表 1
BVMO以及其来源
Table 1.
Name of BVMOs and sources

酶催化合成手性化合物是当前合成化学领域的研究热点之一.其中Baeyer-Villiger单加氧酶(Baeyer-Villi- ger monooxygenase, BVMO)是一种重要的生物催化剂, 可以催化各种有机酮/醛化合物的Baeyer-Villiger (BV)氧化反应, 以及一些含硫、硒、硼等杂原子底物的氧化反应. BVMO拥有广泛的多样性, 根据其黄素辅因子性质不同主要分为Type Ⅰ型和Ⅱ型BVMO.其中Type Ⅰ型属于B家族黄素单加氧酶, 依赖黄素腺嘌呤二核苷酸(FAD)作为辅因子和NAPDH作为还原剂, 目前重组可用的BVMO大多属于这一类; Type Ⅱ型属于C家族黄素单加氧酶, 依赖NADP和黄素单核苷酸(FMN)[1]. BVMO不但拥有优异的立体选择性和区域选择性, 还具有反应条件温和、高效、对环境友好等生物催化剂的共同优点.因此BVMO无可争议地成为绿色不对称Baeyer-Villiger氧化反应的最佳催化剂, 广泛用于手性氧化产物的合成[2]. BVMO主要参与生物体内的次级代谢, 说明在无细胞条件下也能进行催化, 有工业应用的潜质.随着生物信息技术和基因挖掘技术的发展, 来自微生物基因组的众多新型BVMO被发现和表征, 极大地丰富了BVMO酶的种类和应用范围; 同时, 随着蛋白质工程技术的不断进步, 利用理性设计、定向进化等方法将已知的野生型BVMO (表 1)改造成特定催化属性的突变型BVMO, 也是目前较为热门的研究方向, 有力地推动了BVMO研究和合成应用的发展[3].以不同底物结构的Baeyer-Villiger氧化反应为主线, 综述了近5年来国内外对野生型以及蛋白质工程改造的BVMO催化氧化反应研究的新进展.
 下载:
导出CSV
下载:
导出CSV
| Entry | BVMO名称 | 来源 | 中文名 |
| 1 | BVMOLepto | Leptospira biflex | |
| 2 | CPMOComa | Comamonas testosterone | 环戊酮单加氧酶 |
| 3 4 | BVMOCm BVMOPp | Cyanidioschyzon merolae Physcomitrella patens | |
| 5 | hFMO5 | Human | |
| 6 | PAMO | Thermobifida fusca | 苯丙酮单加氧酶 |
| 7 | HAPMO | Pseudomonas fluorescens ACB | 4-羟基苯乙酮单加氧酶 |
| 8 | BVMO9 | Rhodococcus jostii RHA1 | |
| 9 | BVMO15 | Rhodococcus jostii RHA1 | |
| 10 | STMO | Rhodococcus rhodochrous | 类固醇单加氧酶 |
| 11 | BVMOAFL456 | Aspergillus flavu NRRL3357 | |
| 12 | BVMOAFL838 | Aspergillus flavu NRRL3357 | |
| 13 | BVMOAFL210 | Aspergillus flavu NRRL3357 | |
| 14 | BVMOAFL619 | Aspergillus flavu NRRL3357 | |
| 15 | BVMO4 | Dietzia sp. D5 | |
| 16 | CHMOJS666 | Polaromonas sp. strain JS666 | 环己酮单加氧酶 |
| 17 | CPDMO | Pseudomonas sp. HI-70 | 环十五酮单加氧酶 |
| 18 | CAMO | Cylindrocarpon radicicola ATCC 11011 | 环烷酮单加氧酶 |
| 19 | OTEMO | Pseudomonas putida NCIMB 10007 | |
| 20 | CHMOArthro | Arthrobacter sp | 环己酮单加氧酶 |
| 21 | CHMOAcineto | Acinetobacter sp | 环己酮单加氧酶 |
| 22 | CHMOPhi1 | Rhodococcus sp. Phi1 | 环己酮单加氧酶 |
| 23 | BVMO-P1-D08 | — | |
| 24 | BVMO-CDX-003 | — | |
| 25 | BVMO-P3-C07 | — | |
| 26 | BVMO-P1-C06 | — | |
| 27 | 2, 5-DKCMO | Pseudomonas putida | 2, 5二酮莰烷单加氧酶 |
| 28 | BVMOOcean | Oceanicola batsensis DSM 15984 | |
| 29 | BVMOParvi | Parvibaculum lavamentivorans DSM 13023 | |
| 30 | 3, 6-DKCMO | Pseudomonas putida | 3, 6二酮莰烷单加氧酶 |
| 31 | CDMO | Rhodococcus ruber SC1 | 环十二酮单加氧酶 |
| 32 | CHMOTm | Thermocrispum municipale DSM 44069 | 环己酮单加氧酶 |
| 33 | MtmOIV | Streptomyces argillaceus | 光神霉素单加氧酶 |
| 34 | PenE | Streptomyces exfoliatus | |
| 35 | PntE | Streptomyces arenae | |
| 36 | PtIE | Streptomyces avermitilis | |
| 37 | BVMOAR | Acinetobacter radioresistens | |
| 38 | FMO CcsB | Aspergillus clavatus NRRL 1 | |
| 39 | BVMOBo | Bradyrhizobium oligotrophicum | |
| 40 | BVMOAm | Aeromicrobium marinum | |
| 41 | SMO | Pseudomonas putida CA-3 | 苯乙烯单加氧酶 |
| 42 | YMOA | Yarrowia lipolytica | |
| 43 | CHMOTm | Thermocrispum municipale DSM 44069 | |
| 44 | BVMOAf1 | A. fumigatus Af293 |
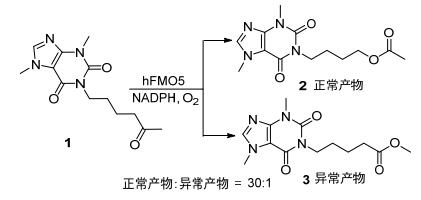
BVMO催化直链酮底物的BV氧化, 对以生物催化的方式生产石油工业相关产品有重大意义.直链酮底物的烷基链长度会影响BVMO对底物的接受程度, 常见的环己酮单加氧酶(CHMO)在氧化至多3个碳原子的烷基链时, 表现出高效率和高选择性[2]. Rial等[4]在2017年发现BVMOLepto(来源于Leptospira biflex)能对无取代直链酮和有支链的直链酮进行生物氧化, 优先生成正常的内酯产物.它可以高效转化小于6个碳的直链酮, 对于碳链较长的底物(例如2-甲基-4辛酮、2-壬酮、4-壬酮), 只有在较低底物浓度(0.1mg/mL)时才有较高的转化率.而来自真核生物的BVMOCm(来源于Cyanidioschyzon merolae)和BVMOPp(来源于Physcomitrella patens)对2-壬酮和3-壬酮等长链酮能实现完全转化[5].己酮可可碱1是一种广泛使用的药物, hFMO5(来源于Human)对这种具有羰基的药物有很强的体外氧合活性, 可以将其氧化成相应的乙酸酯, 反应具有高度的区域选择性(30:1)[6] (Scheme 1).
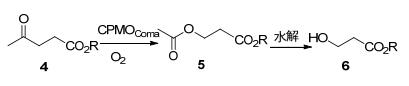
为了提高催化效率, 工程化改造的BVMO也被用于氧化直链酮底物.例如, Mihovilovic等[7]在2015年开发了一种利用BVMO将乙酰丙酸酯4氧化为3-羟基丙酸6衍生物的方法.其中CPMOComa(来源于Comamonas testosterone, 环戊酮单加氧酶)的F156L突变株对乙酰丙酸酯(甲、乙、丁酯)类底物的转化率有明显的提升, 是生物转化扩大生产过程中的最佳选择(Scheme 2).为了改善CHMOAcineto(来源于Acinetobacter sp.的环己酮单加氧酶)转化2-丁酮的催化活性以及对丙酸甲酯的区域选择性, Fraaije等[8]对酶上底物结合位点附近的多个残基和NADP+结合位点进行饱和突变, CHMOAcineto的最佳突变株T56S/I491A对2-丁酮的转化率为73%, 产物中丙酸甲酯的得率为43%, 均优于野生型(26%).后续的研究表明, 以2-丁醇为原料用醇脱氢酶产生丁酮并以BVMO转化为甲酸丁酯和乙酸乙酯的方法也是可行的[9]. Carvalho等[10]在使用PAMO(来源于Thermobifida fusca的苯丙酮单加氧酶)四突变株(P253F/G254A/ R258M/L443F)对2-辛酮进行转化时, 其活性远超野生型, 证明了活性位点的重塑可以改善PAMO对长链脂肪族底物的结合.
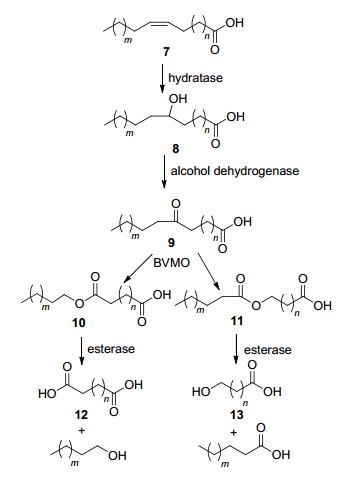
此外, BVMO催化直链酮的氧化反应也被组合进细胞内的多步级联反应中, 用于生产重要的工业酸. 2013年, Park等[11]利用多步酶法以油酸7为原料催化合成ω-羟基羧酸13.以水合酶和醇脱氢酶将不饱和脂肪酸前体中的双键氧化为酮, 再使用不同区域选择性的BVMO催化氧化成酯, 水解后得到α, ω-二羧酸12或ω-羟基脂肪酸13 (Scheme 3). 2016年他们[12]通过使用新的脂肪酸双键水合酶和大肠杆菌细胞, 提高了脂肪酸向细胞内级联酶转运的速率, 从而实现了一锅法将植物油进行氧化裂解, 最终转化为C9羧酸的级联反应, C9羧酸是一种潜在的抗真菌剂. 2018年Seo等[13]利用1, 9-壬二酸仲醇脱氢酶、BVMO、长链伯醇脱氢酶和醛脱氢酶成功将脂肪酸通过酶/全细胞环境转化为工业相关的C9羧酸(9-羟基壬酸、正壬酸等).
HAPMO(来源于Pseudomonas fluorescens ACB的4-羟基苯乙酮单加氧酶)是最早发现对含有芳香基团的酮类有催化活性的BVMO[14], 其催化3-苯基2-丁酮(14)动力学拆分时产物15的ee值可达99% (Scheme 4). 2011年, Fraaije等[15]从微生物Rhodococcus jostii RHA1的基因组中找到22种Type Ⅰ型BVMO, 其中BVMO9(来源于Rhodococcus jostii RHA1)和BVMO15(来源于Rhodococcus jostii RHA1)与HAPMO的底物范围相类似, 对多种芳香酮都表现出高活性.

含芳香基团的酮类底物中, 其酮基团和芳香环的距离对BVMO的催化活性也有影响. 2014年Opperman等[16]使用BVMOAFL456、BVMOAFL838、BVMOAFL210和BVMOAFL619(来源于Aspergillus flavu NRRL3357)转化4-苯基-2-丁酮16等底物, 其中BVMOAFL456和BVMOAFL619的活性随着羰基和苯环距离的增加而下降, 而另外两种BVMO的活性变化则相反.此外他们还发现, 如果苯环的对位上有羟基或甲氧基取代, 也会导致反应转化率下降(Scheme 5).
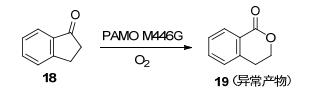
与HAPMO相比, PAMO同样可以转化芳香酮底物, 在反应活性、立体选择性及热稳定性上都更有优势[17], 但PAMO的底物范围狭窄, 一般只能接受体积小的底物, 对较大的底物其反应活性和立体选择性都比较差[18].蛋白质工程可以克服PAMO的这种缺点, 从而扩大其底物范围.例如, PAMO的M446G突变能接受数种野生型PAMO无法转化的大体积底物, 如1-茚酮(18).该突变株在催化1-茚酮的BV氧化反应时, 有很高的区域选择性, 只生成异常内酯1-异苯并二氢吡喃酮(19) (Scheme 6)[19].此外在PAMO的P253和L443这两个氨基酸残基上引入苯丙氨酸, 也能增加PAMO对一些含有芳环的底物的活性[20].由于STMO(来源于Rhodococcus rhodochrous的类固醇单加氧酶)的活性位点结构和PAMO非常相似, 因此, Fraaije和Mattevi等[21]推测并验证, STMO对PAMO的天然底物苯丙酮也能起到催化氧化的作用, 针对STMO上疏水和亲水残基, 并结合其他几种BVMO相关结构特征进行定向进化, 筛选出STMO的V72I突变对苯丙酮的反应活性是野生型的3倍.
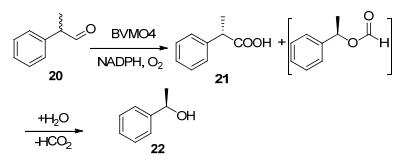
除了芳香酮的BV氧化, BVMO还能催化含有苯基的醛氧化反应. BVMO4(来源于Dietzia sp. D5)对含有苯基的醛类具有较高的活性, 可以将2-苯基丙醛(20)转化为2-苯基丙酸(21)和相应的酯产物, 酯产物最终水解生成1-苯基乙醇(22), 底物低浓度时生成等量的酯和酸, 高浓度时反应趋向于生成酯, 而使用4倍量的酶时, 产物趋向于生成酸(Scheme 7).除此之外, BVMO4还能氧化长链的脂肪醛, 例如辛醛和癸醛[22].
单萜是高性能再生聚合物的单体原料[23], 在BVMO生物氧化研究中, 往往用于酶的区域选择性研究(Scheme 8).许多野生型的BVMO本身就拥有很高的区域选择性, 往往只生成一种区域异构体.例如, CHMOJS666(来源于Polaromonas sp. strain JS666的环己酮单加氧酶)能缓慢将(+)-香芹酮23完全转化为相应的异常内酯(0:100)[24]. CPDMO(来源于Pseudomonas sp. HI-70的环十五酮单加氧酶)在转化这类底物时, 几乎只产生一种区域异构体, 并有着优异的立体选择性(ee>99%).其中(-)-薄荷酮24是首次被BVMO转化, 只生成正常的产物[25]. CAMO(来源于Cylindrocarpon radicicola ATCC 11011的环烷酮单加氧酶)和OTEMO(来源于Pseudomonas putida NCIMB 10007)在转化这类底物时, 后者有更强的区域选择性, OTEMO在转化(-)-二氢香芹酮(25)和(+)-薄荷酮(26)时只产生正常内酯产物[26].

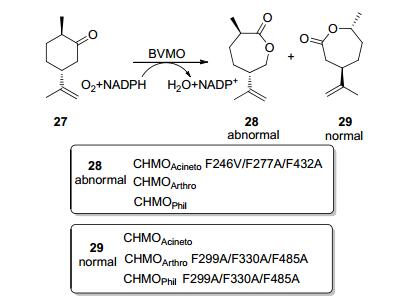
在许多情况下, BVMO缺乏完美的区域选择性. BVMO催化氧化的区域选择性主要取决于活性位点的手性环境和迁移取代基的电子效应, 对活性位点的残基进行突变, 可以改进BVMO的区域选择性. Bornscheuer等[27]利用理性设计得到CHMOArthro的F299A/F330A/F485A突变株来转化(+)二氢香芹酮27, 使底物倒置结合, 产物中正常和异常内酯的比例为99:1, 而野生型酶只生成异常产物.为了证明这种设计的适用性, 他们[27]又将突变引入有相似活性位点的CHMOAcineto, 从中筛选出CHMOAcineto F246V/F277A/F432A突变株, 这种突变株只产生异常内酯(0:100), 再次完全反转了相应野生型酶的区域选择性(Scheme 9).类似的区域选择性反转的结果在CHMOPhi1(来源于Rhodococcus sp. Phi1)酶中也得以实现, 野生型CHMOPhi1催化时只形成异常产物, 而CHMOPhi1 F299A/F330A/F485A突变株则只生成正常产物[23].
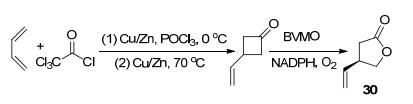
环丁酮类底物是少数几种仅使用过渡金属催化BV氧化即可获得高立体选择性产物的底物[28], 相应的酯产物具有广泛的生物学特性, 可用作香料和香气成分以及生物燃料, 也是合成很多具有复杂结构的天然产物的重要中间体.使用BVMO转化前手性环丁酮, 过程更环保, 适用性也更高.来自美国Codexis公司的6种BVMO[29]和CHMOJS666[24]可以催化3-取代环丁酮的去对称反应获得γ-丁内酯衍生物, 其中BVMO-CDX- 003[29]转化3-取代环丁酮时获得R构型产物(ee=98%), 而BVMO-P3-C07[29]和BVMO-P1-C06[29]则主要生成S构型产物[29]. Mihovilovic和Bornscheuer等[26]在2016年以1, 3-丁二烯为起点, 用CAMO和OTEMO催化3-取代前手性环丁酮的去对称氧化反应, 最终生成(R)-(-)- Taniguchi内酯(30) (Scheme 10).
环己酮及其衍生物是非常重要的化工原料, 也是目前BVMO研究中使用最广泛的底物之一.
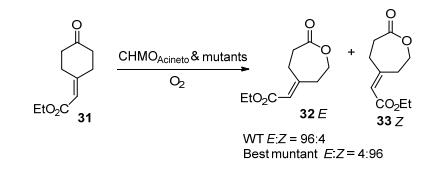
4-取代环己酮本身并无手性, 但在BVMO催化的去对称氧化过程中能生成手性产物.通过对BVMO酶的特定位点进行定向进化可以增强或逆转BVMO的立体选择性. 2004年由Reetz等[30]首次报道了相关研究.他们利用CHMO突变株F432S催化4-羟基环己酮, 反应的立体选择性由野生型的ee=9% (R)翻转至ee=79% (S).之后他们通过4-甲基-环己酮[31]和4-羟基-环己酮的QM/MM计算研究探讨了BVMO的催化机理, 发现反应的立体选择性可能与去质子化的黄素氢过氧化物以及阴离子形式的Criegee中间体有关[32].许多天然与非天然有机烯烃化合物, 其相应的双键具有E-或Z-构型. Reetz等发现野生型CHMOAcineto在全细胞环境下催化4-亚甲基环己酮衍生物31的去对称反应时, 产物中E构型和Z构型比例为96:4.之后他们通过3轮的ISM (Iterative Saturation Mutagenesis, 迭代饱和突变)进化策略, 筛选出最佳CHMOAcineto的突变株F432I/T433G/ L143M/F505C, 它能完全反转这类底物的立体选择性(E:Z=4:96), 同时其转化率可达98%[33] (Scheme 11). PAMO野生型无法接受4-取代环己酮作为底物, 通过定向进化筛选出来的PAMO (I67A/R258A/A442P/ S444A/P286R/L443F/Y502F)突变株可以催化4-(溴代亚甲基)-环己酮, 4-甲基环己酮的去对称反应[34], PAMO的Q93N/P94D突变株在转化4-烷基环己酮时也展现出很高的对映选择性[35].此外例如CAMO[26]、CHMOJS666[24]、CPDMO[25]以及OTEMO[26]这些野生型BVMO也都能催化4-取代环己酮的去对称反应, 并具有优异的立体选择性, 其中OTEMO在转化4-甲酸乙酯-环己酮时, 可以获得ee值>96%的立体选择性, 超越所有已知野生型BVMO[26].
通过BVMO催化2-取代环己酮的动力学拆分反应, 能提供高纯度的手性内酯产物.与上述4-取代环己酮类似, 通过对活性位点周围的特定残基进行定向进化可以控制拆分反应的立体选择性. Reetz等[35]在2010年根据变构效应对PAMO远离底物结合口袋的位点进行饱和突变, 经筛选400个突变株得到PAMO Q93N/P94D突变株, 这种突变株在催化2-取代环己酮的不对称动力学氧化拆分反应时有优异的立体选择性. Berghuis等[36]在2014年发现CHMO W492A突变株在转化2-取代环己酮时比起野生型有更高的(S)-构型立体选择性.与Fre(来源于Escherichia coli的黄素还原酶)共同表达的2, 5-DKCMO (2, 5二酮莰烷单加氧酶)在转化2-苯基环己酮时, 其选择性因子E>200(R)[37]. CHMO的热稳定型突变株DS4 (Y411C, A463C)对2-甲基环己酮进行拆分时能得到98% (R)的底物ee值, 远超野生型的立体选择性[38].此外CPDMO[25]、CAMO[26]以及OTEMO[26]这些野生型BVMO也能催化2-取代环己酮的动力学拆分反应, 后2种酶的转化产物优势构型相同, 但CAMO的立体选择性更佳.
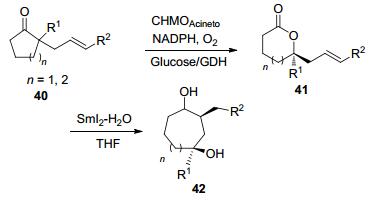
ε-己内酯36是一种非常重要的工业原料, 广泛用于聚合物生产, 每年产量可达数万吨.化学法通常用过氧乙酸作为氧化剂进行生产, 但存在有毒性并在生态学和安全性上有诸多缺点.近几年Gröger, Bornscheuer和Liese等课题组聚焦于此, 发明了一系列以环己醇为起点生产ε-己内酯36的级联反应(Scheme 12).通过使用PDH(多元醇脱氢酶)[39]、ADH(醇脱氢酶)[40]和LK-ADH(来源于Lactobacillus kefir的醇脱氢酶)等酶催化剂[41], 利用空气中的氧气可以将环己醇34氧化为环己酮35, 同时将NADP+还原为NADPH, 再使用CHMO将环己酮氧化为ε-己内酯衍生物.该过程不需要额外的共底物进行辅因子再生. 2015年, 他们[42]又利用CAL-A(南极假丝酵母的脂肪酶A)使ε-己内酯衍生物开环聚合, 有效地解决了产物抑制的问题. 2017年, Rudroff等[43]建立了Michaelis-Menten参数的模型, 证明了这个级联反应的瓶颈在于反应中BVMO的活性.而Kohl等[44]在这类级联反应中对CHMO和ADH使用共表达策略, 比起单独表达能实现更高效的辅因子循环, 从而获得更高的转化率.
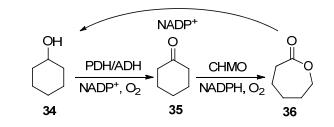
一般来说, 烯酮不会被BVMO转化, Alphand等[45]在2014年发现2种新型BVMO可以转化烯酮为不饱和内酯. BVMOOcean(来源于Oceanicola batsensis DSM 15984)可以在环己(庚、辛)烯酮38的羰基与非烯属碳原子之间插入氧原子, 生成共轭烯内酯37, 而BVMOParv (来源于Parvibaculum lavamentivorans DSM 13023)则可以在羰基和双键之间插入氧原子生成烯醇内酯39 (Scheme 13).另外这两种酶在全细胞环境中转化3-甲基环己酮和4-甲基环己酮获得高光学纯度的内酯产物(ee 98%).
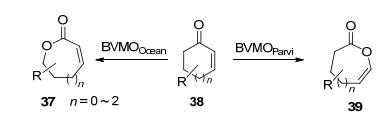
α, α-二烷基环酮的BV转化具备生成季碳手性中心内酯的潜力. Procter等[46]发明了一种以CHMOAcineto作为催化剂, 对带有α-季碳手性中心的外消旋五元或六元环酮40进行动力学拆分的反应.这种反应同时具备高立体选择性和化学选择性, 可以用于生产含有季碳手性中心的不饱和内酯, 并通过SmI2介导的环化作用生成高立体选择性的1, 4-环庚(辛)二醇42 (ee>99%) (Scheme 14).
莰酮是一种双环单萜类的化合物, 在自然界存在2种对映异构体, (+)莰酮43来源于樟树, (-)莰酮44可在植物精油中发现.目前已知有三种BVMOs参与莰酮降解途径, 分别是2, 5-DKCMO (2, 5-二酮莰烷单加氧酶)、3, 6-DKCMO酶(3, 6-二酮莰烷单加氧酶)以及OTEMO.前2者属于Type Ⅱ型的BVMO, 这类BVMO的辅因子是比起NADPH便宜很多的NADH, 因此有较高的工业应用价值[1].其中2, 5-DKCMO能转化(-)莰酮44, 但转化率不如(+)莰酮43, 而3, 6-DKCMO在转化这2种底物时差异不大, 对(-)莰酮44转化率要稍好一些[47].而OTEMO对(+)莰酮43的转化率高于(-)莰酮44[37].除此以外, CDMO(来源于Rhodococcus ruber SC1的环十二酮单加氧酶)[48]和CAMO[49]也能转化这类底物, 前者还能转化降樟脑45, 且有优异立体选择性.
对BVMO和相关辅酶基因采取共表达策略, 是一种增强反应活性的手段.例如, Lau等[37]在2013年将来自2, 5-DKCMO和3, 6-DKCMO的3个关于莰酮降解的功能性基因和Fre的基因串联, 在无细胞环境下转化(+)莰酮43/(-)莰酮44, 发现2, 5-DKCMO的反应活性增加了近20倍.而将Fre基因与2, 5-/3, 6-DKCMO共表达, 能显著增强这2种酶对莰酮和降樟脑的转化能力, 但如将2种基因融合到同一个阅读框架进行表达, 则会降低转化率[50].
|
|
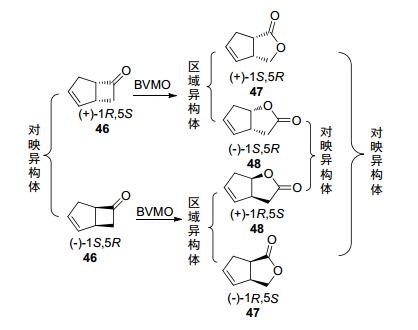
消旋双环[3.2.0]-庚-2-烯-6-酮(46)是一种常用于研究BVMO的立体和区域选择性的经典底物(Scheme 15).该底物不容易发生副反应, 以插入氧原子位置不同而产生普通/异常产物的比例来衡量酶的区域选择能力(表 2).
 下载:
导出CSV
下载:
导出CSV
| Entry | BVMO | 46 | Conv./% | n:aa | Ref. |
| 1 | CHMOAcineto | rac-46 | >99 | 60:40 | [38] |
| 2 | CHMO DS2 (N286C, A293C) | rac-46 | 87 | 60:40 | [38] |
| 3 | CHMO DS3 (T415C, A463C) | rac-46 | >99 | 60:40 | [38] |
| 4 | CHMO DS4 (Y411C, A463C) | rac-46 | 28 | 65:35 | [38] |
| 5 | CHMO T415C | rac-46 | >99 | 62:38 | [38] |
| 6 | CHMOTm | rac-46 | 100 | 49:51 | [54] |
| 7 | BVMOAFL210 | rac-46 | 56 | 87:13 | [16] |
| 8 | BVMOAFL456 | rac-46 | 53 | 94:0 | [16] |
| 9 | BVMOAFL838 | rac-46 | — | 62:13 | [16] |
| 10 | PAMO | rac-46 | — | 73:27 | [19] |
| 11 | PAMO Q152F | rac-46 | — | 30:70 | [19] |
| 12 | PAMO A435Y | rac-46 | — | 29:71 | [19] |
| 13 | PAMO A442G | rac-46 | — | 30:70 | [19] |
| 14 | PAMO Q152F/S441A/A442G | rac-46 | — | 23:77 | [19] |
| 15 | STMO | rac-46 | — | 65:35 | [55] |
| 16 | PASTMO | rac-46 | — | 66:34 | [55] |
| 17 | PACHMO | rac-46 | — | 39:61 | [55] |
| 18 | PAMEMO1 | rac-46 | — | 76:24 | [55] |
| 19 | 2, 5-DKCMO-1-Fred | rac-46 | 100 | 57:43 | [37] |
| 20 | 2, 5-DKCMO-2-Fred | rac-46 | 100 | 52:48 | [37] |
| 21 | 3, 6-DKCMO-Fred | rac-46 | 53 | 40:60 | [37] |
| 22 | 2.5-DKCMO | rac-46 | — | 57:43 | [37] |
| 23 | OTEMO | rac-46 | >90 | 70:30 | [26] |
| 24 | CAMO | rac-46 | >90 | 53:47 | [26] |
| 25 | CPMO | rac-46 | >90 | 97:3 | [26] |
| 26 | BVMOCm | (+)-46 | 100 | 73:27 | [5] |
| BVMOCm | (-)-46 | 100 | 88:12 | [5] | |
| 27 | BVMOPp | (+)-46 | 100 | 76:27 | [5] |
| BVMOPp | (-)-46 | 66 | 62:4 | [5] | |
| 28 | BVMOPp Y160H | (+)-46 | 100 | 72:28 | [5] |
| BVMOPp Y160H | (-)-46 | 100 | 95:5 | [5] | |
| 29 | OTEMO | (-)-46 | — | 51:49 | [52] |
| 30 | OTEMO F255V/F443V | rac-46 | 95 | 91:9 | [52] |
| 31 | OTEMO W501A | (-)-46 | — | 3:97 | [52] |
| 32 | BVMOLepto | rac-46 | 94 | 50:50 | [4] |
| a n: Normal product, a: Abnormal product. | |||||
野生型PAMO在转化这种底物时, 产物中正常内酯48和异常内酯47的比例约为3:1.而PAMO的A435Y、A442G和Q152F这3种突变株[19]则展示了与野生型相反的区域选择性(48:47=1:3), 其中PAMO A435Y转化所得的异常产物47还有着优异的立体选择性(ee 90%).而CHMO[51]、PAMO的V54I和I67T突变株[19]、CAMO[26]以及BVMOLepto(来源于Leptospira biflex)[4]等酶则是等量产生2种区域异构产物(1:1).此外, OTEMO的F255A/F443V突变株在转化这种底物时能产生90%的正常产物48, 而W501V突变株能产生98%的异常产物47, 这2种突变株大幅提升了OTEMO的区域选择性(野生型为1:1)[52].现如今测序和生物信息技术逐渐发展成熟, 大量新型的BVMO已经被发现, 这种底物也用于判断微生物是否有BVMO活性. Alphand等[53]在2015年使用来自46个属的107个真菌来转化该底物, 发现其中86种有活性, 证明在真菌中BVMO基因序列是普遍存在的.
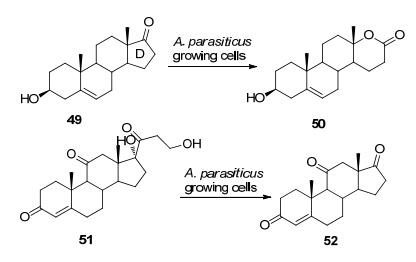
多环酮及大分子底物往往是生产抗生素以及激素的原料或前体.研究这类化合物的生物转化, 可以为药物研发和生产提供技术支持. Kurina-Sanz等[56]在2016年发现一种曲霉属的微生物(A. parasiticus)可以转化脱氢表雄甾酮(DHEA) 49, 通过D环的BV氧化反应形成内酯产物, 也可以通过生物氧化途径降解可的松51的侧链, 完全转化为肾上腺甾酮52, 这说明该微生物中应该有BVMO活性(Scheme 16).
Rohr等[57]在2013通过分析对比MtmOIV(来源于Streptomyces argillaceus ATCC 12956的光神霉素单加氧酶)的晶体结构后, 将底物识别和催化相关的位点逐一突变为丙氨酸, 在转化前光神霉素B (Premithramycin B) 53时, 发现P84A和F89A突变相比野生型分别提高了1.5和5倍的催化活性, 而P84A/F89A突变的催化活性只能提高3倍, 这说明合并多个位点的优势突变其催化特性未必优于单个位点的优势突变.
|
|
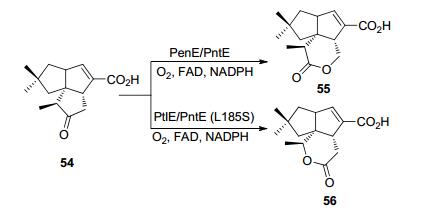
Zhu等[58]在2015年利用1-脱氧-11-氧代戊酸(1- deoxy-11-oxopentalenic acid) 54来考察PenE(来源于Streptomyces exfoliatus)、PntE(来源于Streptomyces arenae)以及PtlE(来源于Streptomyces arenae)的区域选择性, 前2种BVMO主要生成异常内酯产物(戊烯内酯D, 55), 而PtlE则生成正常内酯(新戊烯内酯D, 56).此外他们利用理性设计得到PntE的13种突变株, 发现其中最佳的突变株L185S能逆转野生型PntE的区域选择性, 生成正常内酯(Scheme 17).
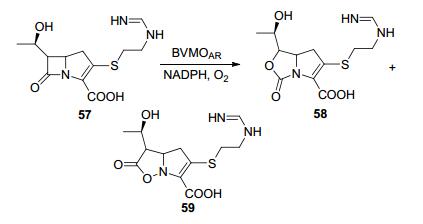
亚胺培南(Imipenem) 57是一种碳青霉烯类的抗生素, 通过检测NADPH消耗实验, 确认了BVMOAR(来源于Acinetobacter radioresistens)可以转化这种D环上带有N和S元素的底物.亚胺培南会因碳青霉烯环的BV氧化而插入氧原子, 从而失去抗生素特性, 它是除四环素以外第二种可以用BVMO灭活的抗菌素(Scheme 18)[59].
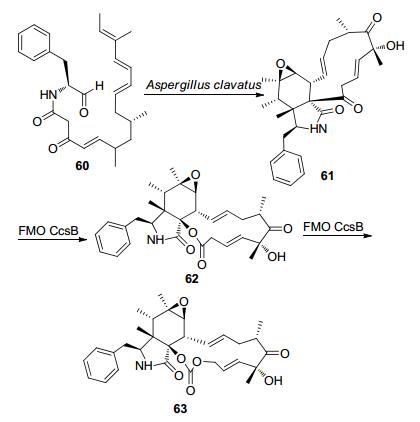
细胞松弛素E (Cytochalasin E) 63是一种血管生成抑制剂, 2011年Tang等[60]在微生物Aspergillus clavatus NRRL 1的序列中找到可能参与细胞松弛素生物合成的ccs基因簇, 其序列和CPDMO表现出高度的同一性(41%)且有BVMO的指纹序列, 于是假定FMO CcsB(来源于Aspergillus clavatus NRRL 1 ccs生物基因簇)的催化能力与CPDMO相似, 这种BVMO可能导致两个连续的BV氧化, 从而生成细胞松弛素E 63 (Scheme 19). 2014年, 他们[61]通过FMO CcsB催化细胞松弛素Z16的转化, 证实了上述假设. FMO CcsB的发现提供了将酮转化为碳酸盐的潜在合成策略.
BVMO除了催化羰基的亲核氧化以外还能实现杂原子的亲电氧化[62], 主要以含硼、硒、硫等杂原子的有机化合物为主.酶催化杂原子底物有机反应一般分为2种, 在杂原子处发生酶促转化, 以及在有机底物的其他官能团中发生酶促转化, 本综述中主要讨论前者.
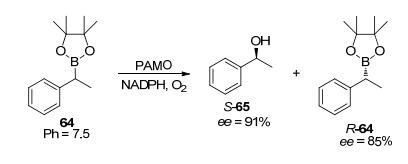
早在1985年, Walsh等[63]就发现了Type Ⅰ型的BVMO可以转化含硼有机化合物. Gonzalo等[64]相继发现了PAMO可以将苯基硼酸氧化成苯酚, HAPMO可以氧化多种含硼苯乙酮衍生物、乙烯基硼化合物以及外消旋含硼化合物[62]. PAMO在转化3-取代的含硼苯乙酮衍生物时, 硼氧化要优先于酮的BV氧化和环氧化[65]. Fraaije等[66]在2011年开发了一种以非手性芳族、乙烯基硼化合物以及外消旋硼化合物作为底物, 利用BVMO对其中的碳硼键进行氧化并通过消除硼酸来得到相应醇的方法.在利用PAMO及其M446G突变株进行催化时, 所有底物都产生了硼氧化反应, 仅4-取代的底物出现了酮的BV氧化, 而使用HAPMO则同时观察到2种氧化反应的发生, CHMO倾向于碳硼键氧化, 但反应活性低下.另外PAMO在有机含硼化合物64的动力学拆分中, 在无细胞环境且pH为7.5的条件下, 产物65的ee值可达91% (S) (Scheme 20).关于有机硼化合物生物转化的报道相对比较少, 主要是因为这些化合物可能对酶产生抑制作用[67].
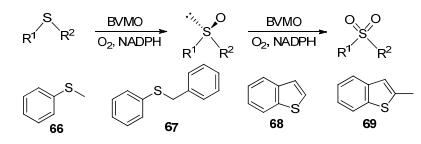
通过化学方法很难实现硫醚的选择性亚砜化, 因此酶介导的选择性亚砜化在过去几十年来都是有机化学家关注的热点之一[65].该反应的主要问题是如何避免进一步氧化生成无手性的砜(Scheme 21)[68].苯烷基硫醚作为含硫的模型底物, 能够通过BVMO转化为相应的亚砜和砜, 广泛地用于BVMO研究中.例如, BVMOAFL210[16]、BVMOAFL619[16]和BVMO4[22]对这类底物都有较强的活性, 但其催化属性各有不同.其中BVMOAFL210能进一步将产生的亚砜氧化为相应的砜, 而BVMO4则优先选择未取代的芳族硫化物, 除此以外也能转化在对位(甲基, 氟)或间位(氯)上含有小取代基的底物[22]. Bornscheuer等[69]在2017年发现一种新的BVMO, 即YMOA(来源于Yarrowia lipolytica), 它对硫醚和亚砜都有转化能力, 甚至可以转化DMSO.对其进行理性设计得到的YMOA V121T突变株能够增加砜产物的形成, 而YMOA C480F和YMOA A483L突变株则只产生亚砜产物.表明可以通过对BVMO的理性设计来控制硫醚类底物在反应中的氧化程度.
苯甲基硫醚66体积相对较小, 一些野生型BVMO对这种底物本身就有很强的活性和立体选择性.例如, CHMOTm(来源于Thermocrispum municipale DSM 44069的环己酮单加氧酶)在转化这种底物时主要生成R构型产物(ee 97%)[70], 而BVMOAf1(来源于A. fumigatus Af293)在转化这种底物时产率较高(3 h后转化88%), 只生成S构型亚砜(ee=99%)[71]. CHMOAcineto和FDH(甲酸脱氢酶)共表达时, 显著提高了细胞对NADPH的再生能力, 获得高立体选择性的亚砜产物(ee 99%)[72].但有些BVMO, 如PAMO, 对硫醚氧化的选择性并不好, 一般都得到低ee值甚至消旋的产物[22], 因此也涌现出许多通过定向进化和理性设计来改进BVMO对硫醚类底物立体选择性的研究.例如, Reetz等[73]在2014年将ISM策略用于PAMO的定向进化, 提高其对硫醚类底物的立体选择性.在筛选结果中发现PAMO I67Q/P440F/ A442N/L443I突变株转化苯甲基硫醚66和其他硫醚时能获得高立体选择性的R构型亚砜产物(ee>95%).值得注意的是, 构成这4倍体突变的4种单倍体PAMO突变株都是S构型选择性的, 而这个4倍体突变的转化产物则是R构型, 再次证明了同一催化属性的突变集中后其作用未必是叠加的.另外以PAMO为骨架进行亚域交换产生的嵌合酶PASTMO[55]和PAMEMO1[55], 与野生型STMO相比, 在转化苯甲基硫醚66时也体现出了更高的立体选择性.
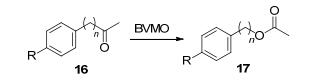
通过调整BVMO底物结合位点附近的氨基酸残基, 可以让改造后的酶转化原本野生型无法接受的大体积底物, 从而扩大底物谱.例如, PAMO M446G[19]、PAMO M446A[74]以及PAMO I67S突变株[74], 这三种PAMO突变株通过将活性位点附近的氨基酸残基突变为体积较小的氨基酸, 减小了空间位阻, 使其能够转化像苄基苯基硫醚67这样的大体积底物, 并有着优异的立体选择性[55]. SMO(来源于Pseudomonas putida CA-3的苯乙烯单加氧酶)对硫醚类底物本身就有较高的立体选择性, 但其对苯并[b]噻吩68和2-甲基苯并[b]噻吩69这2种底物的反应速率极低, 可能是因为这2种底物有较强的刚性和更大的杂环结构.而这种酶的工程酶SMO R3-11上的β链在酶构型上有显著的变化, 改变了活性位点, 扩大了结合口袋, 其转化这2种底物的活性分别是野生型SMO的10.1倍和5.6倍[75].此外, 利用Type Ⅰ BVMO指纹序列对微生物基因组进行基因挖掘发现的BVMOBo(来源于Bradyrhizobium oligotrophicum)和BVMOAm(来源于Aeromicrobium marinum)也对多种体积庞大的前手性硫醚(如吡唑硫醚等), 具有出色的立体选择性和活性[76].
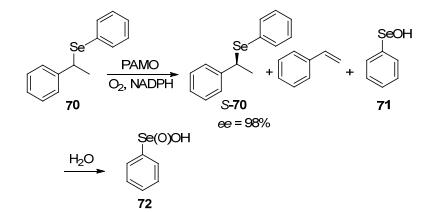
受到硫化物立体选择性氧化的启发, 并考虑到硫和硒类似的化学性质, BVMO研究也进一步拓展到含硒有机化合物. Brondani等[77]在2011年发现PAMO在转化一些对位和间位含硒的苯乙酮衍生物时, 发现底物的酮部分保留完整, 主要生成相应的硒亚砜.次年, 他们通过PAMO催化芳环中没有取代基的有机硒苯乙基化合物70动力学拆分反应, 获得了ee值高达98%的S构型底物(Scheme 22)[78].
综上所述, 随着近年来BVMO相关研究的不断深入, 其催化的反应类型不断得到扩展, 底物结构也日渐丰富, BVMO的催化活性和立体选择性都得到了显著提高.特别是, 通过BVMO的特征序列从各类微生物DNA中快速筛选可能存在的BVMO序列, 并通过克隆、重组等方法获取的新型BVMO不断涌现.随着越来越多BVMO的晶体结构被构建, 基于晶体结构分析以及生物计算的理性设计已经成为研究的主要方向, 用于改进野生型BVMO.虽然BVMO在选择性、反应条件绿色化和催化效率等方面优于传统的化学氧化, 但要在工业领域大范围应用还存在诸多困难.由于BVMO对辅因子的依赖性, 在有机合成中利用BVMO变得复杂, 特别是在无细胞环境下利用纯酶进行反应时, NAD(P)H的消耗成为一项不得不考虑的成本因素, 发展新的辅酶再生系统, 利用相对低廉的辅助底物来再生NAD(P)H将是非常重要的课题; 而通过全细胞环境介导的生物转化中, 减少反应的副产物, 寻找更廉价的诱导剂以及进一步提高酶的活性, 也是今后探索的方向; 除此之外, 还存在酶稳定性不足以及底物/产物抑制作用等其他问题.因此, 随着环境友好型工艺取代传统化学工艺的迫切性不断增加, 未来如何将实验室规模的BVMO研究成果放大到大规模工业生产中将是一个非常有价值的挑战.
Balke, K.; Kadow, M.; Mallin, H.; Sass, S.; Bornscheuer, U. T. Org. Biomol. Chem. 2012, 10, 6249. doi: 10.1039/c2ob25704a
Leisch, H.; Morley, K.; Lau, P. C. Chem. Rev. (Washington, DC, U. S.) 2011, 111, 4165.
(a) Dong, J.; Fernandez-Fueyo, E.; Hollmann, F.; Paul, C. E.; Pesic, M.; Schmidt, S.; Wang, Y.; Younes, S.; Zhang, W. Angew. Chem., Int. Ed. 2018, 57, 9238.
(b) Balke, K.; Beier, A.; Bornscheuer, U. T. Biotechnol. Adv. 2018,
36, 247.
(c) Liang, Y.; Wei, J.; Qiu, X.; Jiao, N. Chem. Rev. 2018, 118, 4912.
Ceccoli, R. D.; Bianchi, D. A.; Fink, M. J.; Mihovilovic, M. D.; Rial, D. V. AMB Express 2017, 7, 87. doi: 10.1186/s13568-017-0390-5
Beneventi, E.; Niero, M.; Motterle, R.; Fraaije, M.; Bergantino, E. J. Mol. Catal. B:Enzym. 2013, 98, 145. doi: 10.1016/j.molcatb.2013.10.006
Fiorentini, F.; Romero, E.; Fraaije, M. W.; Faber, K.; Hall, M.; Mattevi, A. ACS Chem Biol. 2017, 12, 2379. doi: 10.1021/acschembio.7b00470
Fink, M. J.; Mihovilovic, M. D. Chem. Commun. (Cambridge, U. K.) 2015, 51, 2874.
van Beek, H. L.; Romero, E.; Fraaije, M. W. ACS Chem Biol. 2017, 12, 291. doi: 10.1021/acschembio.6b00965
Pereira, J. P. C.; van der Wielen, L. A. M.; Straathof, A. J. J. Bioresour. Technol. 2018, 256, 187. doi: 10.1016/j.biortech.2018.01.118
Carvalho, A. T. P.; Dourado, D.; Skvortsov, T.; de Abreu, M.; Ferguson, L. J.; Quinn, D. J.; Moody, T. S.; Huang, M. Phys. Chem. Chem. Phys. 2018, 20, 2558. doi: 10.1039/C7CP07172H
Song, J. W.; Jeon, E. Y.; Song, D. H.; Jang, H. Y.; Bornscheuer, U. T.; Oh, D. K.; Park, J. B. Angew. Chem., Int. Ed. 2013, 52, 2534. doi: 10.1002/anie.v52.9
Jeon, E.-Y.; Seo, J.-H.; Kang, W.-R.; Kim, M.-J.; Lee, J.-H.; Oh, D.-K.; Park, J.-B. ACS Catal. 2016, 6, 7547. doi: 10.1021/acscatal.6b01884
Seo, E. J.; Yeon, Y. J.; Seo, J. H.; Lee, J. H.; Bongol, J. P.; Oh, Y.; Park, J. M.; Lim, S. M.; Lee, C. G.; Park, J. B. Bioresour. Technol. 2018, 251, 288. doi: 10.1016/j.biortech.2017.12.036
(a) Rehdorf, J.; Zimmer, C. L.; Bornscheuer, U. T. Appl. Environ. Microbiol. 2009, 75, 3106.
(b) Geitner, K.; Rehdorf, J.; Snajdrova, R.; Bornscheuer, U. T. Appl. Microbiol. Biotechnol. 2010, 88, 1087.
Riebel, A.; Dudek, H. M.; de Gonzalo, G.; Stepniak, P.; Rychlewski, L.; Fraaije, M. W. Appl. Microbiol. Biotechnol. 2012, 95, 1479. doi: 10.1007/s00253-011-3823-0
Ferroni, F. M.; Smit, M. S.; Opperman, D. J. J. Mol. Catal. B:Enzym. 2014, 107, 47. doi: 10.1016/j.molcatb.2014.05.015
(a) Fraaije, M. W.; Wu, J.; Heuts, D. P.; van Hellemond, E. W.; Spelberg, J. H.; Janssen, D. B. Appl. Microbiol. Biotechnol. 2005,
66, 393.
(b) de Gonzalo, G.; Mihovilovic, M. D.; Fraaije, M. W. ChemBioChem 2010,
11, 2208.
Pazmino, D. E. T.; Snajdrova, R.; Rial, D. V.; Mihovilovic, M. D.; Fraaije, M. W. Adv. Synth. Catal. 2007, 349, 1361. doi: 10.1002/(ISSN)1615-4169
Dudek, H. M.; de Gonzalo, G.; Pazmino, D. E.; Stepniak, P.; Wyrwicz, L. S.; Rychlewski, L.; Fraaije, M. W. Appl. Environ. Microbiol. 2011, 77, 5730. doi: 10.1128/AEM.00687-11
Dudek, H. M.; Fink, M. J.; Shivange, A. V.; Dennig, A.; Mihovilovic, M. D.; Schwaneberg, U.; Fraaije, M. W. Appl. Microbiol. Biotechnol. 2014, 98, 4009. doi: 10.1007/s00253-013-5364-1
Franceschini, S.; van Beek, H. L.; Pennetta, A.; Martinoli, C.; Fraaije, M. W.; Mattevi, A. J. Biol. Chem. 2012, 287, 22626. doi: 10.1074/jbc.M112.372177
Bisagni, S.; Summers, B.; Kara, S.; Hatti-Kaul, R.; Grogan, G.; Mamo, G.; Hollmann, F. Top. Catal. 2013, 57, 366.
Messiha, H. L.; Ahmed, S. T.; Karuppiah, V.; Suardiaz, R.; Ascue Avalos, G. A.; Fey, N.; Yeates, S.; Toogood, H. S.; Mulholland, A. J.; Scrutton, N. S. Biochemistry 2018, 57, 1997. doi: 10.1021/acs.biochem.8b00169
Alexander, A. K.; Biedermann, D.; Fink, M. J.; Mihovilovic, M. D.; Mattes, T. E. J. Mol. Catal. B:Enzym. 2012, 78, 105. doi: 10.1016/j.molcatb.2012.03.002
Fink, M. J.; Fischer, T. C.; Rudroff, F.; Dudek, H.; Fraaije, M. W.; Mihovilovic, M. D. J. Mol. Catal. B:Enzym. 2011, 73, 9. doi: 10.1016/j.molcatb.2011.07.003
Rudroff, F.; Fink, M. J.; Pydi, R.; Bornscheuer, U. T.; Mihovilovic, M. D. Monatsh. Chem. 2017, 148, 157. doi: 10.1007/s00706-016-1873-9
Balke, K.; Schmidt, S.; Genz, M.; Bornscheuer, U. T. ACS Chem Biol. 2016, 11, 38. doi: 10.1021/acschembio.5b00723
Zhang, Z. G.; Parra, L. P.; Reetz, M. T. Chem.-Eur. J. 2012, 18, 10160. doi: 10.1002/chem.201202163
Rodríguez-Mata, M.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; García-Cerrada, S.; Mendiola, J.; de Frutos, Ó.; Collado, I. Tetrahedron 2016, 72, 7268. doi: 10.1016/j.tet.2015.12.071
Reetz, M. T.; Brunner, B.; Schneider, T.; Schulz, F.; Clouthier, C. M.; Kayser, M. M. Angew. Chem., Int. Ed. 2004, 43, 4075. doi: 10.1002/(ISSN)1521-3773
Clouthier, C. M.; Kayser, M. M.; Reetz, M. T. J. Org. Chem. 2006, 71, 8431. doi: 10.1021/jo0613636
Polyak, I.; Reetz, M. T.; Thiel, W. J. Phys. Chem. B 2013, 117, 4993. doi: 10.1021/jp4018019
Zhang, Z.-G.; Roiban, G.-D.; Acevedo, J. P.; Polyak, I.; Reetz, M. T. Adv. Synth. Catal. 2013, 355, 99. doi: 10.1002/adsc.201200759
Parra, L. P.; Agudo, R.; Reetz, M. T. ChemBioChem 2013, 14, 2301. doi: 10.1002/cbic.v14.17
Wu, S.; Acevedo, J. P.; Reetz, M. T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2775. doi: 10.1073/pnas.0911656107
Yachnin, B. J.; McEvoy, M. B.; MacCuish, R. J.; Morley, K. L.; Lau, P. C.; Berghuis, A. M. ACS Chem Biol. 2014, 9, 2843. doi: 10.1021/cb500442e
Iwaki, H.; Grosse, S.; Bergeron, H.; Leisch, H.; Morley, K.; Hasegawa, Y.; Lau, P. C. Appl. Environ. Microbiol. 2013, 79, 3282. doi: 10.1128/AEM.03958-12
Schmidt, S.; Genz, M.; Balke, K.; Bornscheuer, U. T. J. Biotechnol. 2015, 214, 199.
Mallin, H.; Wulf, H.; Bornscheuer, U. T. Enzyme Microb. Technol. 2013, 53, 283. doi: 10.1016/j.enzmictec.2013.01.007
Staudt, S.; Bornscheuer, U. T.; Menyes, U.; Hummel, W.; Groger, H. Enzyme Microb. Technol. 2013, 53, 288. doi: 10.1016/j.enzmictec.2013.03.011
Oberleitner, N.; Peters, C.; Rudroff, F.; Bornscheuer, U. T.; Mihovilovic, M. D. J. Biotechnol. 2014, 192, 393.
Schmidt, S.; Scherkus, C.; Muschiol, J.; Menyes, U.; Winkler, T.; Hummel, W.; Groger, H.; Liese, A.; Herz, H. G.; Bornscheuer, U. T. Angew. Chem., Int. Ed. 2015, 54, 2784. doi: 10.1002/anie.201410633
Milker, S.; Fink, M. J.; Oberleitner, N.; Ressmann, A. K.; Bornscheuer, U. T.; Mihovilovic, M. D.; Rudroff, F. ChemCatChem 2017, 9, 3420. doi: 10.1002/cctc.201700573
Kohl, A.; Srinivasamurthy, V.; Bottcher, D.; Kabisch, J.; Bornscheuer, U. T. Enzyme Microb. Technol. 2018, 108, 53. doi: 10.1016/j.enzmictec.2017.09.003
Reignier, T.; de Berardinis, V.; Petit, J. L.; Mariage, A.; Hamze, K.; Duquesne, K.; Alphand, V. Chem. Commun. (Cambridge, U. K.) 2014, 50, 7793.
Morrill, C.; Jensen, C.; Just-Baringo, X.; Grogan, G.; Turner, N. J.; Procter, D. J. Angew. Chem., Int. Ed. 2018, 57, 3692. doi: 10.1002/anie.201800121
Kadow, M.; Loschinski, K.; Sass, S.; Schmidt, M.; Bornscheuer, U. T. Appl. Microbiol. Biotechnol. 2012, 96, 419. doi: 10.1007/s00253-011-3859-1
Fink, M. J.; Rial, D. V.; Kapitanova, P.; Lengar, A.; Rehdorf, J.; Cheng, Q.; Rudroff, F.; Mihovilovic, M. D. Adv. Synth. Catal. 2012, 354, 3491. doi: 10.1002/adsc.v354.18
Leipold, F.; Wardenga, R.; Bornscheuer, U. T. Appl. Microbiol. Biotechnol. 2012, 94, 705. doi: 10.1007/s00253-011-3670-z
Kadow, M.; Balke, K.; Willetts, A.; Bornscheuer, U. T.; Backvall, J. E. Appl. Microbiol. Biotechnol. 2014, 98, 3975. doi: 10.1007/s00253-013-5338-3
Furst, M. J.; Savino, S.; Dudek, H. M.; Gomez Castellanos, J. R.; Gutierrez de Souza, C.; Rovida, S.; Fraaije, M. W.; Mattevi, A. J. Am. Chem. Soc. 2017, 139, 627. doi: 10.1021/jacs.6b12246
Balke, K.; Baumgen, M.; Bornscheuer, U. T. ChemBioChem 2017, 18, 1627. doi: 10.1002/cbic.201700223
Butinar, L.; Mohorcic, M.; Deyris, V.; Duquesne, K.; Iacazio, G.; Claeys-Bruno, M.; Friedrich, J.; Alphand, V. Phytochemistry 2015, 117, 144. doi: 10.1016/j.phytochem.2015.06.009
Romero, E.; Castellanos, J. R.; Mattevi, A.; Fraaije, M. W. Angew. Chem., Int. Ed. 2016, 55, 15852. doi: 10.1002/anie.201608951
van Beek, H. L.; de Gonzalo, G.; Fraaije, M. W. Chem. Commun. (Cambridge, U. K.) 2012, 48, 3288.
Mascotti, M. L.; Palazzolo, M. A.; Bisogno, F. R.; Kurina-Sanz, M. Steroids 2016, 109, 44. doi: 10.1016/j.steroids.2016.03.018
Bosserman, M. A.; Downey, T.; Noinaj, N.; Buchanan, S. K.; Rohr, J. ACS Chem Biol. 2013, 8, 2466. doi: 10.1021/cb400399b
Chen, K.; Wu, S.; Zhu, L.; Zhang, C.; Xiang, W.; Deng, Z.; Ikeda, H.; Cane, D. E.; Zhu, D. Biochemistry 2016, 55, 6696. doi: 10.1021/acs.biochem.6b01040
Minerdi, D.; Zgrablic, I.; Castrignano, S.; Catucci, G.; Medana, C.; Terlizzi, M. E.; Gribaudo, G.; Gilardi, G.; Sadeghi, S. J. Antimicrob. Agents Chemother. 2016, 60, 64. doi: 10.1128/AAC.01088-15
Qiao, K.; Chooi, Y. H.; Tang, Y. Metab. Eng. 2011, 13, 723. doi: 10.1016/j.ymben.2011.09.008
Hu, Y.; Dietrich, D.; Xu, W.; Patel, A.; Thuss, J. A.; Wang, J.; Yin, W. B.; Qiao, K.; Houk, K. N.; Vederas, J. C.; Tang, Y. Nat. Chem. Biol. 2014, 10, 552. doi: 10.1038/nchembio.1527
de Gonzalo, G.; Torres Pazmiño, D. E.; Ottolina, G.; Fraaije, M. W.; Carrea, G. Tetrahedron:Asymmetry 2006, 17, 130. doi: 10.1016/j.tetasy.2005.11.024
(a) Branchaud, B. P.; Walsh, C. T. J. Am. Chem. Soc. 1985,
107, 2153.
(b) Walsh, C. T.; Chen, Y. C. J. Angew. Chem., Int. Ed. Engl. 1988,
27, 333.
Gonzalo, G. D.; Pazmiño, D. E. T.; Ottolina, G.; Fraaije, M. W.; Carrea, G. Tetrahedron:Asymmetry 2005, 16, 3077. doi: 10.1016/j.tetasy.2005.08.004
Ceccoli, R. D.; Bianchi, D. A.; Rial, D. V. Front. Microbiol. 2014, 5, 25.
Brondani, P. B.; de Gonzalo, G.; Fraaije, M. W.; Andrade, L. H. Adv. Synth. Catal. 2011, 353, 2169. doi: 10.1002/adsc.v353.11/12
Das, B. C.; Thapa, P.; Karki, R.; Schinke, C.; Das, S.; Kambhampati, S.; Banerjee, S. K.; Van Veldhuizen, P.; Verma, A.; Weiss, L. M.; Evans, T. Future Med. Chem. 2013, 5, 653. doi: 10.4155/fmc.13.38
Alphand, V.; Wohlgemuth, R. Curr. Org. Chem. 2010, 14, 1928. doi: 10.2174/138527210792927519
Bordewick, S.; Beier, A.; Balke, K.; Bornscheuer, U. T. Enzyme Microb. Technol. 2018, 109, 31. doi: 10.1016/j.enzmictec.2017.09.008
de Gonzalo, G.; Franconetti, A. Enzyme Microb. Technol. 2018, 113, 24. doi: 10.1016/j.enzmictec.2018.02.006
Mascotti, M. L.; Juri Ayub, M.; Dudek, H.; Sanz, M. K.; Fraaije, M. W. AMB Express 2013, 3, 33. doi: 10.1186/2191-0855-3-33
Zhai, X. H.; Ma, Y. H.; Lai, D. Y.; Zhou, S.; Chen, Z. M. J. Ind. Microbiol. Biotechnol. 2013, 40, 797. doi: 10.1007/s10295-013-1288-0
Zhang, Z. G.; Lonsdale, R.; Sanchis, J.; Reetz, M. T. J. Am. Chem. Soc. 2014, 136, 17262. doi: 10.1021/ja5098034
Dudek, H. M.; Popken, P.; van Bloois, E.; Duetz, W. A.; Fraaije, M. W. J. Biomol. Screening. 2013, 18, 678. doi: 10.1177/1087057113480390
Nikodinovic-Runic, J.; Coulombel, L.; Francuski, D.; Sharma, N. D.; Boyd, D. R.; Ferrall, R. M.; O'Connor, K. E. Appl. Microbiol. Biotechnol. 2013, 97, 4849. doi: 10.1007/s00253-012-4332-5
Zhang, Y.; Liu, F.; Xu, N.; Wu, Y. Q.; Zheng, Y. C.; Zhao, Q.; Lin, G.; Yu, H. L.; Xu, J. H. Appl. Environ. Microbiol. 2018, 84.
Andrade, L. H.; Pedrozo, E. C.; Leite, H. G.; Brondani, P. B. J. Mol. Catal. B:Enzym. 2011, 73, 63. doi: 10.1016/j.molcatb.2011.07.018
Brondani, P. B.; Guilmoto, N. M. A. F.; Dudek, H. M.; Fraaije, M. W.; Andrade, L. H. Tetrahedron 2012, 68, 10431. doi: 10.1016/j.tet.2012.09.087

图式 1 hFMO5催化己酮可可碱的区域选择性BV氧化反应
Scheme 1 Regioselective BV oxidation reaction of pentoxifylline catalyzed by hFMO5

图式 2 CPMOComa催化乙酰丙酸烷基酯的BV氧化反应
Scheme 2 Bayer-Villiger oxidation reaction of alkyl levulinate catalyzed by CPMOComa

图式 3 通过多步酶催化反应将不饱和脂肪酸转化为α, ω-二羧酸或ω-羟基脂肪酸
Scheme 3 Conversion of unsaturated fatty acids to α, ω-dicar- boxylic acids or ω-hydroxy fatty acids by multi-step enzymatic reactions

图式 4 HAPMO催化3-苯基2-丁酮的动力学拆分反应
Scheme 4 Kinetic resolution reaction of 3-phenyl 2-butanone catalyzed by HAPMO

图式 5 BVMO催化芳香酮的Baeyer-Villiger氧化
Scheme 5 Bayer-Villiger oxidation reaction of aromatic ketone catalyzed by BVMO

图式 6 通过PAMO M446G催化1-茚酮的BV氧化反应
Scheme 6 Bayer-Villiger oxidation reaction of 1-indone catalyzed by PAMO M446G

图式 7 BVMO催化2-苯基丙醛的BV氧化
Scheme 7 Bayer-Villiger oxidation reaction of 2-phenylpro- pionaldehyde catalyzed by BVMOs

图式 8 BVMO催化萜烯酮的区域选择性氧化
Scheme 8 Regioselective oxidation reaction of terpeneone catalyzed by BVMO

图式 9 几种CHMO及其突变株催化(+)二氢香芹酮的BV氧化反应
Scheme 9 Bayer-Villiger oxidation reaction of (+)-dihydro- carvone catalyzed by CHMO and variants

图式 10 以1, 3-丁二烯为起点通过BVMO生成Taniguchi内酯
Scheme 10 Synthesis of taniguchi lactone by BVMO with 1, 3- butadiene as the starting material

图式 11 由CHMOAcineto及其突变株催化4-亚甲基环己酮衍生物的去对称反应
Scheme 11 Diastereoselective desymmetrization of 4-methyl- ene cyclohexanones by WT CHMOAcineto and mutants

图式 12 以环己醇为原料利用醇脱氢酶和CHMO生产ε-己内酯的多步酶法级联反应
Scheme 12 Multi-step cascade reaction using cyclohexanol as starting material to produce ε-caprolactone by ADH and CHMO

图式 13 BVMOOcean和BVMOParv对α, β不饱和环酮的生物转化
Scheme 13 Biotransformation of α, β-unsaturated cyclic ketones catalyzed by BVMOOcean and BVMOParvi

图式 14 具有α-季碳手性中心的外消旋环酮在CHMOAcineto和SmI2的催化下生成1, 4-环庚(辛)二醇
Scheme 14 A biocatalytic-chemical approach to complex, medium-sized cycloheptane/cyclooctanone-1, 4-diol

图式 15 BVMO催化双环[3,2,0]庚-2-烯-6-酮发生BV氧化反应
Scheme 15 Bayer-Villiger oxidation reaction of bicyclo[3,2,0]- hept-2-en-6-one catalyzed by BVMOs

图式 16 微生物A. parasiticus催化DHEA与可的松的转化
Scheme 16 Transformation of DHEA and Cortisone catalyzed by A. parasiticus growing cells.

图式 17 PtlE和PntE及其突变株催化1-脱氧-11-氧代戊酸的区域选择性氧化
Scheme 17 Regioselective oxidation of 1-deoxy-11-oxopen- talenicacid catalyzed by wild type PtlE, PntE and L185S mutant

图式 18 BVMO催化亚胺培南的区域选择氧化反应
Scheme 18 Regioselective oxidation of imipenem catalyzed by BVMO

图式 19 BVMOs参与细胞松弛素E的生物合成途径
Scheme 19 Involvement of BVMOs in the biosynthetic pathways of cytochalasin E

图式 20 PAMO催化含硼化合物的动力学拆分
Scheme 20 Enzymatic kinetic resolution of racemic boron- compounds catalyzed by PAMO

图式 21 BVMO催化硫醚类化合物的氧化反应
Scheme 21 Heteroatom oxidation reaction of thioethers catalyzed by BVMOs

图式 22 PAMO催化的含硒芳香化合物的动力学拆分反应
Scheme 22 Oxidative kinetic resolution of selenium-containing aromatic compounds catalyzed by BVMOs
表 1 BVMO以及其来源
Table 1. Name of BVMOs and sources
| Entry | BVMO名称 | 来源 | 中文名 |
| 1 | BVMOLepto | Leptospira biflex | |
| 2 | CPMOComa | Comamonas testosterone | 环戊酮单加氧酶 |
| 3 4 | BVMOCm BVMOPp | Cyanidioschyzon merolae Physcomitrella patens | |
| 5 | hFMO5 | Human | |
| 6 | PAMO | Thermobifida fusca | 苯丙酮单加氧酶 |
| 7 | HAPMO | Pseudomonas fluorescens ACB | 4-羟基苯乙酮单加氧酶 |
| 8 | BVMO9 | Rhodococcus jostii RHA1 | |
| 9 | BVMO15 | Rhodococcus jostii RHA1 | |
| 10 | STMO | Rhodococcus rhodochrous | 类固醇单加氧酶 |
| 11 | BVMOAFL456 | Aspergillus flavu NRRL3357 | |
| 12 | BVMOAFL838 | Aspergillus flavu NRRL3357 | |
| 13 | BVMOAFL210 | Aspergillus flavu NRRL3357 | |
| 14 | BVMOAFL619 | Aspergillus flavu NRRL3357 | |
| 15 | BVMO4 | Dietzia sp. D5 | |
| 16 | CHMOJS666 | Polaromonas sp. strain JS666 | 环己酮单加氧酶 |
| 17 | CPDMO | Pseudomonas sp. HI-70 | 环十五酮单加氧酶 |
| 18 | CAMO | Cylindrocarpon radicicola ATCC 11011 | 环烷酮单加氧酶 |
| 19 | OTEMO | Pseudomonas putida NCIMB 10007 | |
| 20 | CHMOArthro | Arthrobacter sp | 环己酮单加氧酶 |
| 21 | CHMOAcineto | Acinetobacter sp | 环己酮单加氧酶 |
| 22 | CHMOPhi1 | Rhodococcus sp. Phi1 | 环己酮单加氧酶 |
| 23 | BVMO-P1-D08 | — | |
| 24 | BVMO-CDX-003 | — | |
| 25 | BVMO-P3-C07 | — | |
| 26 | BVMO-P1-C06 | — | |
| 27 | 2, 5-DKCMO | Pseudomonas putida | 2, 5二酮莰烷单加氧酶 |
| 28 | BVMOOcean | Oceanicola batsensis DSM 15984 | |
| 29 | BVMOParvi | Parvibaculum lavamentivorans DSM 13023 | |
| 30 | 3, 6-DKCMO | Pseudomonas putida | 3, 6二酮莰烷单加氧酶 |
| 31 | CDMO | Rhodococcus ruber SC1 | 环十二酮单加氧酶 |
| 32 | CHMOTm | Thermocrispum municipale DSM 44069 | 环己酮单加氧酶 |
| 33 | MtmOIV | Streptomyces argillaceus | 光神霉素单加氧酶 |
| 34 | PenE | Streptomyces exfoliatus | |
| 35 | PntE | Streptomyces arenae | |
| 36 | PtIE | Streptomyces avermitilis | |
| 37 | BVMOAR | Acinetobacter radioresistens | |
| 38 | FMO CcsB | Aspergillus clavatus NRRL 1 | |
| 39 | BVMOBo | Bradyrhizobium oligotrophicum | |
| 40 | BVMOAm | Aeromicrobium marinum | |
| 41 | SMO | Pseudomonas putida CA-3 | 苯乙烯单加氧酶 |
| 42 | YMOA | Yarrowia lipolytica | |
| 43 | CHMOTm | Thermocrispum municipale DSM 44069 | |
| 44 | BVMOAf1 | A. fumigatus Af293 |
 下载: 导出CSV
下载: 导出CSV
表 2 各种BVMO催化[3.2.0]-庚-2-烯-6-酮的BV氧化反应区域选择性
Table 2. Regioselectivity of Bayer-Villiger oxidation of [3.2.0]-hept-2-en-6-one catalyzed by BVMOs
| Entry | BVMO | 46 | Conv./% | n:aa | Ref. |
| 1 | CHMOAcineto | rac-46 | >99 | 60:40 | [38] |
| 2 | CHMO DS2 (N286C, A293C) | rac-46 | 87 | 60:40 | [38] |
| 3 | CHMO DS3 (T415C, A463C) | rac-46 | >99 | 60:40 | [38] |
| 4 | CHMO DS4 (Y411C, A463C) | rac-46 | 28 | 65:35 | [38] |
| 5 | CHMO T415C | rac-46 | >99 | 62:38 | [38] |
| 6 | CHMOTm | rac-46 | 100 | 49:51 | [54] |
| 7 | BVMOAFL210 | rac-46 | 56 | 87:13 | [16] |
| 8 | BVMOAFL456 | rac-46 | 53 | 94:0 | [16] |
| 9 | BVMOAFL838 | rac-46 | — | 62:13 | [16] |
| 10 | PAMO | rac-46 | — | 73:27 | [19] |
| 11 | PAMO Q152F | rac-46 | — | 30:70 | [19] |
| 12 | PAMO A435Y | rac-46 | — | 29:71 | [19] |
| 13 | PAMO A442G | rac-46 | — | 30:70 | [19] |
| 14 | PAMO Q152F/S441A/A442G | rac-46 | — | 23:77 | [19] |
| 15 | STMO | rac-46 | — | 65:35 | [55] |
| 16 | PASTMO | rac-46 | — | 66:34 | [55] |
| 17 | PACHMO | rac-46 | — | 39:61 | [55] |
| 18 | PAMEMO1 | rac-46 | — | 76:24 | [55] |
| 19 | 2, 5-DKCMO-1-Fred | rac-46 | 100 | 57:43 | [37] |
| 20 | 2, 5-DKCMO-2-Fred | rac-46 | 100 | 52:48 | [37] |
| 21 | 3, 6-DKCMO-Fred | rac-46 | 53 | 40:60 | [37] |
| 22 | 2.5-DKCMO | rac-46 | — | 57:43 | [37] |
| 23 | OTEMO | rac-46 | >90 | 70:30 | [26] |
| 24 | CAMO | rac-46 | >90 | 53:47 | [26] |
| 25 | CPMO | rac-46 | >90 | 97:3 | [26] |
| 26 | BVMOCm | (+)-46 | 100 | 73:27 | [5] |
| BVMOCm | (-)-46 | 100 | 88:12 | [5] | |
| 27 | BVMOPp | (+)-46 | 100 | 76:27 | [5] |
| BVMOPp | (-)-46 | 66 | 62:4 | [5] | |
| 28 | BVMOPp Y160H | (+)-46 | 100 | 72:28 | [5] |
| BVMOPp Y160H | (-)-46 | 100 | 95:5 | [5] | |
| 29 | OTEMO | (-)-46 | — | 51:49 | [52] |
| 30 | OTEMO F255V/F443V | rac-46 | 95 | 91:9 | [52] |
| 31 | OTEMO W501A | (-)-46 | — | 3:97 | [52] |
| 32 | BVMOLepto | rac-46 | 94 | 50:50 | [4] |
| a n: Normal product, a: Abnormal product. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: