水剂, 与溴代葡萄糖糖进行糖苷化反应获得糖苷产物, 最后经过去保护基反应得到黄芩苷(42), 总产率为27% (Scheme 6).在反应中, 通过使用Koenig- Knorr法成功地构建关键的糖苷键, 同时也通过该法使黄芩素苷元与不同的糖基供体进行糖苷化反应, 得到了四种黄芩苷衍生物, 糖苷化产率达85%~94%.在上述反应中, 体现了糖苷化反应的局限性和上述合成路线Koenig-Knorr法的一般适用性.因此, 这种传统的Koenig-Knorr法在其他糖苷化不适合的条件下仍然具有适用性.
相转移催化法是在经典Koenig-Knorr法上发展起来, 常以邻位酰基等保护的溴代糖为糖基给体, 是合成黄酮醇3-糖苷的首选方法[26].该法具有操作简便, 反应速度快, 反应条件温和, 后处理简单和立体选择性强, 提高了产率, 避免了使用价格昂贵的重金属盐, 以及银盐法中卤代糖基不稳定等优点.其中, 四丁基溴化铵(TBAB)是目前最常用的相转移催化剂.
Demetzos等[60]在CHCl3/KOH体系中, 使用相转移催化剂三乙硫醇胺(TMEA)催化乙酰基保护的溴代糖与7, 4-二-O-苄基槲皮素反应, 得到系列槲皮素3-O-糖苷, 产率为10%~60%.进一步地, 在无水丙酮/水碳酸钾体系中, 以TBAB为相转移催化剂催化乙酰基溴代葡萄糖与5, 6, 7, 8, 3, 4'-六甲氧基黄酮醇进行糖苷化反应, 然后在30%氨水碱性条件下脱乙酰化得到5, 6, 7, 8, 3', 4'-六甲氧基黄酮-3-O-β-葡萄糖苷, 产率为85.0%[61].在相转移催化法的反应过程中, 使用碱性相对KOH更弱的K2CO3, 降低碱的浓度的同时也减少了溴代糖水解副反应的发生, 提高了产率.
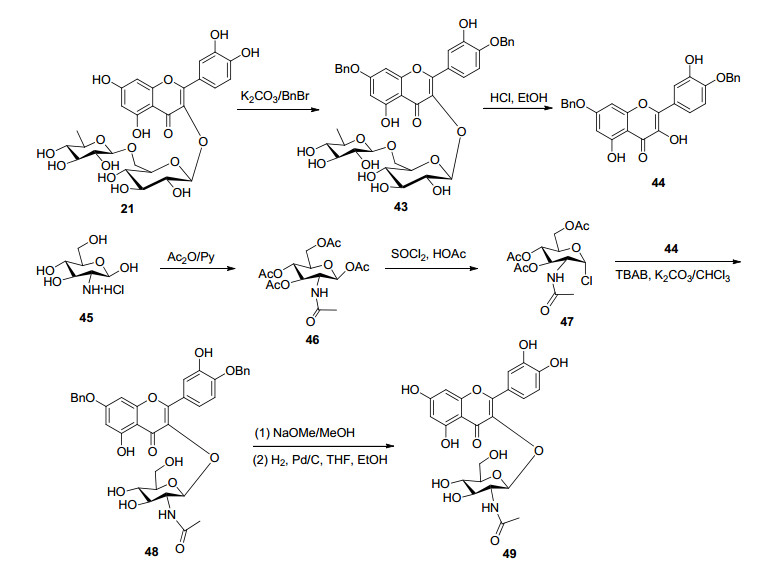
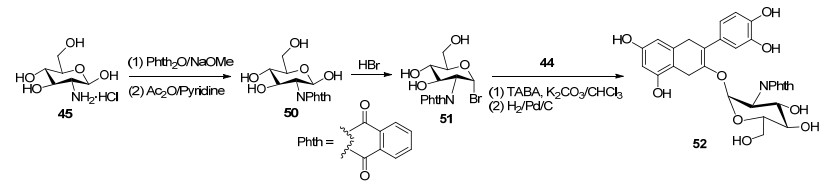
在合成槲皮素3-O-(2-氨基-2-脱氧)-D-葡萄糖苷研究中, 首先以芦丁(21)为原料, 经过选择性苄基化、酸水解反应得到7, 4'-二苄基槲皮素(44), 并以此为苷元进行对传统的Koening-Knorr法、相转移催化法、糖基三氯乙酰亚胺酯三种糖苷缩合反应研究, 结果发现只有在CHCl3/K2CO3体系, 以TBAB为相转移催化剂, 苷元才能与乙酰基保护的卤代2-氨基-2-脱氧-D-葡萄糖糖基供体发生糖苷化反应获得目标产物.其中, 以乙酰基保护的氯代2-乙酰氨基-2-脱氧-D-葡萄糖(47)为糖基供体反应得到槲皮素3-O-1, 2-反式糖苷(49) (Scheme 7).然而, 以乙酰基保护的溴代N-邻苯二甲酰亚胺基-2-脱氧-D-葡萄糖(51)为糖基供体得到槲皮素3-O-1, 2-顺式糖苷(52)[62] (Scheme 8).此外, 通过类似的反应合成了槲皮素3-O、3'-O和4'-O-N-乙酰氨基葡萄糖苷3种黄酮苷[63].在碳水化合物化学中, 由于糖基供体的2-O-酰基不具有邻基参与作用, 从而造成1, 2-顺式糖苷的立体选择性合成成为很有挑战性的工作[64].邻苯二甲酰亚胺是经典的相邻参与基, 通常邻苯二甲酰亚胺糖基卤化物的糖基化反应倾向于生成1, 2-反式-2-氨基糖苷.然而, 在上述反应中, 以乙酰基保护的N-邻苯二甲酰亚胺基-2-脱氧-D-葡萄糖为糖基供体生成1, 2-顺式糖苷.这可能是由于发生SN2反应, 在CHCl3/K2CO3/ TBAB体系中, 没有强有力的启动子可以使溴离子从糖基供体中脱离形成反应性双环酰氧基鎓离子中间体.此外, 在碱性CHCl3/K2CO3/TBAB体系中, 槲皮素-C-3- OH可能更具亲核性, 并且其对糖基供体端位碳的有效攻击主要来自卤素原子的相反方向.上述在相转移催化法中, 以N-邻苯二甲酰亚胺基保护的糖基供体合成槲皮素3-O-1, 2-顺式-2-氨基糖苷的方法, 值得进一步深入研究其机理, 为合成1, 2-顺式糖苷提供参考.
由于有邻基参与效应(neighboring group participation), 糖基三氯乙酰亚胺酯法具有高立体选择性和高收率的优点, 从而受到化学家们的重视.近年来在寡糖和糖苷的合成中得到了广泛的应用, 逐渐成为合成糖苷键的首选方法, 但该方法在黄酮苷的合成中产率较低, 应用较少.目前, 催化三氯乙酰亚胺酯与受体连接的活化剂主要有AgOTf[65]、BF3·Et2O[31]等路易斯酸.
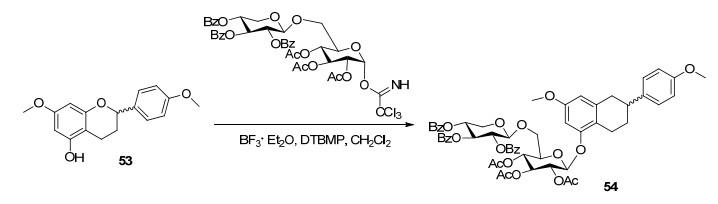
由于5-OH与4-C=O形成氢键作用, 使得5-羟基- 7, 4'-二甲氧基黄酮直接与卤代糖或糖基三氯乙酰亚胺酯的二糖糖基供体进行糖苷化反应得不到5位糖基化产物.因此, Arai等[66]在合成藿香苷时, 首先设法合成α/β构型5-羟基-7, 4'-二甲氧基黄烷(53)糖基受体关键中间体.由于与氟代糖进行糖苷化反应, 得到极难分离的5-O-糖苷和6-C-糖苷的混合产物.因此, 以糖基三氯乙酰亚胺酸酯替代氟代糖, 在路易斯酸BF3·Et2O催化下进行糖苷化反应得到了α/β构型黄烷5-O-糖苷关键反应中间体(54), 最后经过DDQ氧化脱氢、碱水解去保护基等步骤反应得到藿香苷, 总收率为9.5%(反应关键步骤Scheme 9).
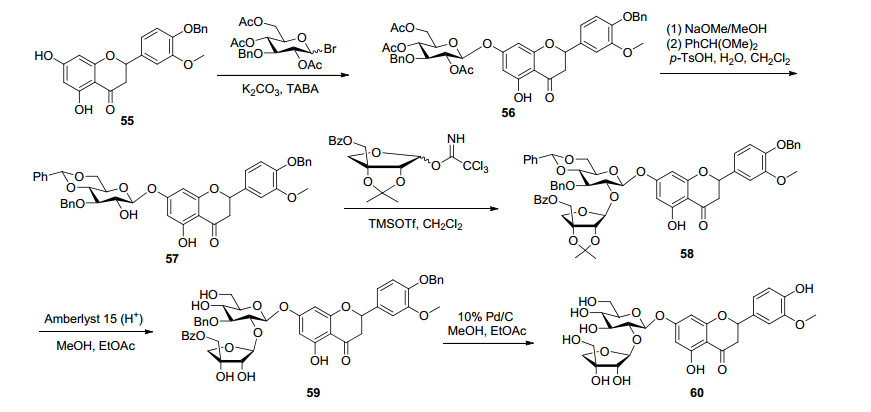
文献报道[67], 通过相转移催化法和糖基三氯乙酰亚胺酯法相结合, 合成了黄酮二糖苷高圣草素7-O-β-D-芹菜糖(1/2)-β-D-葡萄糖苷[又称槲寄生新苷Ⅲ (viscumneoside Ⅲ)](60).首先在CHCl3/KOH体系中, 使用相转移催化剂TBAB催化α/β构型苄基乙酰基保护的溴代葡萄糖与4'-苄氧基-3'-甲氧基-5, 7-二羟基二氢黄酮(55)进行糖苷化反应, 然后经过去乙酰基、亚苄基取代等步骤得到关键中间体4'-O-苄氧基高圣草素-7-O-3-苄基-4, 5-亚苄基-β-D-葡萄糖苷(60), 糖基化产率为80%.由于乙酰化保护的溴代芹菜糖不稳定, 在DCM/4Å分子筛体系中, 以TMSOTf或BF3·Et2O为催化剂和α/β构型端头乙酰化的溴代芹菜糖糖基供体进行糖苷化反应, 得不到糖苷产物.因此, 将糖基供体替换成α/β构型糖基三氯乙酰亚胺酯进行糖苷化反应, 再去保护基后得到了目标产物高圣草素-7-O-β-D-芹菜糖(1/2)-β-D-葡萄糖苷(60), 糖基化产率60% (Scheme 10).在二糖苷合成反应中, 当溴代糖不稳定以及反应性非常低的端基为乙酰基的糖基给体不适用于糖苷化合成时, 具有高反应性和可靠性的糖基三氯乙酰亚胺酯也是一个可供选择的糖基供体.
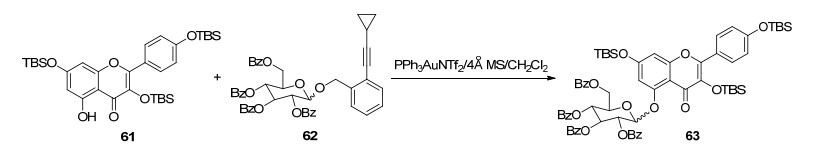
传统的糖苷化方法合成黄酮5-O-糖苷产率特别低, 甚至不反应, 原因是由于5-OH与4-C=O形成分子内氢键, 使得5-OH反应活性非常低[66].而金催化的邻炔基苯甲酸糖基酯法由于它们参与形成强的分子内氢键, 使得黄酮类5-OH反应活性大大提高, 从而实现了黄酮5-OH高选择性、高效糖基化.虽然Au(Ⅰ)离子容易被还原为沉淀, 使金催化剂失活, 且来源昂贵, 但是由于其具有黄酮5-OH高选择性、高效糖基化的特点, 使其成为一种合成黄酮5-O-糖苷的可选择方法. Sun等[68~70]通过在PPh3AuNTf2催化下, 以邻炔基苯甲酸糖基酯(62)作为糖基供体, 与选择性苄基、叔丁基二甲基硅氧基等保护的山奈酚、槲皮素、芹菜素5-OH进行糖基化反应, 获得了山奈酚、槲皮素、芹菜素5-O-糖苷, 糖苷化收率高达65%~99% (Scheme 11).
天然的黄酮碳苷的糖基通常与黄酮的C-6和(或)C-8位置连接, 如异牡荆素、牡荆素、牡荆苷、荭草苷等.目前合成黄酮碳苷链主要是两种方法.第一种是糖基供体直接与黄酮苷元及其衍生物糖基化, 主要应用于半合成; 第二种是首先构建糖苷键后再合成黄酮部分, 主要应用于全合成.目前最常用合成黄酮碳苷的糖苷链的方法是通过糖基供体与酚形成氧苷, 然后在路易斯酸的作用下重排为碳苷, 这种反应简称为O→C重排反应.
通过O→C糖苷重排方法合成黄酮苷具有相对可靠的区域选择性优点, 即在酚羟基的邻位选择性地形成糖苷.黄酮碳苷全合成一般是以羟基苯乙酮及其衍生物为原料如2, 4, 6-三羟基苯乙酮及其衍生物, 在路易斯酸如TfOH的作用下, 与糖基供体糖苷化反应生成羟基苯乙酮及其衍生物碳糖苷, 然后再与苯甲醛及其衍生物或苯甲酰氯及其衍生物通过经典的BK-VK法、AFO法或其他方法合成黄酮骨架部分.
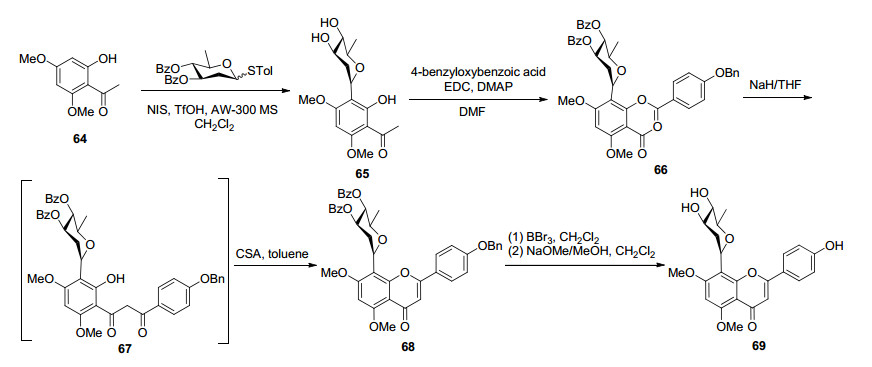
对于黄酮8-C-糖苷的合成, 一般是以2, 4, 6-三羟基苯乙酮为原料, 进行区域选择性保护2或2, 4位酚羟基, 如苄基化、甲氧基化等.刺果素苷是天然黄酮-8-C-糖苷化合物, 具有良好抗肿瘤[71, 72]、抗炎[73]等活性, 然而其在植物中含量极低, 从干燥的竹节草根和根茎提取分离获得的刺果素苷产率仅为0.26%[71], 其化学合成受到了化学家重视.文献报道, 首先通过在NIS/TfOH酸性条件下, 2-羟基-4, 6-二甲氧基苯乙酮(64)与α/β构型洋地黄毒糖糖基供体通过Fries型O→C重排反应构建C-芳基糖苷键, 得到关键中间体α-羟基芳基C-糖苷(65), 然后以EDC/DMAP介导、催化与4-苄氧基苯甲酸进行酯化反应, 再通过BK-VK重排得到β-丙二酮中间体(67), 以CSA介导环化脱水反应构建黄酮结构单元, 最后去苄基化和苯甲酰基化合成了刺果素苷(69), 总收率为8.3%[74] (Scheme 12).
而对于黄酮6-C-糖苷的合成, 一般是区域选择性保护2, 4, 6-三羟基苯乙酮4位或4, 6位酚羟基. Wang等[75]通过在Sc(OTf)3/PhMe/4Å分子筛体系中, 以2, 6-二羟基-4-苄基-苯乙酮为原料, 通过类似上述合成刺果素苷的反应步骤, 获得了具有良好降血糖活性的杨桃黄酮A.
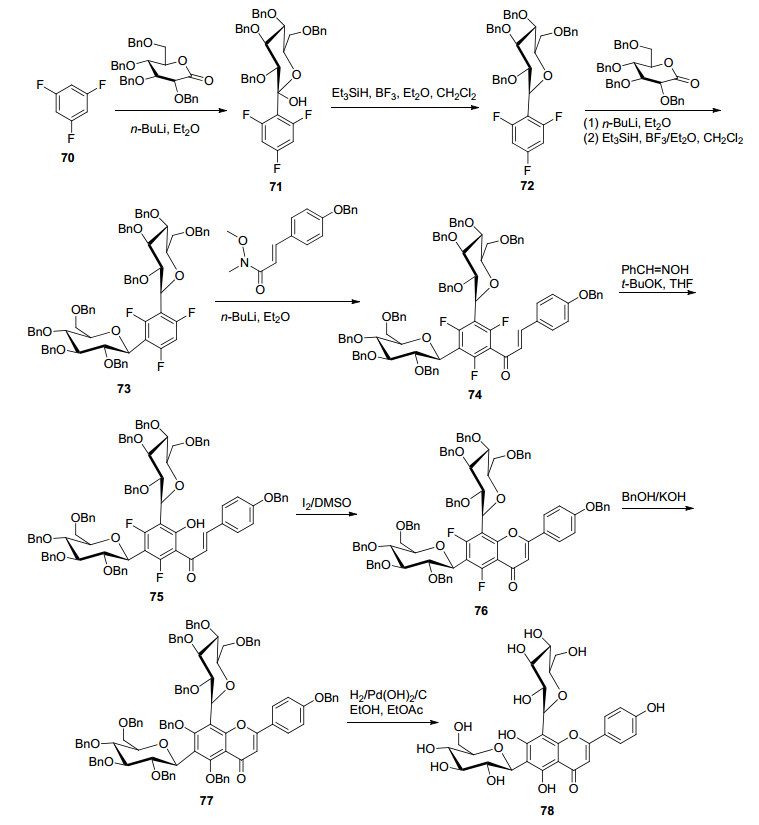
牡荆苷-2是天然黄酮6, 8-二糖苷, 具有良好抗肿瘤[76]、抗炎[77]等活性.文献报道了一种环境友好且简单合成牡荆苷-2的方法.首先在Sc(OTf)3水溶液下, 通过无保护的D-葡萄糖和2, 4, 6-三羟基苯乙酮一步反应, 获得的2, 4, 6-三羟基苯乙酮-3, 5-二-C-β-D-葡萄糖苷关键中间体与2, 4, 6-三羟基苯乙酮-3-C-β-D-葡萄糖苷副产物, 再经过选择性苄基化、与对氧苄基苯甲醛发生AFO反应等步骤获得了牡荆苷-2[78, 79].但该方法糖基化区域选择性低、产率较低, 且在二糖苷合成时, 只适用于具有相同糖基的合成. 2018年, 文献报道一种利用1, 3, 5-三氟苯(70)为原料构建二-C-糖苷关键中间体(73), 后期再通过氧官能团对氟原子进行芳香亲核取代合成黄酮6, 8-二- C-糖苷的独特方法.首先在t-BuLi·Et2O体系中, 将三氟苯锂盐化, 与D-葡糖酸内酯糖基供体进行糖基化, 然后再进行硅烷还原得到单C-糖苷, 重复相同的过程得到二-C-糖苷.接着, 与α, β-不饱和Weinreb酰胺构建查耳酮后, 再通过氧亲核试剂亲核取代氟化物构建黄酮骨架, 然后再脱苄基得到牡荆苷-2 (78)[80](Scheme 13).
与黄酮氧苷的半合成相比, 黄酮碳苷的半合成研究相对较少, 主要是碳-碳键的构建比碳-氧键的构建困难, 直接糖基化的产率很低.文献报道[79], 在CH3CN/ H2O体系中, 以Sc(OTf)3为催化剂, 直接以无保护的α/β构型柚皮素和D-葡萄糖进行糖基化反应, 得到了α/β构型柚皮素6, 8-C-二葡萄糖苷(收率17.3%), 然后经过乙酰化、DDQ氧化脱氢、去乙酰化等步骤获得了牡荆苷-2.在该反应中, 存在糖基化区域选择性差、产率低, 糖基化副产物多, 糖苷化产物分离困难等缺点, 其中副产物包括单糖和一些尚未鉴定的其他副产物, 单糖苷产物与二糖苷产物相当(单糖苷总产率约15%), 并且二糖苷产物只适用于相同糖基的二糖苷化合物的合成.因此, 对上述方法进行改进, 利用柚皮素7位羟基反应活性最高的特点, 依次通过选择性7位苄基化、乙酰基化、还原4位羰基、选择性去5位乙酰基获得的5位羟基黄烷衍生物糖基受体关键中间体, 再与苄基保护的糖基三氯乙酰亚胺酯糖基供体进行区域选择性6位基化反应获得黄烷6-C-单糖苷关键中间体, 接着, 去7位苄基, 再区域选择性进行8位糖基化获得黄烷6, 8-C-二糖苷关键中间体, 最后再经过对4位C氧化羟基化和羰基化、去乙酰基等反应获得系列黄酮6, 8-C-糖苷[78].通过改进后, 糖基化区域选择性、糖苷化产率得到提高(产率55%~79%), 不仅适用于相同糖基的二糖苷化合物合成, 而且也适用于不同糖基的二糖苷化合物的合成.
黄酮苷的化学合成文献报道较多, 但总体产率不高、选择性及对底物适应性差, 步骤多且产物分离困难, 且许多糖苷化方法适用范围均有一定的局限性, 难以满足糖苷键的多样性.近年来, 随着生物技术的发展, 人们对酶催化生物合成黄酮苷的研究越来越广泛, 酶催化生物合成具有高立体选择性、高效性等优点, 除此之外, 酶催化生物合成糖苷, 不仅避免了繁琐的保护和去保护等复杂的反应步骤, 而且反应条件温和、对环境友好, 所得产物纯度高、收率高但酶不易获得、价格昂贵.目前酶催化生物合成黄酮苷最常用的酶有两种:糖基转移酶(glycosyltransferases, GTs)和糖苷合成酶(glycosyn- thase), 而糖基转移酶又主要包括氧糖基转移酶(O-gly- cosyltransferases, OGTs)和碳糖基转移酶(C-glycosyl- transferases, CGTs).
糖基转移酶催化活性糖基核苷磷酸糖(主要是尿苷二磷酸糖或胸腺嘧啶二磷酸糖)转移到糖基受体, 如蛋白、核酸、寡糖、脂, 以及二级代谢产物如黄酮、抗生素等分子.目前, 由于活性糖供体核苷磷酸糖价格昂贵、所用酶的不易获得等缺点限制了其广泛的使用及工业化大生产[81].
人们对氧糖基转移酶催化合成黄酮苷的研究, 主要是对酶糖基化的区域选择性、底物、活性糖基给体等方面研究.
目前, 大多数氧糖基转移酶是催化活性糖基与黄酮A环的7-O和(或)C环的3-O和(或)B环的3'-O或4'-O连接[82~85].文献报道, 来自地衣芽孢杆菌DSM 13的氧糖基转移酶(YjiC), 以UDP-β-D-葡萄糖为糖基供体, 区域选择性葡萄糖苷化位置为黄酮3-O.此外, 以另外四种不同核苷酸的糖基给体代替UDP-β-D-葡萄糖葡对活性糖基特异性研究发现, 五种不同类型的NDP-D-葡萄糖可作为糖基转移酶反应的供体, 但是在不同的核苷酸葡萄糖基供体中底物转化率不同, 其中ADP-β-D-葡萄糖、GDP-β-D-葡萄糖和UDP-β-D-葡萄糖的转化率(约90%)几乎相等, 而TDP-β-D-葡萄糖约为75%和CDP-β- D-葡萄糖约为67%[86].然而, 有文献报道上述酶(YjiC)催化UDP-β-D-葡萄糖与芹菜素进行糖苷化反应, 得到芹菜素4'-O-葡萄糖苷、芹菜素7-O-葡萄糖苷、芹菜素4', 7-O-二葡萄糖苷[87].上述糖基化的结果不同的原因可能是芹菜素没有3-O, 因此该酶区域选择性糖苷化的位置为7-O和4'-O.由此说明氧糖基转移酶催化的活性位点与底物有关, 当底物黄酮含3-O时, YjiC酶催化糖苷化的区域选择性位置为3-O; 当底物黄酮不含3-O时, 该酶催化糖苷化的区域选择性位置为7-O和4'-O.在对氧糖基转移酶CsUGT76F1催化合成黄酮的研究中发现, 该酶可以催化合成黄酮3-O、7-O、3, 7-O-葡萄糖糖基苷和7-O-鼠李糖糖苷, 而对该酶底物特异性的研究表明, 对黄酮具有广泛的底物特异性, 包括存在于柑橘属物种中的柚皮素、橙皮素、香叶木素、山奈酚和槲皮素等黄酮[88].上述例子说明部分微生物来源的氧糖基转移酶可催化黄酮的多个OH位点, 这是造成氧糖基转移酶底物区域选择性差的主要原因.因此, 人们可根据底物特点及糖基化位点进行氧糖基酶选择, 同时提示能否通过基因工程技术对控制糖基转移酶其他位点进行基因敲除, 或通过某种手段、方法抑制该酶其他活性位点, 以提高对底物的区域选择性糖基化.
2016年首次报道来自真菌毛霉(Mucor hiemalis)的氧糖基转移酶MhGT1, 通过以UDP-葡萄糖作为糖供体, 酚类化合物作为底物, 对MhGT1的底物特异性进行研究发现, 该酶对黄酮类、香豆素类等酚类化合物具有较高氧糖基化的能力, 尤其是对异戊烯化酚类化合物的区域特异性和活性更高[89].异戊烯基黄酮的C-异戊烯基侧链的存在大大加强化合物的亲脂性, 使得化合物更容易穿过脂溶性的细胞膜与靶蛋白相结合, 显著提高黄酮的生物活性.而黄酮苷元水溶性较低, 糖基化可增加水溶性, 因此可利用氧糖基转移酶MhGT1对异戊烯基酚类的区域特异性和活性更高的特点, 进行异戊烯基黄酮苷(如具有良好抗肿瘤活性的淫羊藿苷等)生物合成.
由于大部分植物来源的糖基转移酶是以UPD-葡萄糖作为活性糖基供体, 使得转移酶催化除葡萄糖以外的糖基供体与黄酮连接变得更具有挑战性.后来人们把目标转向对微生物的研究, 如携带植物UGT的大肠杆菌, 通过基因工程改造UGT的糖选择性, 寻找其他的糖基供体.近年来, 通过基因工程的途径合成了多个天然的黄酮苷, 如黄酮O-半乳糖苷[84]、黄酮O-鼠李糖苷[85]、黄酮O-木糖苷[90]等.来自芽孢杆菌DSM 13的氧糖基转移酶GT(YjiC)催化由pgi、zwf和galU基因缺陷的大肠杆菌BL21(DE3)产生的dTDP-1-鼠李糖、dTDP-D-维他命胺、dTDP-4-氨基4, 6-二脱氧-D-半乳糖和dTDP-3-氨基3, 6-二脱氧-D-半乳糖这四种dTDP-脱氧糖与黄酮醇进行糖苷化, 结果只有dTDP-1-鼠李糖可以与黄酮醇3-O发生糖苷化生物转化反应, 得到黄酮3-O-鼠李糖苷[91].此外, 通过基因工程的途径还合成了一些自然界未被发现的黄酮氧苷[92].文献报道, 以UDP-N-乙酰葡糖胺作为糖供体, 在携带糖基转移酶AtUGT78D2的大肠杆菌中进行槲皮素糖苷的生物合成, 得到了新的黄酮苷槲皮素3-O-N-乙酰葡糖胺.并且, pgm和galU基因缺失的大肠杆菌突变菌株与野生型大肠杆菌BL21(DE3)对槲皮素3-O-N-乙酰葡糖胺的生成量相差较大, 其中野生型DE3、pgm突变体和galU突变体产生的槲皮素3-O-NAG与槲皮素3-O-葡萄糖苷的比分别为32:68、65:35和93:7, 由此说明了pgm突变体和galU突变体均可促进产生槲皮素3-O-N-乙酰葡糖胺, 但galU突变体促进效果最明显, 比野生型DE3多三倍, 产生380.7mg/L槲皮素3-O-N-乙酰葡糖胺[93].通过基因工程技术对微生物的改造, 大大地丰富了活性糖基的来源, 从而扩充了黄酮碳苷的种类和数量, 具有重要意义.
人们在碳糖基转移酶催化合成黄酮苷的研究中, 主要是对酶催化糖基化的底物、活性位点、植物来源等方面研究.
研究发现, 在谷物黄酮C-糖苷的生物合成中, 黄酮本身不是C-糖基化的直接底物, 而是由一类黄烷酮2-羟化酶FNS Ⅱ催化形成的2-羟基黄烷酮中间体[94, 95].然而, 在其他植物中发现了可催化黄酮底物进行直接糖基化的糖基转移酶, 如UGT708D1、UF6CGT1等[96, 97].文献报道[97], 来自大豆(Glycine max)的碳葡糖基转移酶UGT708D1和来自龙胆草(Gentiana triflora)的碳葡糖基转移酶UF6CGT1, 催化黄酮白杨素和木犀草素与葡萄糖直接糖基化生成相应6-C-葡萄糖苷.此外, 通过生物工程获得重组大肠杆菌, 进行引入对外源的UPD-葡萄糖生物合成基因、促进葡萄糖扩散蛋白以及发酵罐规模的因素对黄酮底物转化为碳糖苷化的影响研究, 结果发现外源的UPD-葡萄糖生物合成基因、促进葡萄糖扩散蛋白以及扩大发酵罐规模, 均有利于提高C-糖基转移酶催化底物转化为C-糖苷的转化率.
在对来自荞麦(Fagopyrum esculentum M)的碳糖基转移酶UGT708D1和大豆(Glycine max)的碳糖基转移酶UGT708C2直接和间接进行碳糖基化研究中[98], 首先通过基因工程重建酵母, 使其产生一些相关的CGT底物:黄烷酮柚皮素、圣草酚、黄酮芹菜素和木犀草素, 然后再分别将黄烷酮2-羟化酶UGT708C2[95]、OsCYP93G2[99]、SbCYP93G3[100]与碳糖基转移酶UGT708D1或UGT708C2的组合进行黄烷酮柚皮素、圣草酚底物间接糖基化, 分别获得了6-C-葡糖苷黄酮异牡荆素、8-C-葡糖苷黄酮牡荆素和6-C-葡糖苷异荭草素、8-C-葡糖苷荭草苷, 其中产黄烷酮柚皮素底物的酵母菌组生成6-C-葡萄糖苷为主, 而产圣草酚底物的酵母菌组生成8-C-葡萄糖苷为主.而以来自龙胆(Gentiana triflora)的碳糖基转移酶GtUF6CGT1进行芹菜素、木犀草素底物直接糖基化, 仅产生6-C-葡糖苷, 与之前报道结果相一致[96], 且6-C-葡糖苷黄酮异牡荆素的产量超过200 mg/L.上述报道中, 碳糖基转移酶UGT708D1、UGT708C2间接糖基化的糖基化位点及底物对糖基化位点影响的机理仍需进一步研究, 通过基因工程手段, 使重建酵母菌株产生单一糖苷产物.而含碳糖基转移酶GtUF6CGT1的重组酵母菌直接糖基化位点为6-C, 使通过酵母发酵生产异荭草素、异牡荆素成为可能.
在2-羟基黄烷酮碳葡萄糖基转移酶UGT708D1的活性位点的研究中, 发现其三个氨基酸残基His20, Asp85和Arg292位于C-葡糖基化活性位点, 且在进化上是保守的.此外, 用丙氨酸取代Asp85或Arg292破坏了碳葡萄糖基转移酶活性, 但是用丙氨酸取代His20, 使碳葡萄糖基转移酶活性转变为氧葡萄糖基转移酶活性[101].天然植物来源的黄酮C-糖苷的糖链多位于6-C和(或)8-C, 提示植物来源的碳糖基转移酶的活性位点有限, 有希望通过生物工程利用氧糖基转移酶(O-GTs)多样性开发新的碳糖基转移酶[102].植物来源的糖基转移酶的区域活性位置与天然植物来源的黄酮C-糖苷多位于6-C和(或)C-8是相一致的.文献报道, 来自植物山蚂蝗的碳糖基转移酶OsCGT催化2-羟基黄烷酮中间体与UDP-葡糖、UDP-半乳糖和UDP-阿拉伯糖活性糖基转化, 得到了系列的6-C、8-C和6, 8-C糖基黄酮[103]; 来自金桔(Fortunella crassifolia)和柑橘(Citrus unshiu)的碳糖基转移酶FcCGT(UGT708G1)和CuCGT(UGT708G2)参与黄酮6, 8-C-葡萄糖苷生物合成[104].
在2014年前人们仅从水稻和玉米等单子叶植物中发现碳糖基转移酶, 直到2014年Taguchi等[105]报道了从双子叶荞麦(Fagopyrum esculentum M)中鉴定了碳葡萄糖基转移酶基因, 从荞麦幼苗中分离纯化了两种同工酶FeCGTa(UGT708C1)和FeCGTb(UGT708C2)及其cDNA, 丰富了C-糖基转移酶的植物来源.通过对不同的组织器官来源的RNA研究发现, 这两种酶基因在种子萌发期间在子叶中特异性表达.此外, 对酶底物的研究发现, 两种C-糖基酶均表现出对2-羟基黄烷酮、二氢查尔酮、三羟基苯乙酮和其它相关化合物的C-葡糖基化活性, 其底物的化学结构类似于2, 4, 6-三羟基苯乙酮, 这对于结构多样性的黄酮C-糖苷的合成具有巨大的吸引力.
于1998年5月, Withers等[106]最早研究报道糖苷合成酶, 同年11月Planas等[107]报道了第一个内切葡聚糖酶突变的内切糖苷合成酶, 同时, Moracci等[108]报道了嗜热糖苷合成酶, 从此以后, 在国际范围内引起人们对糖苷合成酶的研究.糖苷合酶是通过基因工程的亲核试剂定点诱变构型保持糖苷酶产生的酶, 由于其不能水解产物, 解决了糖苷合成可逆反应造成的产物水解问题, 可以从根本上解决黄酮化学合成产率低的问题, 但主要限于氟代糖作糖基供体.与糖基转移酶催化糖苷化合成相比, 糖苷酶催化糖苷化合成具有底物糖基供体廉价、易得等优点.目前, 糖苷合酶主要用于寡糖、多糖、鞘糖脂等合成[109~111], 但对于黄酮苷的合成报道较少.
2003年首次报道特异腐质霉Cel7B E197S突变体是糖苷合酶催化剂[112]. 2007年Yang等[113]以20种糖基供体和80种黄酮糖基受体对糖苷合酶特异腐质霉Cel7B E197S突变体研究发现, 该酶可催化黄酮化合物, 如黄芩素、木犀草素、非瑟素、槲皮素与唯一有活性的β构型的二乳糖基氟化物(LacF)糖基给体发生反应, 得到相应黄酮4'-O-二糖苷或黄酮6-O-二糖苷, 收率72%~95% (Scheme 14).当黄酮B环存在对二酚羟基时, 特异腐质霉Cel7BE197S突变体糖苷酶催化与乳糖糖基化得到黄酮4'-O-二糖苷; 当B环不含对二酚羟基时, 但含有6位羟基时, 得到黄酮6-O-二糖苷.由此证明了黄酮的6-O和4'-O (B环含对二酚羟基)是该酶催化糖苷化反应的立体选择性和区域选择性的结构位置.
目前两种经典的黄酮苷元的化学合成方法比较成熟, 但仍然存在一些缺点, 如反应须经过保护、去保护等步骤, 使得反应步骤较冗长, 副产物多等缺点, 仍需针对这些缺点进行改良以及开发更好的新技术和方法.虽然糖苷化方法较多, 但许多糖苷化方法适用范围均有一定局限性, 难以满足糖苷键的多样性, 且糖苷化收率低、结构性、区域选择性差等这些问题有待解决, 这对合成化学家来说仍是一个严峻的挑战.而酶催化糖苷合成具有较高的结构、区域选择性, 且可避免繁琐的保护、去保护等复杂步骤, 但由于酶不易获得、价格昂贵、糖基给体底物柔性差等缺点, 限制了其目前应用.新方法、新技术在黄酮苷合成方面的应用, 将是黄酮苷合成研究的发展趋势.相信随着有机化学、药物化学的发展以及新方法、新技术的出现, 将使得黄酮苷化学合成实现无保护基反应、提高化学选择性(chemoselectivity)和区域选择性(Regioselectivity).同时, 随着生物技术的发展, 生物工程对目标基因的改造、对酶来源的挖掘和改造将会解决酶的来源困难、价格昂贵的问题以及使糖基供体柔性变得高, 促进酶催化糖苷化方法应用于黄酮苷的合成.
Zhang, T. T.; Wang, M.; Yang, L.; Jiang, J. G.; Zhao, J. W.; Zhu, W. J. Funct. Foods 2015, 18, 235. doi: 10.1016/j.jff.2015.07.006
Shu, J.; Li, L.; Zhou, M.; Yu, J.; Peng, C.; Shao, F.; Liu, R.; Zhu, G.; Huang, H. Nat. Prod. Res. 2017, 23, 1.
黄秀香, 韦汉龙, 盘玉芬, 食品工业科技, 2013, 34, 140.Huang, X.-X.; Wei, H.-L.; Pan, Y.-F. Food Ferment. Ind. 2013, 34, 140 (in Chinese).
Ekuadzi, E.; Dickson, R.; Fleischer, T.; Annan, K.; Pistorius, D.; Oberer, L.; Gibbons, S. Phytother. Res. 2014, 28, 784. doi: 10.1002/ptr.5053
蒋晓文, 白俊鹏, 田星, 赵庆春, 中草药, 2016, 47, 726.Jiang, X.-W.; Bai, J.-P.; Tian, X.; Zhao, Q.-C. Chin. Trad. Herb. Drugs 2016, 47, 726 (in Chinese).
师琪, 管福琴, 孙浩, 赵友谊, 王鸣, 张建华, 冯煦, 单宇, 食品科技, 2013, (6), 220.Shi, Q.; Guan, F.-Q.; Sun, H.; Zhao, Y.-Y.; Wang, M.; Zhang, J.-H.; Feng, X.; Shan, Y. Food Sci. Technol. 2013, (6), 220 (in Chinese).
Sun, L.; Peng, Q.; Qu, L.; Gong, L.; Si, J. Mol. Med. Rep. 2015, 11, 3094. doi: 10.3892/mmr.2014.3007
Ekuadzi, E.; Dickson, S.; Fleischer.; Annan, K.; Pistorius, D.; Oberer, L.; Gibbons, S. Phytother. Res. 2014, 28, 784. doi: 10.1002/ptr.5053
Nawwar, M. A.; Hashem, A. N.; Hussein, S. A.; Swilam, N. F.; Becker, A.; Haertel, B.; Lindequist, U.; El-Khatib, A.; Linscheid, M. W. Pharmazie 2016, 71, 162.
Kumamoto, H.; Matsubara, Y.; Iizuka, Y.; Okamoto, K.; Yokoi, K. Agric. Biol. Chem. 2014, 49, 2613.
Nguyen, P. H.; Dung, V. V.; Bing, T. Z.; Kim, Y. H.; Min, B. S.; Mi, H. W. Arch. Pharmacal Res. 2014, 37, 1394. doi: 10.1007/s12272-014-0422-5
杜晓光, 耿美玉, 生命科学, 2011, 23, 671.Du, X.-G.; Geng, M.-Y. Sci. Life 2011, 23, 671 (in Chinese).
Montreuil, J. Adv. Carbohydr. Chem. Biochem. 1980, 37, 157. doi: 10.1016/S0065-2318(08)60021-9
Winterburn, P. J.; Phelps, C. F. Nature 1972, 236, 147. doi: 10.1038/236147a0
张勇民, 西北大学学报(自然科学版), 2016, 46, 385.Zhang, Y.-M. J. Northwest Univ. (Nat. Sci. Ed.) 2016, 46, 385 (in Chinese).
Ranjbari, J.; Mokhtarzadeh, A.; Alibakhshi, A.; Tabarzad, M.; Hejazi, M.; Ramezani, M. Curr. Pharm. Des. 2017, 23, 6019.
Hofer, B. Appl. Microbiol. Biotechnol. 2016, 100, 4269. doi: 10.1007/s00253-016-7465-0
黄丹丹, 陈欢欢, 黎梅桂, 黄俊彬, 李运容, 魏刚, 中药新药与临床药理, 2017, 28, 73.Huang, D.-D.; Chen, H.-H.; Li, M.-G.; Huang, J.-B.; Li, Y.-R.; Wei, G. Tradit. Chin. Drug Res. Pharmacol. 2017, 28, 73 (in Chinese).
Obmann, A.; Zehl, M.; Purevsuren, S.; Narantuya, S.; Reznicek, G.; Kletter, C.; Glasl, S. J. Sep. Sci. 2015, 34, 292.
Qiu, L.; Jiao, Y.; Xie, J. Z.; Huang, G. K.; Qiu, S. L.; Miao, J. H.; Yao, X. S. J. Asian Nat. Prod. Res. 2013, 16, 589.
Kondo, T.; Yoshida, K.; Oyama, K. I. Curr. Org. Chem. 2011, 15, 2567. doi: 10.2174/138527211796367354
Wang, Z. L.; Yang, L. Y.; Yang, X. W. Synth. Commun. 2013, 43, 22.
梅青刚, 袁伟成, 王淳, 有机化学, 2015, 35, 70. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344624.shtmlMei, Q.-G.; Yuan, W.-C.; Wang, C. Chin. J. Org. Chem. 2015, 35, 70 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344624.shtml
Sun, J. S.; Laval, S.; Yu, B. Synthesis 2014, 46, 1030. doi: 10.1055/s-0033-1341052
Mohadeszadeh, M.; Iranshahi, M. Mini-Rev. Med. Chem. 2017, 17, 1377.
Kitamura, K.; Ando, Y.; Matsumoto, T.; Suzuki, K. Chem. Rev. 2018, 118, 1495. doi: 10.1021/acs.chemrev.7b00380
Xiao, J.; Muzashvili, T. S.; Georgiev, M. I. Biotechnol. Adv. 2014, 32, 1145. doi: 10.1016/j.biotechadv.2014.04.006
Hofer, B. Appl. Microbiol. Biotechnol. 2016, 100, 4269. doi: 10.1007/s00253-016-7465-0
Ares, J. J.; Outt, P. E.; Kakodkar, S. V.; Buss, R. C.; Geiger, J. C. J. Org. Chem. 1993, 58, 7093.
Song, G. Y.; Ahn, B. Z. Arch. Pharmacal Res. 1994, 17, 434. doi: 10.1007/BF02979121
汪秋安, 廖头根, 汤建国, 范华芳, 湖南大学学报(自科版), 2004, 31, 1.Wang, Q.-A.; Liao, T.-G.; Tang, J.-G.; Fan, H.-F. J. Hunan Univ. (Nat. Sci.) 2004, 31, 1 (in Chinese).
Zacharia, J. T.; Hayashi, M. Carbohydr. Res. 2012, 348, 91. doi: 10.1016/j.carres.2011.11.015
Kerl, T.; Berger, F.; Schmalz, H. G. Chemistry 2016, 22, 2935. doi: 10.1002/chem.201505118
Algarl, J.; Flynn, J. P. Proc. R. Ir. Acad. 1934, 42, 1.
Oyamada. B. Chem. Soc. Jpn. 1934, 55, 2039.
Oyamada, T. Tetrahedron 1935, 64, 988.
周强, 王淳, 李玉萍, 蒲文臣, 应用与环境生物学报, 2017, 24, 232.Zhou, Q.; Wang, C.; Li, Y.-P.; Pu, W.-C. Chin. J. Appl. Environ. Biol. 2017, 24, 232 (in Chinese).
Shen, X.; Zhou, Q.; Xiong, W.; Pu, W.; Zhang, W.; Zhang, G.; Wang, C. Tetrahedron 2017, 73, 4822. doi: 10.1016/j.tet.2017.06.064
Li, X.; Chen, G.; Zhang, X.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q. H. Bioorg. Med. Chem. Lett. 2016, 26, 4241. doi: 10.1016/j.bmcl.2016.07.050
汪秋安, 王盛淳, 李悦, 单杨, 湖南大学学报(自然科学版), 2015, 42, 53.Wang, Q.-A.; Wang, S.-C.; Li, Y.; Shan, Y. J. Hunan Univ. (Nat. Sci.) 2015, 42, 53 (in Chinese).
Zhang, J.; Fu, X. L.; Yang, N.; Wang, Q. A. Sci. World J. 2013, 2013, 649485.
Jian, J.; Fan, J.; Yang, H.; Lan, P.; Li, M.; Liu, P.; Gao, H.; Sun, P. J. Nat. Prod. 2018, 81, 371. doi: 10.1021/acs.jnatprod.7b00791
Yamauchi, K.; Mitsunaga, T.; Batubara, I. Bioorg. Med. Chem. 2014, 22, 937. doi: 10.1016/j.bmc.2013.12.062
Liang, G.; Xu, B.; Wen, Z.; Hu, Z.; Yuan, J.; Chen, H.; Zhang, L. Heterocycles. 2016, 92, 1245. doi: 10.3987/COM-16-13455
薛向南, 化学试剂, 2018, 40, 465.Xue, X.-N. Chem. Reag. 2018, 40, 465 (in Chinese).
Liu, J. D; Chen, L.; Cai, S. C.; Wang, Q. A. Carbohydr. Res. 2012, 357, 41. doi: 10.1016/j.carres.2012.05.013
Park, K. S.; Kim, H.; Mi K. K.; Kim, K.; Chong, Y. J. Korean Soc. Appl. Biol. Chem. 2015, 58, 317.
Yang, W.; Sun, J.; Yang, Z.; Han, W.; Zhang, W. D., Yu B. Tetrahedron Lett. 2012, 43, 2773.
Yang, W.; Sun J.; Lu, W.; Li, Y.; Shan, L.; Han, W.; Zhang, W.; Yu, B. J. Org. Chem. 2010, 75, 6879. doi: 10.1021/jo1014189
Mei, Q.; Wang, C. Z.; Yuan, W.; Zhang, G. Beilstein J. Org. Chem. 2015, 11, 1220. doi: 10.3762/bjoc.11.135
Chen, J.; Huang, W.; Lian, G.; Lin, F. Carbohydr. Res. 2009, 344, 2245. doi: 10.1016/j.carres.2009.08.014
Koenigs, W., Knorr, E. Eur. J. Inorg. Chem. 2010, 34, 957.
赵晶, 张致平, 陈鸿珊, 张兴权, 陈湘红, 药学学报, 1998, 33, 22.Zhao, J.; Zhang, Z.-P.; Chen, H.-S.; Zhang, X.-Q.; Chen, X.-H. Acta Pharm. Sin. 1998, 33, 22 (in Chinese).
Vermes, B.; Farkas, L.; Nógrádi, M.; Wagner, H.; Dirscherl, R. Phytochemistry 1976, 15, 1320. doi: 10.1016/0031-9422(76)85106-0
Farkas, L.; Nógr di, M.; Vermes, B.; Wolfner, A.; Wagner, H.; Hörhammer, L.; Krämer, H. Eur. J. Inorg. Chem. 1969, 102, 2583.
Mezey-Vandor, G.; Farkas, L.; Kanzel, I.; Ndgradi, M. Chem. Ber. 1980, 113, 1945. doi: 10.1002/cber.19801130529
Docampo, M.; Olubu, A.; Wang, X. Q.; Pasinetti, G.; Dixon, R. A. J. Agric. Food Chem. 2017, 65, 7607. doi: 10.1021/acs.jafc.7b02633
Huang, W. H.; Chien, P. Y.; Yang, C. H.; Lee, A. R. Chem. Pharm. Bull. 2003, 34, 339.
Li, Y. F.; Yu, B.; Sun, J. S.; Wang, R. X. Tetrahedron Lett. 2015, 56, 3816. doi: 10.1016/j.tetlet.2015.04.083
Demetzos, C.; Skaltsounis, A. L.; Tillequin, F.; Koch, M. Planta Med. 1990, 207, 131.
汪秋安, 吴峥, 刘莉, 邹亮华, 罗茗, 有机化学, 2010, 30, 1682. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract339373.shtmlWang, Q.-A.; Wu, Z.; Liu, L.; Zou, L.-H.; Luo, M. Chin. J. Org. Chem. 2010, 30, 1682 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract339373.shtml
Cao, Z. L.; Qu, Y. Y.; Zhou, J. X.; Liu, W. W.; Yao, G. W. J. Carbohydr. Chem. 2015, 34, 28. doi: 10.1080/07328303.2014.996290
Cao, Z. L.; Chen, J.; Zhu, D. D.; Yang, Z. N.; Teng, W. Q.; Liu, G. F.; Liu, B.; Tao, C. Z. J. Chem. Res. 2018, 42, 189. doi: 10.3184/174751918X15232706115112
Nakagawa, A.; Tanaka, M.; Hanamura, S.; Takahashi, D.; Toshima, K. Angew. Chem., Int. Ed. 2015, 54, 10935. doi: 10.1002/anie.201504182
Du, Y.; Wei, G.; Lindhardt, R. J. Tetrahedron Lett. 2003, 44, 6887. doi: 10.1016/S0040-4039(03)01706-4
Arai, M. A.; Yamaguchi, Y.; Ishibashi, M. Org. Biomol. Chem. 2017, 15, 5025. doi: 10.1039/C7OB01004D
Zou, L.; Zhang, Z.; Chen, X.; Chen, H.; Zhang, Y.; Li, J.; Liu, Y. Tetrahedron 2018, 74, 2376. doi: 10.1016/j.tet.2018.03.057
Liao, J. X.; Fan, N. L.; Liu, H.; Tu, Y. H.; Sun, J. S. Org. Biomol. Chem. 2015, 14, 1221.
Hu, Y.; Tu, Y. H.; Liu, D. Y.; Liao, J. X.; Sun, J. S. Org. Biomol. Chem. 2016, 14, 4842. doi: 10.1039/C6OB00655H
杨为准, 李荣耀, 韩伟, 张卫东, 孙建松, 有机化学, 2012, 32, 1067. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341192.shtmlYang, W.-Z.; Li, R.-Y.; Han, W.; Zhang, W.-D.; Sun, J.-S. Chin. J. Org. Chem. 2012, 32, 1067 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341192.shtml
Carte, B. K.; Carr, S.; Debrosse, C.; Hemling, M. E.; Mackenzie, L.; Offen, P.; Berry, D. E. ChemInform 1991, 22, 1815.
Lai, C. Y.; Tsai, A. C.; Chen, M. C.; Chang, L. H.; Sun, H. L.; Chang, Y. L.; Chen, C. C.; Teng, C. M.; Pan, S. L. Plos One 2012, 7, e42192. doi: 10.1371/journal.pone.0042192
Hsieh, I.; Chang, S. Y.; Teng, C. M.; Chen, C. C.; Yang, C. R. J. Biomed. Sci. 2011, 18, 28. doi: 10.1186/1423-0127-18-28
Yao, C. H.; Tsai, C. H.; Lee, J. C. J. Nat. Prod. 2016, 79, 1719. doi: 10.1021/acs.jnatprod.5b01051
Wang, Y.; Liu, M.; Liu, L.; Xia, J. H.; Du, Y. G.; Sun, J. S. J. Org. Chem. 2018, 83, 4111. doi: 10.1021/acs.joc.8b00008
Yang, D.; Zhang, X.; Zhang, W.; Rengarajan, T. Drug Des., Dev. Ther. 2018, 12, 1303. doi: 10.2147/DDDT.S149307
Ku, S. K.; Bae, J. S. Can. J. Physiol. Pharmacol. 2016, 94, 287. doi: 10.1139/cjpp-2015-0215
Shie, J. J.; Chen, C. A.; Lin, C. C.; Ku, A. F.; Cheng, T. J.; Fang, J. M.; Wong, C. H. Org. Biomol. Chem. 2010, 8, 4451. doi: 10.1039/c0ob00011f
Sato, S.; Akiya, T.; Nishizawa, H.; Suzuki, T. Carbohydr. Res. 2006, 341, 964. doi: 10.1016/j.carres.2006.02.019
Ho, T. C.; Kamimura, H.; Ohmori, K.; Suzuki, K. Org. Lett. 2016, 18, 4488. doi: 10.1021/acs.orglett.6b02203
毛多斌, 黄顺利, 陈永森, 日用化学工业, 2007, 37, 321. doi: 10.3969/j.issn.1001-1803.2007.05.011Mao, D.-B.; Huang, S.-L.; Chen, Y.-S. China Surf. Deterg. Cosmet. 2007, 37, 321 (in Chinese). doi: 10.3969/j.issn.1001-1803.2007.05.011
Hofer, B. Appl. Microbiol. Biotechnol. 2016, 100, 4269. doi: 10.1007/s00253-016-7465-0
Choung, W. J.; Hwang, S. H.; Ko, D. S.; Kim, S. B.; Kim, S. H.; Jeon. S. H.; Choi, H. D.; Lim, S. S.; Shim, J. H. J. Agric. Food Chem. 2017, 65, 2760. doi: 10.1021/acs.jafc.7b00501
Kim, S. Y.; Lee, H. R.; Park, K. S.; Kim, B. G.; Ahn, J. H. Appl. Microbiol. Biotechnol. 2015, 99, 2233. doi: 10.1007/s00253-014-6282-6
Liang, C.; Zhang, Y.; Jia, Y.; Wang, W.; Li, Y.; Lu, S.; Jin, J. M.; Tang, S. Y. Sci Rep. 2016, 6, 21051. doi: 10.1038/srep21051
Pandey, R. P.; Parajuli, P.; Koirala, N.; Park, J. W.; Sohng, J. K. Appl. Environ. Microbiol. 2013, 79, 6833. doi: 10.1128/AEM.02057-13
Gurung, R. B.; Kim, E. H.; Oh, T. J.; Sohng, J. K. Mol. Cells 2013, 36, 355. doi: 10.1007/s10059-013-0164-0
Liu, X. G.; Lin, C. L.; Ma, X. D.; Yan, Y.; Wang, J. Z.; Zeng, M. Front. Recent Dev. Plant Sci. 2018, 9, 166. doi: 10.3389/fpls.2018.00166
Feng, J.; Zhang, P.; Cui, Y.; Li, K.; Qiao, X.; Zhang, Y. T.; Li, S. M.; Cox, R. J.; Wu, B.; Ye, M. Adv. Synth. Catal. 2017, 359, 955.
Han, S. H.; Kim, B. G.; Yoon, J. A.; Chong, Y.; Ahn, J. H. Appl. Environ. Microbiol. 2014, 80, 2754. doi: 10.1128/AEM.03797-13
Pandey, R. P.; Parajuli, P.; Gurung, R. B.; Sohng, J. K. Enzyme Microb. Technol. 2016, 91, 26. doi: 10.1016/j.enzmictec.2016.05.006
Simkhada, D.; Lee, H. C.; Sohng, J. K. Biotechnol. Bioeng. 2010, 107, 154. doi: 10.1002/bit.22782
Kim, B. G.; Su, H. S.; Ahn, J. H. Appl. Microbiol. Biotechnol. 2012, 93, 2447. doi: 10.1007/s00253-011-3747-8
Brazierhicks, M.; Evans, K. M.; Gershater, M. C.; Puschmann, H.; Steel, P. G.; Edwards, R. J. Biol. Chem. 2009, 284, 17926. doi: 10.1074/jbc.M109.009258
Du, Y.; Chu, H.; Chu, I. K.; Lo, C. Plant Physiol. 2010, 154, 324. doi: 10.1104/pp.110.161042
Sasaki, N.; Nishizaki, Y.; Yamada, E.; Tatsuzawa, F.; Nakatsuka, T.; Takahashi, H.; Nishihara, M. FEBS Lett. 2016, 589, 182.
Shrestha, A.; Pandey, R. P.; Dhakal, D.; Parajuli, P.; Sohng, J. K. Appl. Microbiol. Biotechnol. 2018, 102, 1251. doi: 10.1007/s00253-017-8694-6
Vanegas, K. G.; Larsen, A. B.; Eichenberger, M.; Fischer, D.; Mortensen, U. H.; Naesby, M. Microb. Cell Fact. 2018, 17, 107. doi: 10.1186/s12934-018-0952-5
Du, Y.; Chu, H.; Wang, M.; Chu, I. K.; Lo, C. J. Exp. Bot. 2010, 61, 983. doi: 10.1093/jxb/erp364
Morohashi, K.; Grotewold, E. Plant Cell. 2012, 24, 2745. doi: 10.1105/tpc.112.098004
Hirade, Y.; Kotoku, N.; Terasaka, K.; Saijo-Hamano, Y.; Fukumoto, A.; Mizukami, H. FEBS Lett. 2015, 589, 1778. doi: 10.1016/j.febslet.2015.05.010
Gutmann, A.; Nidetzky, B. Pure Appl. Chem. 2013, 85, 1865. doi: 10.1351/pac-con-12-11-24
Hao, B.; Caulfield, J. C.; Hamilton, M. L.; Pickett, J. A.; Midega, C. A.; Khan, Z. R.; Wang, J. R. Org. Biomol. Chem. 2015, 13, 11663. doi: 10.1039/C5OB01926E
Ito, T.; Fujimoto, S.; Suito, F.; Shimosaka, M.; Taguchi, G. Plant J. Cell Mol. Biol. 2017, 91, 187. doi: 10.1111/tpj.13555
Nagatomo, Y.; Usui, S.; Ito, T.; Kao, A.; Shimosaka, M.; Taguchi, G. Plant J. 2014, 80, 437. doi: 10.1111/tpj.12645
Mackenzie, L. F.; Wang, Q.; Warren, R. A. J.; Withers, S. G. J. Am. Chem. Soc. 1998, 120, 5583. doi: 10.1021/ja980833d
Malet, C.; Planas, A. FEBS Lett. 1998, 440, 208. doi: 10.1016/S0014-5793(98)01448-3
Moracci, M.; Trincone, A.; Perugino, G.; Ciaramella, M.; Rossi, M. Biochemistry 1998, 37, 17262. doi: 10.1021/bi981855f
Hayes, M. R.; Pietruszka, J. Molecules 2017, 22, 1434. doi: 10.3390/molecules22091434
Kobayashi, S.; Shoda, S.; Uyama, H. Adv. Polym. Sci. 2012, 121, 217.
Díez-Municio, M.; Kolida, S.; Herrero, M.; Rastall, R. A.; Moreno, F. J. J. Funct. Foods 2016, 20, 532. doi: 10.1016/j.jff.2015.11.032
Ducros, V. M.; Tarling, C. A.; Zechel, D. L.; Brzozowski, A. M., Frandsen, T. P.; Ossowski, I. V.; Schülein, M.; Withers, S. G.; Davies, G. J. Chem. Biol. 2003, 10, 619. doi: 10.1016/S1074-5521(03)00143-1
Yang, M.; Davies, G. J.; Davis, B. G. Angew. Chem. 2007, 46, 3885. doi: 10.1002/anie.200604177

图式 1 金合欢素7-O-β-D-吡喃半乳糖苷的全合成路线
Scheme 1 Total synthetic route of acacetin-7-O-β-D-galactopyranoside

图式 7 槲皮素3-O-1, 2-反式糖苷的半合成路线
Scheme 7 Semi-synthetic route of quercetin 3-O-1, 2-transglycoside

图式 8 槲皮素槲皮素3-O-1, 2-顺式糖苷的半合成路线
Scheme 8 Semi-synthetic route of quercetin-3-O-1, 2-cis-glucoside

图式 11 金催化的邻炔基苯甲酸糖基酯法合成路线
Scheme 11 Synthetic route of gold-catalyzed o-alkynyl benzoate glycosyl ester

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:










 下载:
下载: