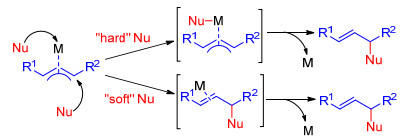
图式 1.
软亲核试剂与硬亲核试剂的两种反应路径
Scheme 1.
Two reaction paths for "soft" nucleophile and "hard" nucleophile

过渡金属催化的烯丙基取代反应是构建碳-碳键、碳-杂原子键的有效方法, 在有机合成中占有重要地位.目前钯[1]、铱[2]、铑[3]、钌[4]和镍[5]等过渡金属催化的烯丙基取代反应已被化学家们研究报道, 其中关于钯和铱催化的烯丙基取代反应研究最为广泛.烯丙基取代的反应历程首先是金属与烯丙基底物的双键配位, 然后与底物中的离去基团发生氧化加成, 生成金属π-烯丙基中间体, 接受亲核试剂进攻, 最后金属与烯烃解离生成产物, 释放催化剂完成催化循环.根据亲核试剂软硬度的不同, 分别经历不同的过程.软亲核试剂(pKa<25, 如稳定的碳负离子或烯醇化物、杂原子等亲核试剂)通常直接进攻π-烯丙基中间体发生取代反应.硬亲核试剂(pKa>25, 如格氏试剂、有机硼、有机锌和有机锡等金属试剂)首先进攻π-烯丙基中间体的金属中心, 然后发生还原消除生成烯丙基取代产物(Scheme 1).
在钯催化的不对称烯丙基取代反应中, 软亲核试剂参与的反应研究广泛且取得了优秀的对映选择性, 而硬亲核试剂参与的反应, 其不对称控制相对困难.镍与钯在元素周期表中位于同一族, 具有相似的催化活性, 然而, 在镍催化的反应中, 硬亲核试剂参与的烯丙基取代反应研究居多.通常除格氏试剂外, 反应性较低的有机金属化合物如硼试剂和锌试剂也可以作为亲核试剂参与反应, 这些试剂具有更好的官能团兼容性, 其稳定性也较高.另一方面, 镍的储量丰富且价格低廉.以摩尔计, 镍成本为钯的0.05%和铂金的0.01%[6].更重要的是, 镍自身具有许多特征, 使其不仅仅作为钯的替代物, 例如, 更小的原子半径, 更强的亲核性, 与亲电试剂更容易发生氧化加成等[7].在过去50年里, 关于镍催化的烯丙基取代反应的报道不断涌现.本文主要根据成键类型及亲核试剂的种类进行划分, 总结近年来镍催化的烯丙基取代反应的进展及其在有机合成中的应用.
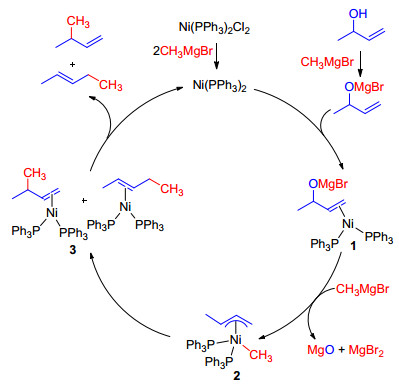
在镍催化烯丙基取代反应中, 格氏试剂是最早使用的亲核试剂.早在20世纪60年代, Felkin等[8]就报道了Ni(PPh3)2Cl2催化的烯丙醇和格氏试剂的取代反应, 该反应在乙醚回流下进行, 通常得到混合产物.例如, α-甲基烯丙醇与甲基溴化镁反应, 以82%的总收率得到比例为71: 29的支链和直链的混合物(Eq. 1).在随后的研究中, 他们认为异构体的产生是因为反应过程中经历了π-烯丙基镍中间体[9], 并提出了可能的反应机理[10]. Ni(PPh3)2Cl2经甲基溴化镁还原后产生零价Ni, 随后与烯烃配位生成中间体1, 1又与甲基溴化镁反应, 经历π-烯丙基镍中间体2, 随后还原消除生成区域异构体3, 最后配位解离分别生成支链和直链产物, 并释放催化剂完成催化循环(Scheme 2).
|
|
(1) |
1980年, Consiglio等[11]发现使用[(-)-(R)-1, 2-双(二苯基膦基)-1-苯基乙烷]镍氯化物可以催化烯丙醇与格氏试剂的不对称烷基化反应, 但只获得很低的对映选择性.他们认为如果使用具有C2对称性的手性双膦配体, 可以减少过渡态中间体非对映体的数目, 进而提高反应的对映选择性.随后, Consiglio小组[12]使用C2对称性的手性配体(S, S)-双(二苯基膦)丁烷(chiraphos), 实现了支链、直链以及环状烯丙基化合物与格氏试剂的不对称取代反应, 为合成具有光学活性的烯烃提供了两种可行的方法:一是使用非手性烯丙基底物与外消旋格氏试剂反应获得手性产物, 如烯丙基苯基醚和1-苯基乙基氯化镁反应, 获得了58.3% ee的取代产物[13](Eq. 2);二是使用外消旋烯丙基化合物与非手性格氏试剂反应获得手性产物, 如1-苯氧基-2-环戊烯和乙基溴化镁反应, 获得了90.4% ee的取代产物[13](Eq. 3).与此相似的, Hiyama小组[14]也使用(S, S)-chiraphos作手性配体, 烯丙基叔丁酯与芳基格氏试剂在0 ℃下反应, 以50%收率和89% ee生成R构型的支链产物, 同时还有收率为25%的直链副产物(Eq. 4).
|
|
(2) |
|
|
(3) |
|
|
(4) |
镍催化不对称烯丙基取代反应的效率和对映选择性高度依赖于所使用的亲核试剂和配体, 实现优秀的对映选择性依旧困难, 但上述初步的研究成果引起了化学家们的关注. 1997年, RajanBabu小组[15]报道了格氏试剂与1, 3-二苯基烯丙基醚的不对称取代反应.在Ni(cod)2/[(S, S)-chiraphos]催化下, 外消旋的3-甲氧基-1, 3-二苯基-1-丙烯与甲基溴化镁反应20 h后, 以74% ee得到甲基取代手性产物(Eq. 5).值得注意的是, 反应中存在一定的动力学拆分效应, 以26%收率和79% ee回收了原料.
|
|
(5) |
为了实现高对映选择性的镍催化不对称烯丙基取代反应, Hoveyda小组[16]报道了不饱和环状缩酮与格氏试剂反应.反应后如经碱处理可以生成手性烯醇醚产物, 如经过酸处理则生成手性酮产物.该反应芳基和烷基格氏试剂都能顺利地发生.特别需要指出的是, 加入额外的PPh3配体对反应的对映选择性有很大影响.对于2-环己烯-1-酮缩二甲醇与正丁基氯化镁的反应, 以Ni(PPh3)2Cl2/[(S, S)-chiraphos]作为催化剂, 需要加入5~20 mol% PPh3.不加入PPh3时仅得到消旋产物, 而加入10 mol% PPh3, 反应可以获得最优85% ee的对映选择性(表 1, Entry 3).
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Entry | x/mol% | Yield/% | ee/% |
| 1 | None | 50 | 0 |
| 2 | 5 | 65 | 82 |
| 3 | 10 | 85 | 85 |
| 4 | 20 | 81 | 76 |
2000年, Uemura小组[17]将平面手性P, N配体应用到烯丙基醚与格氏试剂的取代反应中.当使用二茂铁噁唑啉(L1)作为手性配体时, 3-环己烯醚与芳基格氏试剂反应, 以30%~100%的收率和14%~95% ee得到芳基取代的产物.其中, 3-环己烯苯基醚与2-萘基溴化镁反应可以获得最高95% ee的对映选择性(Eq. 6).
|
|
(6) |
硼酸衍生物方便易得, 对水不敏感, 可以兼容多种官能团, 因此是一类备受青睐的反应试剂. 1995年, Trost和Spagnol小组[18]发展了硼酸衍生物作为亲核试剂, 烯丙基胺作为烯丙基前体的烯丙基取代反应. Ni(cod)2作为镍源, 氢氧化钾作碱, 在甲苯回流条件下, 实现了各种芳基和烯基硼试剂的烯丙基取代反应.当使用1, 1'-联萘-2, 2'-双(二苯基亚膦酸酯)(BINAPO)作配体时, 3-吡咯烷基-1-丁烯与苯硼酸反应, 以69%的总收率得到直链和支链混合物, 反应优先发生在烯丙基取代较多的一端生成支链产物(Eq. 7).
|
|
(7) |
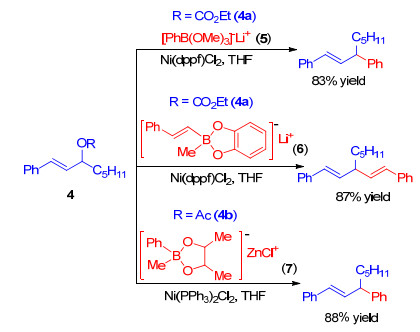
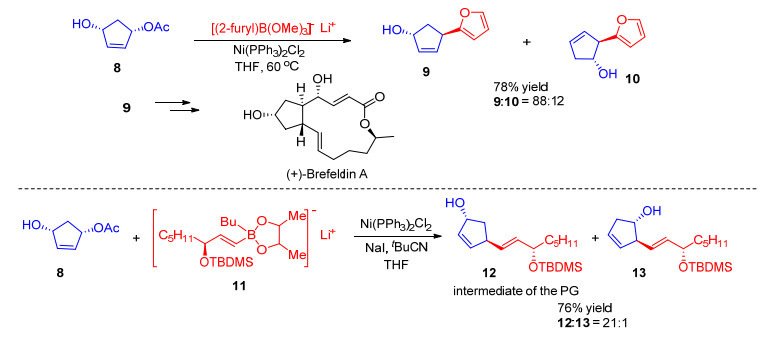
Kobayashi小组[19]也发展了一系列镍催化的有机硼试剂与烯丙醇衍生物的反应.例如, 碳酸酯4a与硼酸盐5在Ni(dppf)Cl2催化下生成苯基取代的产物[19a]; 4a与苯乙烯基硼酸盐6在Ni(dppf)Cl2催化下生成苯乙烯基取代的产物[19a]; 醋酸酯4b与锌硼酸盐7在Ni(PPh3)2Cl2催化下生成苯基取代的产物[19b](Scheme 3).这些反应条件温和, 通常在室温下进行.反应中使用的硼酸盐制备相对简单, 可以由相应的金属试剂分别和乙炔、溴乙炔、醛等反应得到. 1996年, 该小组[19c]运用该策略成功实现了(+)-Brefeldin A的形式全合成.在Ni(PPh3)2Cl2的催化下, 对映纯的环戊烯二醇单乙酸酯8和2-呋喃基硼酸三甲酯锂盐以良好的立体选择性和区域选择性生成呋喃取代产物9和10, 比例为88: 12.以9为原料可以实现(+)-Brefeldin A的形式全合成(Scheme 4). 1998年, 该小组[19d, 19e]将该策略又成功运用到Prostaglandin (PG)中间体的不对称合成中.单乙酸酯8与烯基硼酸盐11反应以21: 1的比例得到1, 3-异构体12与1, 2-异构体13 (Scheme 4).反式1, 3-异构体12是合成多种前列腺素衍生物的通用中间体. 2011年, 该小组[19f]又报道了烯丙醇与烯丙基硼试剂的分子间反应, 可以以优秀的直链选择性和γ选择性得到1, 5-二烯烃.
硼试剂参与的不对称烯丙基取代反应发展较为缓慢.如前所述, Uemura小组[17]率先使用二茂铁噁唑啉P, N配体, 实现了格氏试剂参与的不对称烯丙基取代反应. 2000年, 他们[20]又将该类型配体应用到烯丙基醋酸酯与芳基硼酸的取代反应中(Eq. 8), 以中等到良好的收率得到苯基取代产物, 对映选择性最高达53% ee.邓敏智小组[21]使用末端炔烃与丁基锂和硼酸三甲酯反应制备得到的炔基硼酸盐作为亲核试剂, 在Ni(dppe)Cl2存在下, 炔基硼酸盐与肉桂基碳酸酯发生烯丙基取代反应, 以中等到良好收率(54%~93%)得到1-炔-4-烯产物.若使用(S, S)-chiraphos作为手性配体, 反应能获得13% ee的不对称控制(Eq. 9).
|
|
(8) |
|
|
(9) |
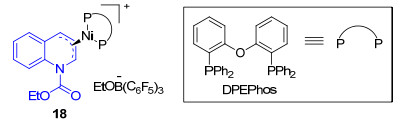
Doyle小组[22]报道了Ni催化的N, O-乙缩醛和芳基硼酸的取代反应.研究表明该反应经历镍-亚铵活化模式[22a].首先在芳基硼酸的作用下, N-乙氧酰基-2-乙氧基-1, 2-二氢喹啉(14)生成亚铵离子15, 15与镍发生氧化加成生成π-烯丙基镍中间体16, 随后接受硼试剂进攻, 最后生成取代产物17 (Scheme 5).在机理研究上, 他们分离得到了π-烯丙基镍中间体18[22b], 并通过单晶X射线衍射进行了表征.通过这种活化模式, 他们获得了一系列2-芳基-1, 2-二氢喹啉产物[22c](Eq. 10).这对研究喹啉类化合物不对称芳基化反应提供了新思路. 2014年, 该小组[22a]使用手性单膦配体L3和[(methallyl)NiCl]2以优秀的对映选择性实现了硼酸与N-乙氧酰基-2-乙氧基-1, 2-二氢喹啉的不对称取代反应, 产物最高取得90% ee对映选择性(Eq. 11).
|
|
(10) |
|
|
(11) |
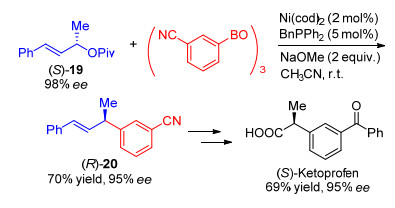
Watson小组[23]使用BnPPh2配体, NaOMe作碱, 实现了1, 3-二取代烯丙基戊酸酯与芳基环硼氧烷手性专一的取代反应(Eq. 12).芳基环硼氧烷在该体系下表现出优秀的官能团兼容性, 能够容忍乙烯基、酯基、醚、氟、氯、三氟甲基和缩醛等官能团.吸电子取代的和给电子取代的芳基环硼氧烷都能够顺利发生反应, 以最高达95%的收率和96% ee的对映选择性获得1, 3-二芳基烯丙基化合物.作者通过(S)-19的芳基取代反应获得95% ee的化合物(R)-20, (R)-20通过后续转化合成抗炎药(S)-Ketoprofen, 证明了该方法的实用性(Scheme 6).
|
|
(12) |
Michaelis小组[24]使用由咪唑盐L4现场生成的氮杂环卡宾作为配体, 应用到镍催化的烯丙基取代反应中. Ni-NHC配合物能够高效催化芳基氧杂硼烷与烯丙基醇反应, 取得57%~98%的收率(Eq. 13).他们通过单晶X射线衍射确认了中间体配合物21的结构, 结果表明双齿NHC配体能够提高催化剂稳定性, 延长催化剂寿命, 从而提高催化反应的效率.
|
|
(13) |
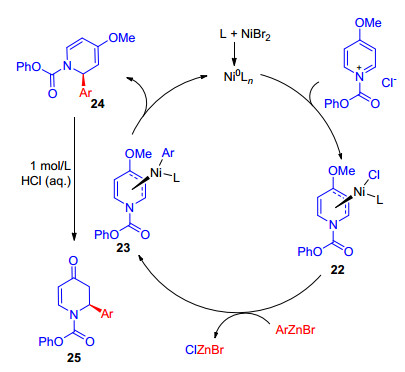
尽管镍催化格氏试剂、有机硼试剂等参与的烯丙基取代反应已经取得重要进展, 但亲核试剂的多样性和官能团兼容性还有待进一步提高.有机锌试剂, 相比有机硼、有机硅和有机锡试剂具有更高的反应活性, 相比格氏试剂具有更好的官能团兼容性. Doyle小组[25]发展了4-甲氧基吡啶盐与芳基锌试剂的不对称取代反应, 获得α-取代哌啶化合物(Eq. 14).这类化合物是生物活性小分子中非常普遍的片段, 也是天然产物和药物合成的重要砌块[26].作者提出了可能的反应机理(Scheme 7), 零价镍与吡啶盐发生氧化加成, 生成二价镍烯丙基中间体22, 22再与芳基溴化锌发生转金属化, 得到中间体23, 经还原消除再生催化剂, 并生成产物烯基醚24. 24在酸性条件下水解生成哌啶酮25.
|
|
(14) |
在之前工作中, 王中夏小组[27]已经证明了P, N, N-Ni配合物可有效催化芳基锌试剂的偶联反应. 2015年, 他们[28]发现P, N, N-Ni配合物还可以高效催化烯丙基甲基醚与芳基氯化锌的取代反应, 使用0.1 mol% C1, 反应可以获得良好到优秀的收率和优秀的区域选择性(Eq. 15).在绝大多数例子中, 反应专一地生成直链反式烯烃产物.反应区域选择性可能是共轭效应和位阻效应共同作用的结果, 芳基锌试剂与π-烯丙基镍中间体反应时共轭效应起主要作用, 增强共轭效应并且尽量减小空间位阻, 因此优先生成直链产物. 2017年, 王中夏小组[29]进一步实现了烯丙醇与芳基或烯基氯化锌的烯丙基取代反应.
|
|
(15) |
1995年, Mortreux小组[30]发现丙二酸二甲酯能够作为亲核试剂参与镍催化烯丙基取代反应.随后, 该课题组[31]发展了丙二酸二甲酯与烯丙基醋酸酯的取代反应. Ni(cod)2/1, 4-双(二苯基膦)丁烷(dppb)或Ni(cod)2/N, N'-双(二苯基膦)-N, N'-二甲基乙二胺(dppmae)能催化反应的进行, 获得直链和支链混合产物(Eq. 16).以环己烯醋酸酯与丙二酸二甲酯为模板反应, 作者对一系列手性配体进行了考察.当使用(S)-1-二苯基膦-5-二苯基膦氧甲基-2-吡咯烷酮[(S)-Ph, Ph-oxoProNOP]作配体时, THF作溶剂, 50 ℃下反应14 h, 取得几乎定量的收率, 并且实现了40% ee的对映选择性(Eq. 17).这是首例软亲核试剂参与的镍催化不对称烯丙基烷基化反应.
|
|
(16) |
|
|
(17) |
Walsh小组[32]首次采用二芳基甲烷作为亲核试剂, 与Boc保护的烯丙醇反应, 成功实现了二芳基甲烷的烯丙基取代反应, 亲核试剂芳基或杂芳基取代甲烷都适用于该反应体系.如β, β'-二吡啶甲烷与cis为主的外消旋5-苯基-2-环己烯基叔丁氧羰基酯反应, 发生取代的碳原子构型保持, 得到cis为主的外消旋产物(Eq. 18).这说明反应是软亲核试剂的进攻路径, 即亲核试剂进攻π-烯丙基中间体而不是进攻金属中心.他们对178种手性膦配体进行了考察, 最终筛选出最优配体L6, 反应可以取得高达92% ee的对映选择性(Eq. 19).对于镍催化的烯丙基取代反应来说, 实现这样优秀的不对称控制是十分难得的.
|
|
(18) |
|
|
(19) |
烯丙基取代反应常用的烯丙基前体一般带有良好的离去基团, 如醋酸酯、碳酸酯和卤代物等.相比这些活化的底物, 如果直接以烯丙醇为烯丙基前体, 则生成的副产物为水.但是, 烯丙醇的反应活性较低, 通常需使用外部活化试剂如砷氧化物[33]、硼烷衍生物[34]和钛添加剂[35]活化烯丙醇.
Mashima小组[36]在2016年报道了Ni(cod)2/(S)-(-)-2, 2'-双(二苯基膦)-5, 5', 6, 6', 7, 7', 8, 8'-八氢-1, 1'-联萘[(S)-H8-BINAP]催化的烯丙醇与β-酮酸酯的不对称烯丙基烷基化反应.该反应不需要添加剂来活化烯丙醇, 反应能够以优秀的对映选择性生成季碳手性中心, 而且以直链的产物为主(Eq. 20).他们提出了可能的反应机理, 零价镍双膦配合物与烯丙醇的烯烃配位生成配合物26, 接着氧化加成生成羟基配位的烯丙基二价镍络合物27, 羟基随后攫取β-酮酸酯的α-H生成π-烯丙基镍中间体28, 28经还原消除生成29, 最后, 烯丙醇与29发生配体交换生成取代产物30并再生26, 完成催化循环(Scheme 8).
|
|
(20) |

同年, Sauthier和Bonin小组[37]报道了Ni(cod)2/dppb催化的烯丙醇与软亲核试剂的双烯丙基化反应.一些软亲核试剂如β-酮酯、β-二酮和丙二酸酯都可应用于该反应体系(Eq. 21).此外, Sauthier小组[38]还报道了镍催化醛的α-烯丙基化反应.在以往钯催化醛的α-烯丙基化反应中, 大多数例子都需要使用如硼烷衍生物[39]、布朗斯特酸[40]和胺(形成烯胺)[41]等活化试剂.该反应不需要活化试剂, 使用1 mol% Ni(cod)2, 2 mol% dppf作催化剂, 在80 ℃的甲醇溶液中, α-取代的醛或α, β-不饱和醛与烯丙醇的反应可以顺利进行, 水是唯一副产物(Eq. 22).
|
|
(21) |
|
|
(22) |
最近, 杨东旭和王锐[42]发展了以手性二胺作配体的不对称烯丙基烷基化反应.咪唑酮与硝基取代的烯丙基醋酸酯反应, 得到一系列手性α-烯丙基取代的酮类化合物, 给出中等到良好的收率及优秀的对映选择性(Eq. 23). 2018年, Stoltz小组[43]首次报道了镍催化的α-酰基内酯或内酰胺的不对称烯丙基取代反应. α-酰基内酯和内酰胺可以用作构建季碳手性中心的亲核试剂, α-酰基取代基也为产物的进一步转化提供了可能.此外, 内酯的开环反应还可以转化非环状含季碳手性中心化合物.内酰胺的消除反应可以实现哌啶环的直接官能团化, 而哌啶环又是含氮药物分子中最常见的杂环.因此发展内酯和内酰胺的烯丙基取代反应极具合成价值. Stoltz小组利用商业可得的手性双膦配体(R)-2, 2', 6, 6'-四甲氧基-4, 4'-双(二苯基膦)-3, 3'-联吡啶[(R)-P-phos]实现了烯丙醇与α-酰基内酯和内酰胺的不对称烯丙基取代反应, 以最高达90% ee的对映选择性生成季碳手性中心产物(Eq. 24), 该类产物可转化为多官能团化的手性砌块.
|
|
(23) |
|
|
(24) |
1995年, Ikeda小组[44]研究了烯丙基卤化物与炔基锡试剂的烯丙基取代反应.在THF回流下, Ni(acac)2/PPh3催化得到1, 4-烯炔产物, 其他膦配体, 如P(OEt)3、P(OPh)3也能催化该反应(Eq. 25).
|
|
(25) |
在镍催化的反应中, 烯烃用作亲核试剂早有报道[45], 但使用简单烯烃如乙烯、丙烯的烯丙基取代反应在最近几年才被报道. 2010年, Jamison小组[46]报道了首例简单烯烃参与的烯丙基取代反应.在三乙基硅基三氟甲磺酸酯(TESOTf)和三乙胺存在下, 由Ni(cod)2/三(邻甲氧基苯基)膦[P(o-anisyl)3]催化肉桂醇衍生物与简单烯烃反应, 可得到以1, 4-二烯为主的产物(Eq. 26).这种方法适用范围广, 乙烯与各种肉桂醇衍生物反应良好, 包括带有易离去基团的肉桂醇衍生物, 例如醋酸酯、烯丙基氯和碳酸酯; 带有不易离去基团的肉桂醇衍生物, 例如烷基醚、三甲基硅醚, 甚至肉桂醇也能顺利反应.这是使用简单烯烃参与的烯丙基取代反应的重要进展.
|
|
(26) |
2011年, Jamison小组[47]发展了烯烃和苄氯的分子间苄基化反应.以往的烯烃组分大多是带有官能团的取代烯烃, 如丙烯酸酯、苯乙烯和N-乙烯基酰胺, 而脂肪族烯烃用于苄基化尚未实现.而且, 通常在100~130 ℃下进行, 产物容易发生烯烃异构化. 1, 1-二取代烯烃在许多生物活性分子中普遍存在, 还是非常有用的中间体[48], 但与1, 2-二取代烯烃合成相比, 使用简单烯烃直接构建1, 1-二取代烯烃的方法要少的多[49]. Jamison小组发展了镍催化简单烯烃与苄氯的反应, 各种取代的脂肪族烯烃在该体系下都能够顺利发生反应, 反应在室温下进行, 生成1, 1-二取代的烯烃衍生物(Eq. 27).他们分离得到了Ni(η3-CH2C6H5)(PCy2Ph)Cl中间体33, 其结构通过单晶X射线衍射得以验证, 并对其反应的机理进行了假设(Scheme 9).络合物32与苄氯氧化加成生成η3-CH2C6H5镍中间体33, 在三乙基硅基三氟甲磺酸酯作用下, 氯被置换为三氟甲磺酸根, 生成中间体34, 烯烃插入生成中间体35, 35发生β-H消除, 得到1, 1-二取代的产物36和络合物37, 37在三乙胺存在下再生32.
|
|
(27) |
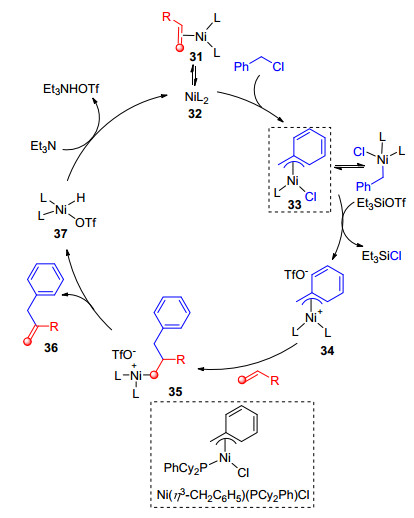
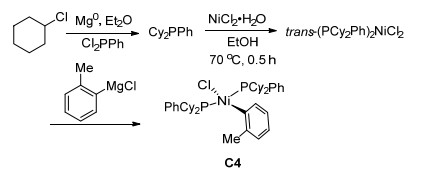
在前面的工作中, 多使用Ni(cod)2作为零价镍来源, 虽然已经有很好的实验室合成方法, 但它需要Schlenk管或手套箱操作, 需要在惰性气氛下储存和使用. Jamison小组[50]希望通过设计空气稳定的催化剂, 摆脱对惰性气氛技术的依赖, 使得反应在操作上更加方便简单.于是他们合成了新型催化剂C4, 以环己基氯为起始原料, 三步反应即可得到(Scheme 10).这种催化剂不仅合成简单, 催化活性高, 并且能够在空气中稳定存在.他们将这种催化剂用于烯烃和苄基氯化物的取代反应, 不仅不需要惰性气氛操作, 而且以高收率和多数大于95: 5的区域选择性生成烯丙基苯衍生物(Eq. 28).
|
|
(28) |
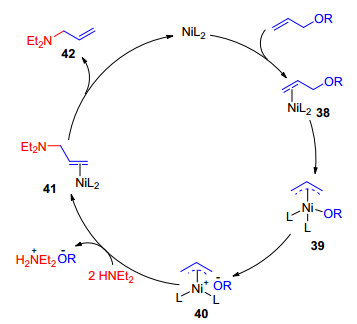
早在1973年, Furukawa等[51]研究了镍催化烯丙醇或烯丙基醚与吗啉的反应, 反应可以顺利生成相应的烯丙基胺(Eq. 29).后来, Moberg小组[52]和Yamamoto小组[53]分别对该反应的机理进行了研究, 认为镍与烯丙醇氧化加成生成π-烯丙基中间体, 然后接受亲核试剂进攻. 1998年, Mortreux小组[54]设计了二乙胺与烯丙基化合物的取代反应(Eq. 30), 考察了底物、溶剂和盐等反应参数对反应选择性及动力学的影响.他们提出了可能的反应机理.催化剂与烯丙基醚配位形成中间体38, 并发生氧化加成形成中间体39, 离去基团作为中间体40的抗衡阴离子, 接下来二级胺进攻40, 配体解离生成相应的产物42, 同时再生催化物种, 完成催化循环(Scheme 11).
|
|
(29) |
|
|
(30) |
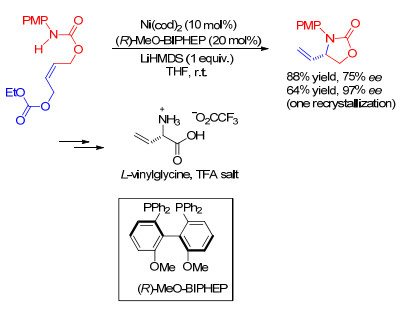
镍催化的不对称烯丙基胺化反应近年来才有报道. 2004年, Berkowitz小组[55]在分子内烯丙基胺化反应中应用原位酶筛选(ISES)来评估催化剂, 相对的ISES速率能反映产物形成的相对速率.在指定条件下, PMP为N-保护基, Ni(cod)2为催化剂前体, 测试了一系列中心手性、轴手性和平面手性的双齿配体.当使用轴手性配体(R)-2, 2'-双(二苯基膦)-6, 6'-二甲氧-1, 1'-联苯[(R)-MeO-BIPHEP]时, 可以取得88%收率和75% ee对映选择性(Scheme 12).这是首例零价镍催化的不对称烯丙基胺化反应.围绕这种新颖的不对称胺化反应, 作者合成了L-乙烯基甘氨酸衍生物.
2015年, Carpentier, Ogoshi和Mashima等[56]发展了Ni(cod)2/dppf催化的烯丙醇直接胺化反应. Ni(cod)2用量可以降低至0.5 mol%, 配体dppf仅使用1 mol%.通过添加nBu4NOAc和3 Å分子筛, 以高产率获得相应的烯丙基胺.值得注意的是, nBu4NOAc作用显著.在相同条件下, 不使用nBu4NOAc, 产物收率为33%, 加入2.5 mol% nBu4NOAc产物收率可提高至82%(表 2).为了阐明nBu4NOAc所起的作用, 作者分离和表征了一些重要中间体, 与先前在钯和铂催化中广泛认同的阳离子四配位络合物不同, 在该反应体系下醋酸根离子与η3-烯丙基镍配位, 形成电中性的五配位的络合物(η3-allyl)Ni-(dppf)OAc 43, 有助于亲核进攻[57].
下载:
导出CSV
 |
||
| Entry | x/mol% | Yield/% |
| 1 | None | 33 |
| 2 | 2.5 | 82 |
 |
||
2015年, Sauthier小组[58]报道了二[1, 2-双(二苯基膦甲基苯]镍[Ni(dppmb)2]催化酰胺的N-烯丙基化反应.反应在中性条件下进行, 不需要添加剂来活化烯丙醇, 但由于使用过量的烯丙醇生成双烯丙基取代的副产物(Eq. 31).同时, 烯丙醇会发生自身醚化的竞争反应, 形成二烯丙基醚.
|
|
(31) |
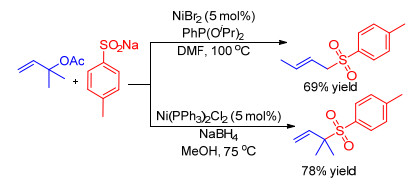
使用硫亲核试剂的镍催化烯丙基取代反应最早是在1983年由Julia和Cuvigny[59]报道.对甲苯亚磺酸钠与烯丙基醋酸酯反应, 分别以NiBr2/PhP(OiPr)2和Ni(PPh3)2Cl2作为催化剂, 可以选择性地生成直链或支链烯丙基砜产物(Scheme 13).
2007年, Takeda小组[60]利用空气稳定的Ni[P-(OEt)3]4催化剂, 实现了烯丙基醋酸酯与硫醇的烯丙基硫醚化反应, 得到构型保持的烯丙基硫化物.如使用Z式烯丙基醋酸酯则得到Z式取代的产物(Eq. 32), 反之使用E式烯丙基醋酸酯则得到E式取代的产物(Eq. 33).烯丙基醋酸酯与醇或酚同样能够顺利发生反应, 也得到区域专一性和立体专一性的产物(Eq. 34).
|
|
(32) |
|
|
(33) |
|
|
(34) |
在过去50年里, 镍催化的烯丙基取代反应研究取得了不错的进展.有机化学家们使用格氏试剂作亲核试剂的的早期研究, 奠定了镍催化烯丙基取代反应的研究基础.随后陆续实现了有机硼、有机锌试剂等硬亲核试剂, β-二酮酯、二芳基甲烷等软亲核试剂以及二级胺等杂原子亲核试剂参与的烯丙基取代反应.一些高选择性(区域、立体和对映选择性)烯丙基取代反应也得以实现.这些研究成果为构建碳-碳键、碳-杂原子键提供了强有力的方法, 同时这些方法在天然产物及药物活性分子中间体的合成中发挥了举足轻重的作用.
尽管如此, 镍催化的烯丙基取代反应仍存在一些亟待解决的问题, 发展更加高效高选择性的催化反应依然是研究重点, 主要包括以下几个方面: (1)进一步阐明反应的机理以及探索反应选择性控制的规律, 为设计新反应和新配体提供理论依据; (2)发展高对映选择性的催化新反应; (3)发展高效的新催化体系(包括新配体和镍源)以提高催化效率; (4)进一步拓展镍催化烯丙基取代反应在有机合成中的应用.
不难看出, 镍催化的烯丙基取代反应已表现出独特的发展潜力, 是一个有待开发的广阔领域, 也已经取得了一些研究成果.我们预计未来会有大量关于此类反应的报道, 这些报道将进一步证明这一方法的重要性, 并推动该领域的发展.
For selected reviews, see: (a) Helmchen, G. J. Organomet. Chem. 1999, 576, 203.
(b) Tenaglia, A.; Heumann, A. Angew. Chem., Int. Ed. 1999, 38, 2180.
(c) Trost, B. M. Chem. Pharm. Bull. 2002, 50, 1.
(d) Kazmaier, U. Curr. Org. Chem. 2003, 7, 317.
(e) Trost, B. M. J. Org. Chem. 2004, 69, 5813.
(f) Jensen, T.; Fristrup, P. Chem.-Eur. J. 2009, 15, 9632.
(g) Milhau, L.; Guiry, P. J. Top. Organomet. Chem. 2012, 38, 95.
For selected reviews, see: (a) Takeuchi, R.; Kezuka, S. Synthesis 2006, 3349.
(b) Helmchen, G.; Dahnz, A.; Döbon, P.; Schelwies, M.; Weihofen, R. Chem. Commun. 2007, 675.
(c) Hartwig, J. F.; Stanley, L. M. Acc. Chem. Res. 2010, 43, 1461.
(d) Hartwig, J. F.; Pouy, M. J. Top. Organomet. Chem. 2011, 34, 169.
(e) Liu, W.-B.; Xia, J.-B.; You, S.-L. Top. Organomet. Chem. 2012, 38, 155.
(f) Tosatti, P.; Nelson, A.; Marsden, S. P. Org. Biomol. Chem. 2012, 10, 3147.
(g) Helmchen, G. In Molecular Catalysis, Eds.: Gade, L. H.; Hofmann, P., Wiley-VCH, Weinheim, 2014, pp. 235~254.
(h) Zhuo, C.-X.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2014, 47, 2558.
(i) Hethcox, J. C.; Shockley, S. E.; Stoltz, B. M. ACS Catal. 2016, 6, 6207. (j) Qu, J.; Helmchen, G. Acc. Chem. Res. 2017, 50, 2539.
For selected reviews, see: (a) Evans, P. A.; Nelson, J. D. J. Am. Chem. Soc. 1998, 120, 5581.
(b) Selvakumar, K.; Valentini, M.; Pregosin, P. S.; Albinati, A. Organometallics 1999, 18, 4591.
(c) Hayashi, T.; Okada, A.; Suzuka, T.; Kawatsura, M. Org. Lett. 2003, 5, 1713.
(d) Ashfeld, B. L.; Miller, K. A.; Martin, S. F. Org. Lett. 2004, 6, 1321.
For selected reviews, see: (a) Minami, I.; Shimizu, I.; Tsuji, J. J. Organomet. Chem. 1985, 296, 269.
(b) Zhang, S.-W.; Mitsudo, T.; Kondo, T.; Watanabe, Y. J. Organomet. Chem. 1993, 450, 197.
(c) Matsushima, Y.; Onitsuka, K.; Kondo, T.; Mitsudo, T.; Takahashi, S. J. Am. Chem. Soc. 2001, 123, 10405.
(d) Trost, B. M.; Fraisse, P. L.; Ball, Z. T. Angew. Chem., Int. Ed. 2002, 41, 1059.
(e) Mbaye, M. D.; Demerseman, B.; Renaud, J.-L.; Toupet, L.; Bruneau, C. Angew. Chem., Int. Ed. 2003, 42, 5066.
(f) Hermatschweiler, R.; Fernandez, I.; Breher, F.; Pregosin, P. S.; Veiros, L. F.; Calhorda, M. J. Angew. Chem., Int. Ed. 2005, 44, 4397.
(g) Kawatsura, M.; Ata, F.; Wada, S.; Hayase, S.; Uno, H.; Itoh, T. Chem. Commun. 2007, 298.
For selected reviews, see: (a) Consiglio, G.; Waymouth, R. M. Chem. Rev. 1989, 89, 257.
(b) Ho, C.-Y.; Schleicher, K. D.; Chan, C.-W.; Jamison, T. F. Synlett 2009, 2565.
(c) Pigge, F. C. Synthesis 2010, 1745.
Henrion, M.; Ritleng, V.; Chetcuti, M. J. ACS Catal. 2015, 5, 1283. doi: 10.1021/cs5014927
For selected examples, see:
(a) Schaub, T.; Backes, M.; Radius, U. J. Am. Chem. Soc. 2006, 128, 15964.
(b) Rosen, B. M.; Quasdorf, K. W.; Wilson, D. A.; Zhang, N.; Resmerita, A.-M.; Garg, N. K.; Percec, V. Chem. Rev. 2011, 111, 1346.
(c) Li, B.-J.; Yu, D.-G.; Sun, C.-L.; Shi, Z.-J. Chem.-Eur. J. 2011, 17, 1728.
(d) Tobisu, M.; Xu, T.; Shimasaki, T.; Chatani, N. J. Am. Chem. Soc. 2011, 133, 19505.
Chuit, C.; Felkin, H.; Frajerman, C.; Roussi, G.; Swierczewski, G. Chem. Commun. 1968, 1604.
Felkin, H.; Swierczewski, G. Tetrahedron Lett. 1972, 15, 1433.
Felkin, H.; Joly-Goudket, M.; Davies, S. G. Tetrahedron Lett. 1981, 22, 1157. doi: 10.1016/S0040-4039(01)90263-1
Consiglio, G.; Morandini, F.; Piccolol, O. Helv. Chim. Acta 1980, 63, 987. doi: 10.1002/(ISSN)1522-2675
Consiglio, G.; Morandini, F.; Piccolo, O. J. Chem. Soc., Chem. Commun. 1983, 112.
Consiglio, G.; Piccolo, O.; Roncetti, L.; Morandini, F. Tetrahedron 1986, 42, 2043. doi: 10.1016/S0040-4020(01)87621-3
Hiyama, T.; Wakasa, N. Tetrahedron Lett. 1985, 26, 3259. doi: 10.1016/S0040-4039(00)98166-8
Nomura, N.; RajanBabu, T. V. Tetrahedron Lett. 1997, 38, 1713. doi: 10.1016/S0040-4039(97)00178-0
Gomez-Bengoa, E.; Heron, N. M.; Didiuk, M. T.; Luchaco, C. A.; Hoveyda, A. H. J. Am. Chem. Soc. 1998, 120, 7649. doi: 10.1021/ja980499l
Chung, K.-G.; Miyake, Y.; Uemura, S. J. Chem. Soc., Perkin Trans. 1 2000, 2725.
Trost, B. M.; Spagnol, M. D. J. Chem. Soc., Perkin Trans. 1 1995, 2083.
(a) Kobayashi, Y.; Ikeda, E. J. Chem. Soc., Chem. Commun. 1994, 1789.
(b) Kobayashi, Y.; Tokoro, Y.; Watatani, K. Eur. J. Org. Chem. 2000, 3825.
(c) Kobayashi, Y.; Watatani, K.; Kikori, Y.; Mizojiri, R. Tetrahedron Lett. 1996, 37, 6125.
(d) Kobayashi, Y.; Takahisa, E.; Usmani, S. B. Tetrahedron Lett. 1998, 39, 597.
(e) Kobayashi, Y.; Takahisa, E.; Usmani, S. B. Tetrahedron Lett. 1998, 39, 601.
(f) Jimenez-Aquino, A.; Flegeau, E. F.; Schneider, U.; Kobayashi, S. Chem. Commun. 2011, 47, 9456.
Chung, K.-G.; Miyake, Y.; Uemura, S. J. Chem. Soc., Perkin Trans. 1 2000, 15.
Chen, H.; Deng, M.-Z. J. Organomet. Chem. 2000, 603, 189. doi: 10.1016/S0022-328X(00)00164-9
(a) Shields, J. D.; Ahneman, D. T.; Graham, T. J. A.; Doyle, A. G. Org. Lett. 2014, 16, 142.
(b) Sylvester, K. T.; Wu, K.; Doyle, A. G. J. Am. Chem. Soc. 2012, 134, 16967.
(c) Graham, T. J. A.; Shields, J. D.; Doyle, A. G. Chem. Sci. 2011, 2, 980.
Srinivas, H. D.; Zhou, Q.; Watson, M. P. Org. Lett. 2014, 16, 3596. doi: 10.1021/ol5016724
Nazari, S. H.; Bourdeau, J. E.; Talley, M. R.; Valdivia-Berroeta, G. A.; Smith, S. J.; Michaelis, D. J. ACS Catal. 2018, 8, 86. doi: 10.1021/acscatal.7b03079
Chau, S. T.; Lutz, J. P.; Wu, K.; Doyle, A. G. Angew. Chem., Int. Ed. 2013, 52, 9153. doi: 10.1002/anie.201303994
For some recent examples synthesis of biologically active 4-piperidines, see: (a) Tawara, J. N.; Lorenz, P. F.; Stermitz, R. J. Nat. Prod. 1999, 62, 321.
(b) Watson, P. S.; Jiang, B.; Scott, B. Org. Lett. 2000, 2, 3679.
(c) Brooks, C. A.; Comins, D. L. Tetrahedron Lett. 2000, 41, 3551.
(a) Wu, D.; Wang, Z.-X. Org. Biomol. Chem. 2014, 12, 6414.
(b) Wu, D.; Tao, J.-L.; Wang, Z.-X. Org. Chem. Front. 2015, 2, 265.
Tao, J.-L.; Yang, B.; Wang, Z.-X. J. Org. Chem. 2015, 80, 12627. doi: 10.1021/acs.joc.5b02151
Yang, B.; Wang, Z.-X. J. Org. Chem. 2017, 82, 4542. doi: 10.1021/acs.joc.6b02564
Bricout, H.; Carpentier, J.-F.; Mortreux, A. J. Chem. Soc., Chem. Commun. 1995, 1863.
Bricout, H.; Carpentier, J.-F.; Mortreux, A. Tetrahedron Lett. 1996, 37, 6105. doi: 10.1016/0040-4039(96)01302-0
Sha, S.-C.; Jiang, H.; Mao, J.; Bellomo, A.; Jeong, S. A.; Walsh, P. J. Angew. Chem., Int. Ed. 2016, 55, 1070. doi: 10.1002/anie.201507494
For selected examples, see:
(a) Lu, X.; Lu, L. J. Organomet. Chem. 1986, 307, 285.
(b) Lu, X.; Lu, L.; Sun, J. J. Mol. Catal. 1987, 41, 245.
(c) Lu, X.; Jiang, X.; Tao, X. J. Organomet. Chem. 1988, 344, 109.
For selected examples, see:
(a) Starý, I.; Stará, I. G.; Kočovský, P. Tetrahedron Lett. 1993, 34, 179.
(b) Starý, I.; Stará, I. G.; Kočovský, P. Tetrahedron 1994, 50, 529.
(c) Tamaru, Y.; Horino, Y.; Araki, M.; Tanaka, S.; Kimura, M. Tetrahedron Lett. 2000, 41, 5705.
(d) Takacs, J. M.; Jiang, X.-T.; Leonov, A. P. Tetrahedron Lett. 2003, 44, 7075.
(e) Kimura, M.; Mukai, R.; Tanigawa, N.; Tanaka, S.; Tamaru, Y. Tetrahedron 2003, 59, 7767.
Itoh, K.; Hamaguchi, N.; Miura, M.; Nomura, M. J. Chem. Soc., Perkin Trans. 1 1992, 1, 2833.
Kita, Y.; Kavthe, R. D.; Oda, H.; Mashima, K. Angew. Chem., Int. Ed. 2016, 55, 1098. doi: 10.1002/anie.201508757
Blieck, R.; Azizi, M. S.; Mifleur, A.; Roger, M.; Persyn, C.; Sauthier, M.; Bonin, H. Eur. J. Org. Chem. 2016, 1194.
Bernhard, Y.; Thomson, B.; Ferey, V.; Sauthier, M. Angew. Chem., Int. Ed. 2017, 56, 7460. doi: 10.1002/anie.201703486
For selected examples, see:
(a) Kimura, M.; Horino, Y.; Mukai, R.; Tanaka, S.; Tamaru, Y. J. Am. Chem. Soc. 2001, 123, 10401.
(b) Kimura, M.; Shimizu, M.; Shibata, K.; Tazoe, M.; Tamaru, Y. Angew. Chem., Int. Ed. 2003, 42, 3392.
Jiang, G.; List, B. Adv. Synth. Catal. 2011, 353, 1667. doi: 10.1002/adsc.201100260
For selected examples, see:
(a) Ibrahem, I.; Clrdova, A. Angew. Chem., Int. Ed. 2006, 45, 1952.
(b) Mukherjee, S.; List, B. J. Am. Chem. Soc. 2007, 129, 11336.
(c) Jiang, G.; List, B. Angew. Chem., Int. Ed. 2011, 50, 9471.
Wang, J.; Wang, P.; Wang, L.; Li, D.; Wang, K.; Wang, Y.; Zhu, H.; Yang, D.; Wang, R. Org. Lett. 2017, 19, 4826. doi: 10.1021/acs.orglett.7b02264
Ngamnithiporn, A.; Jette, C. I.; Bachman, S.; Virgil, S. C.; Stoltz, B. M. Chem. Sci. 2018, 9, 2547. doi: 10.1039/C7SC05216B
Cui, D.-M.; Hashimoto, N.; Ikeda, S.-I.; Sato, Y. J. Org. Chem. 1995, 60, 5752. doi: 10.1021/jo00123a006
For selected examples, see:(a) Ng, S.-S.; Ho, C.-Y.; Schleicher, K. D.; Jamison, T. F. Pure Appl. Chem. 2008, 80, 929.
(b) Ogoshi, S.; Haba, T.; Ohashi, M. J. Am. Chem. Soc. 2009, 131, 10350.
Matsubara, R.; Jamison, T. F. J. Am. Chem. Soc. 2010, 132, 6880. doi: 10.1021/ja101186p
Matsubara, R.; Gutierrez, A. C.; Jamison, T. F. J. Am. Chem. Soc. 2011, 133, 19020. doi: 10.1021/ja209235d
For selected examples, see: (a) Miller, J. A.; Negishi, E.-I. Tetrahedron Lett. 1984, 25, 5863.
(b) Sabarre, A.; Love, J. Org. Lett. 2008, 10, 3941.
For selected examples, see:
(a) Milstein, D.; Stille, J. K. J. Am. Chem. Soc. 1979, 101, 4992.
(b) Kamlage, S.; Sefkow, M.; Peter, M. G. J. Org. Chem. 1999, 64, 2938.
(c) Zhang, S.; Marshall, D.; Liebeskind, L. S. J. Org. Chem. 1999, 64, 2796.
(d) Perez, I.; Sestelo, J. P.; Sarandeses, L. A. J. Am. Chem. Soc. 2001, 123, 4155.
Standley, E. A.; Jamison, T. F. J. Am. Chem. Soc. 2013, 135, 1585. doi: 10.1021/ja3116718
Furukawa, J.; Kui, J.; Yamamoto, K.; Tojo, T. Tetrahedron 1973, 29, 3149. doi: 10.1016/S0040-4020(01)93457-X
Moberg, C. Tetrahedron Lett. 1980, 21, 4539. doi: 10.1016/S0040-4039(00)74545-X
Yamamoto, T.; Ishizu, J.; Yamamoto, A. J. Am. Chem. Soc. 1981, 103, 6863. doi: 10.1021/ja00413a014
Bricout, H.; Carpentier, J.-F.; Mortreux, A. Tetrahedron 1998, 54, 1073. doi: 10.1016/S0040-4020(97)10208-3
Berkowitz, D. B.; Maiti, G. Org. Lett. 2004, 6, 2661. doi: 10.1021/ol049159x
Kita, Y.; Sakaguchi, H.; Hoshimoto, Y.; Nakauchi, D.; Nakahara, Y.; Carpentier, J.-F.; Ogoshi, S.; Mashima, K. Chem.-Eur. J. 2015, 21, 14571. doi: 10.1002/chem.201502329
For selected examples, see:
(a) Brunkan, N. M.; Jones, W. D. J. Organomet. Chem. 2003, 683, 77.
(b) Brunkan, N. M.; Brestensky, D. M.; Jones, W. D. J. Am. Chem. Soc. 2004, 126, 3627.
(c) Chaumonnot, A.; Lamy, F.; Sabo-Etienne, S.; Donnadieu, B.; Chaudret, B.; Barthelat, J.-C.; Galland, J.-C. Organometallics 2004, 23, 3363.
(d) Acosta-Ramírez, A.; Muñoz-Hernández, M.; Jones, W. D.; Garcia, J. J. J. Organomet. Chem. 2006, 691, 3895.
Azizi, M. S.; Edder, Y.; Karim, A.; Sauthier, M. Eur. J. Org. Chem. 2016, 3796.
Cuvigny, T.; Julia, M. J. Organomet. Chem. 1983, 250, C21. doi: 10.1016/0022-328X(83)85087-6
Yatsumonji, Y.; Ishida, Y.; Tsubouchi, A.; Takeda, T. Org. Lett. 2007, 9, 4603. doi: 10.1021/ol702122d

图式 1 软亲核试剂与硬亲核试剂的两种反应路径
Scheme 1 Two reaction paths for "soft" nucleophile and "hard" nucleophile

图式 2 烯丙醇与格氏试剂可能的反应机理
Scheme 2 Proposed mechanism of the reaction of allylic alcohols with Grignard reagent

图式 3 有机硼试剂与烯丙醇衍生物的反应
Scheme 3 Reactions of organoboron reagents with allylic alcohol derivatives

图式 7 镍催化吡啶盐的对映选择性芳基化可能的催化循环
Scheme 7 Proposed catalytic cycle of nickel-catalyzed enantioselective arylation of pyridinium ion

图式 8 镍催化β-酮酸酯与烯丙醇可能的反应机理
Scheme 8 Plausible reaction mechanism of nickel-catalyzed reaction of β-ketoesters with allylic alcohols

图式 9 镍催化苄基氯化物和简单烯烃可能的反应机理
Scheme 9 Possible mechanism of nickel-catalyzed reactions of benzyl chlorides with simple olefins

图式 10 合成催化剂trans-(PCy2Ph)2Ni(o-tolyl)Cl
Scheme 10 Synthesis of trans-(PCy2Ph)2Ni(o-tolyl)Cl

图式 11 镍催化二乙胺与烯丙基衍生物可能的反应机理
Scheme 11 Plausible reaction mechanism of nickel-catalyzed reaction of allylic derivatives with diethylamines

图式 12 镍催化分子内不对称烯丙基胺化反应
Scheme 12 Nickel-catalyzed intramolecular asymmetric allylic amination reaction

图式 13 镍催化烯丙基醋酸酯与对甲苯亚磺酸钠的取代反应
Scheme 13 Nickel-catalyzed substitution of allylic acetates with p-tolSO2Na
表 1 PPh3对于反应对映选择性的影响
Table 1. Influence of PPh3 on enantioselectivity
|
|||
| Entry | x/mol% | Yield/% | ee/% |
| 1 | None | 50 | 0 |
| 2 | 5 | 65 | 82 |
| 3 | 10 | 85 | 85 |
| 4 | 20 | 81 | 76 |
 下载: 导出CSV
下载: 导出CSV
表 2 四丁基醋酸铵对反应收率的影响
Table 2. Effect of nBu4NOAc on reaction yield
|
||
| Entry | x/mol% | Yield/% |
| 1 | None | 33 |
| 2 | 2.5 | 82 |
|
||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们





































 下载:
下载: