咪唑并[1, 2-a ]吡啶[1 是由五元环咪唑和六元环吡啶稠合的氮杂双环化合物, 因此同时具有它们各自的特性, 并被广泛的应用于药物结构中, 由此产生了许多具有各种生物活性的咪唑并[1, 2-a ]吡啶衍生物.这些活性包括抗肿瘤、抗病毒、镇定催眠、抗炎、抗溃疡等.其中咪唑并[1, 2-a ]吡啶类化合物已成为上市药物, 如抗溃疡药Soraprazan[2 和Zolimidine[3 、血管扩张药Olp-rinone[4 、安眠药Alpidem[5 、抗焦虑药Alpidem[6 和Saripidem[7 等.因此, 咪唑并[1, 2-a ]吡啶是一类研究价值很高的化合物.
对于咪唑并[1, 2-a ]吡啶的报道可以追溯到20世纪20年代, 之后的几十年间, 人们对于咪唑并[1, 2-a ]吡啶的性质逐渐了解.至今为止, 有关咪唑并[1, 2-a ]吡啶的合成被大量报道.随着合成研究的不断探索, 以及对绿色化学和资源节约的追求, 许多新型有机合成技术被应用到化学合成中, 如微波合成和固相合成.另外寻找实用、原子经济性和绿色的合成方法是化学家们努力的方向.接下来本文将从一锅“多组分”反应、微波反应和固相合成这三种类型概述近几年咪唑[1, 2-a ]吡啶类化合物绿色合成的方法.
1.
一锅多组分合成咪唑并[1, 2-a ]吡啶
“一锅”多组分[8 反应, 即是在反应器中将三种或三种以上的反应物原料投入, 并给以一定的反应条件让其发生反应, 有反应操作简单、反应原料易得及不经分离中间体等优点, 被合成方面的学者们所关注.一锅多组分反应合成咪唑并[1, 2-a ]吡啶衍生物正被广泛地研究与应用.
1.1
以2-氨基吡啶, 醛类化合物和异腈类化合物合成咪唑并[1, 2-a ]吡啶
2010年, Bazgir等[9 采用2-氨基吡啶、二茂铁甲醛以及异腈三组分反应, 在2位上引入二茂铁基, 得到具有潜在生物活性的二茂铁基咪唑并[1, 2-a ]吡啶(Eq. 1), 此合成策略是基于异腈化合物的三组分有效合成二茂铁基咪唑并[1, 2-a ]吡啶的第一个例子, 相较用乙醇作溶剂的三组分反应, 此反应具有高效且后处理简单等优点.
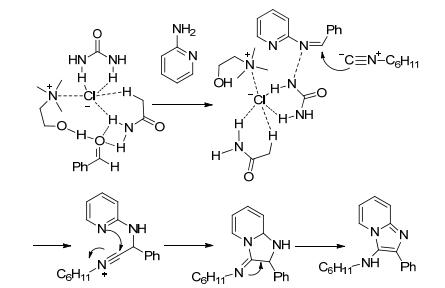
2014年, Sanaeishoar等[10 使用溶胶-凝胶(sal-gel)方法制备了LaMnO3 钙钛矿纳米颗粒, 该纳米颗粒成功催化了2-氨基吡啶、苯甲醛和异腈化合物的一锅法三组分反应, 得到N -环己基-2-苯基咪唑并[1, 2-a ]吡啶-3-胺(Eq. 2).首先, 2-氨基吡啶与催化剂表面的路易斯酸性位点配位, 所得的配合物的NH对醛的活性羰基进行攻击得到季铵盐中间体.消除水得到亚胺盐, 然后与异腈化合物进行亲核加成形成异腈基中间体, 异腈基中间体环化成亚氨基中间体.该中间体互变异构得到咪唑并吡啶基本骨架中间体, 最后通过消除生成最终产物, 并释放来催化剂(Scheme 1 a ]吡啶, 产率高达96%.这种绿色和可重复使用的钙钛矿型氧化物对咪唑并[1, 2-a ]吡啶的合成显示出优异的催化活性, 并且循环回收可达五次.
图式 1
2015年, Shaabani等[11 报道了在0 ℃下将氯磺酸滴加到羟基多壁碳纳米管的正己烷溶液中, 简单地制备了多壁碳纳米管硫酸(MWCNTs-OSO3 H).以此作为固体碳基酸催化剂, 在室温下催化2-氨基吡啶、醛和异腈化合物进行合环反应, 得到带有三个取代基的咪唑并[1, 2-a ]吡啶衍生物(Eq. 3), 产率高达80%~90%.反应具有条件温和、反应时间短和产率高等优点, 所用的催化剂可通过简单过滤回收并重复利用.
过低的共熔溶剂现在变得越来越流行, 因为它们绿色、安全, 与普通有机溶剂相比, 节省原料和能源. 2015年, Azizi等[12 在氯化胆碱基础的低共熔溶剂中, 实现了2-氨基吡啶、苯甲醛和环己基异腈化合物的一锅法多组分反应, 得到了N -(6-1, 3, 5-三炔-1-基)-2-苯基咪唑并[1, 2-a ]吡啶-3-胺化合物(Eq. 4).首先, 低共熔溶剂中羰基氧与羟基氢和氨基氢形成氢键活化醛的C=O, 然后与2-氨基吡啶发生亲核加成生成亚胺中间体.低共熔溶剂可以激活亚胺亲核攻击环己基异腈化合物, 发生加成反应产生双环加合物, 然后通过1, 3-H移位进行内部重排, 得到目标产物(Scheme 2
图式 2
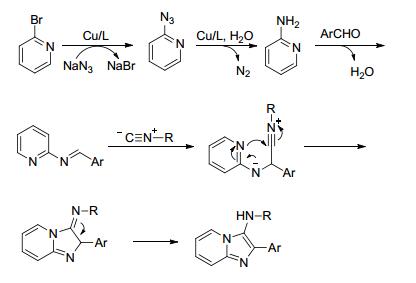
2016年, Allahabadi等[13 发表了碘化铜催化2-溴吡啶、叠氮化钠、芳醛和异腈化合物的多组分反应, 得到了2, 3-二取代咪唑并[1, 2-a ]吡啶(Eq. 5).首先, 2-溴吡啶和叠氮化钠反应得到2-叠氮吡啶, 随后脱N2 还原生成2-氨基吡啶, 然后与醛缩合生成亚胺中间体, 接着该亚胺中间体和异腈化合物亲核加成形成异腈基中间体, 异腈基中间体继续分子内环化得到2, 3-二取代咪唑并[1, 2-a ]吡啶(Scheme 3 a ]吡啶的第一例文献报道, 此反应是一种新颖高效的四组分反应.
图式 3
2018年, Rostami等[14 使用杯[n ]芳烃(calix[n ]arene-SO3 H)表面活性剂在水中形成疏水腔, 起到反应器的作用.在该体系中实现了2-氨基吡啶、苯甲醛和异腈化合物的一锅法多组分反应, 合成了2-苯基咪唑并[1, 2-a ]吡啶-3-胺化合物(Eq. 6).首先, 2-氨基吡啶和苯甲醛生成亚胺中间体, 然后与异腈化物进行[1+4]环加成反应得到最终产物(Scheme 4 [4 芳烃和杯[6 芳烃作为可重复使用的催化剂.具有原料简单易得, 反应时间短, 收率高等优点.
图式 4
2018年, Kurva等[15 通过超声辐照(USI)辅助Groebke多组分反应, 采用氯化铵(10 mol%)作为催化剂, 催化2-氨基吡啶、醛类化合物和异腈化合物合成了一系列新型3-咪唑并[1, 2-a ]吡啶咔唑结合型双杂环化合物(Eq. 7), 产率高达90%~96%.这种新型方案具有高效、温和、绿色经济和溶剂易于获得等优点.
1.2
以2-氨基吡啶、醛类化合物和末端炔基化合物为原料合成咪唑并[1, 2-a ]吡啶
2010年, Gevorgyan小组[16 使用取代2-氨基吡啶、醛类和适当的末端炔(N , N -二异丙基丙炔酰胺、N , N -二甲基丙炔酰胺)通过铜催化的三组分偶联反应合成药物Alpidem (Eq. 8), 这是一种高效合成此类药物的新型的TCC方法[T指温度-120 ℃, CC指两种铜类催化剂-CuCl、Cu(OTf)2 ].在此之前, 这些药物的合成通常需要通过多步反应来实现.
2011年, Reddy等[17 将2-氨基吡啶、取代苯甲醛、末端炔烃衍生物三组分偶联得到咪唑并[1, 2-a ]吡啶衍生物, 体系中使用InBr3 /Et3 N进行催化(Eq. 9).反应通过从醛和2-氨基吡啶原位形成亚胺来进行, 然后加入炔烃, 经InBr3 (Ⅲ)活化, Et3 N去质子化形成所需的咪唑并[1, 2-a ]吡啶.在该体系中三乙胺起到了缓冲的作用, 为反应提供了更温和的条件.另外, 三溴化铟与传统路易斯酸相比, 不需无水条件, 有易循环利用和操作简便等优点.
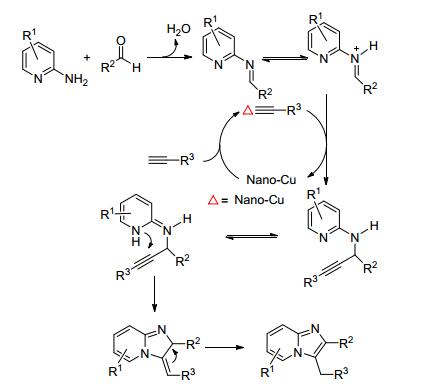
2014年, Zong等[18 开发出一种直接有效的方法, 以Cu-纳米颗粒催化2-氨基吡啶、醛和末端炔烃的一锅法多组分反应, 合成了咪唑并[1, 2-a ]吡啶化合物(Eq. 10).首先, Cu-纳米颗粒活化炔烃的C—H键, 炔基-Cu中间体进一步与由醛和2-氨基吡啶原位产生的亚胺离子进行亲核加成反应, 得到相应的炔丙胺和Cu-纳米颗粒.随后, 炔丙胺发生分子内环化形成咪唑并[1, 2-a ]吡啶化合物(Scheme 5 a ]吡啶, 收率良好(高达85%).
图式 5
2015年, Lu等[19 制备了超顺磁性CuFeO2 纳米粒子, 采用柠檬酸-二甲基脲混合溶液作为绿色溶剂, 研究了CuFeO2 纳米粒子在2-氨基吡啶、醛和炔烃的一锅法缩合反应中的催化活性.结果显示在65 ℃以极好的产率(90%)得到了一系列咪唑并[1, 2-a ]吡啶化合物(Eq. 11).催化体系可以重复使用六次, 同时保持其催化性能.
2014年, Maleki[20 使用多孔纳米复合材料Fe3 O4 @ SiO2 催化2-氨基吡啶、 醛和末端炔烃合成咪唑并[1, 2-a ]吡啶衍生物(Eq. 12).首先, 在K2 CO3 存在条件下, 乙醇溶液中回流, 2-氨基吡啶和醛的缩合形成亚胺.然后, 由末端炔烃的碳负离子形式的亲核攻击亚胺形成复合物.复合物通过分子内亲核反应环化产生中间体.该中间体最后异构化得到咪唑并[1, 2-a ]吡啶衍生物(Scheme 6 3 O4 @SiO2 可用磁铁回收利用, 用乙醇洗涤后晾干, 无需进一步纯化, 可再次循环利用, 符合当代绿色化学的要求.
图式 6
2015年, Rassokhina等[21 开发了一种以2-氨基吡啶、苯甲醛和丙炔酸酯衍生物为原料, 使用Cu(OAc)2 催化的三组分级联丙酰化反应生成咪唑并[1, 2-a ]吡啶化合物的方法(Eq. 13).此反应方法规避了无氧操作的要求, 简化了实验操作, 具有廉价易得的反应原料、官能团耐受、产率高达95%等优点.
2015年, Balijapalli等[22 建立以CuO-CuAl2 O4 复合物和D -葡萄糖为催化剂的最佳反应条件, 建立了以2-氨基吡啶、 芳基醛和苯乙炔为原料合成咪唑并[1, 2-a ]吡啶(Eq. 14)的简单而有效的途径, 即通过一锅法串联方法实现.
2016年, Mandlimath等[23 研制出简便有效的多表面纳米催化剂ZnAl2 O4 .使用Cu/nano ZnAl2 O4 催化一锅反应, 活化炔烃的C—H键, 与2-氨基吡啶和取代苯甲醛生成亚胺中间体的缩合反应, 高效合成2, 3-二取代咪唑并[1, 2-a ]吡啶化合物(Eq. 15).该合成方案具有简单、无溶剂和不需要任何添加剂的优点, 同时催化剂廉价且可重复使用, 效果优于那些昂贵的贵金属催化剂.另外与Balijapalli等报道结果相比, 使用Cu/nano ZnAl2 O4 催化剂, 达到相同的最大产率所需的反应时间要缩短很多.
2017年, Hussain等[24 应用二维(2D)材料, 如石墨烯和功能化六方氮化硼(h-BN), 负载铜催化剂(CuO/rGO, CuO/h-BN, Cu(0)/rGO, Cu(0)/h-BN, CuS/rGO和CuS/h-BN), 探索了它们对2-氨基吡啶、醛和末端炔之间的一锅三组分反应合成咪唑并[1, 2-a ]吡啶的催化活性(Eq. 16), 最终得出CuO/rGO纳米复合材料是高活性催化剂.与以前报道相比, 该制备咪唑并[1, 2-a ]吡啶的方案具有反应时间更短、官能团耐受性好、可回收催化剂和无添加剂的优点.
1.3
以2-氨基吡啶、苯甲醛类衍生物和三甲基甲硅烷基氰化物(TMSCN)为原料合成咪唑并[1, 2-a ]吡啶
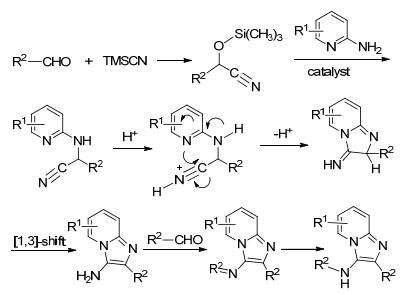
以2-氨基吡啶、苯甲醛类衍生物和TMSCN为原料合成咪唑并[1, 2-a]吡啶的机理如Scheme 7 [1 3 质子转移产生咪唑并[1, 2-a ]吡啶基本骨架, 再与前面的醛亲核反应生成N -取代烷基的咪唑并[1, 2-a ]吡啶, 最后再加氢得到最终产物三取代的咪唑并[1, 2-a ]吡啶类化合物.
图式 7
2011年, Venkatesham等[25 利用(溴二甲基锍)溴化物催化2-氨基吡啶、芳香醛和TMSCN的一锅多组分反应合成N -亚苄基-2-苯基咪唑并[1, 2-a ]吡啶(Eq. 17).该反应具有无溶剂、通用和所需时间短等优点.
2014年, Abdollahi-Alibeik和Rezaeipoor-Anari[26 在MCM-41负载的三氟化硼(BF3 /MCM-41)存在下, 进行2-氨基吡啶、 醛和TMSCN的多组分反应, 合成了3-亚氨基芳基咪唑并[1, 2-a ]吡啶衍生物101 (Eq. 18).该催化剂可重复使用多次, 活性适度降低.
1.4
其他原料合成咪唑并[1, 2-a ]吡啶
2012年, Li[27 使用空气作为氧化剂, 铜作催化剂直接催化2-氨基吡啶与β -酮酯的C—N偶联, 生成咪唑并[1, 2-a ]吡啶-3-羧酸酯(Eq. 19).此反应简单, 经济且环境友好, 反应条件温和, 但其产率不高, 反应时间过长, 需要进一步改善.
2012年, Wen等[28 报道了一种绿色且高效的区域选择性合成咪唑并[1, 2-a ]吡啶衍生物的方法(Eq. 20), 即使用Et3 N作为催化剂, 在无溶剂条件下, 以杂环烯酮(HKAs)、β -氧代二酯(ODE)和醛作为结构单元环化而成.该绿色合成工艺显示出高区域选择性、简明的一锅法、较短的反应时间、易于纯化和避免使用过渡金属等优点.该方法可以高产率地获得多取代的咪唑并[1, 2-a ]吡啶.
2014年, Cao等[29 报道了使用2-氨基吡啶、炔醛、醇或胺作为反应底物多组分一锅反应(Eq. 21), 合成了3位上引入杂原子烷基片段的骨架结构, AcOH促进2-氨基吡啶和醛的分子间脱水, 得到的产物在EtOH中互变异构为1, 3-二烯中间体, 然后迈克尔加成得到咪唑并[1, 2-a ]吡啶骨架中间体, 随后, 在AcOH的帮助下进行质子转移得到所需产物(Scheme 8 a ]吡啶衍生物.
图式 8
2015年, Kundu和Basu[30 成功地将绿色碳催化剂氧化石墨烯(GO)用于一锅多组分法, 催化2-氨基吡啶、苯乙酮和硫醇类化合物的反应, 选择性地合成了生物学上重要的3-亚磺酰基咪唑并[1, 2-a ]吡啶(Eq. 22).这种反应具有高选择性、无金属、耐受各种官能团、可以回收和再利用碳催化剂等优点.此GO催化的多组分串联反应的应用是第一次报道.这种可持续且易于获得的碳质材料有望在合成药物化学和材料科学中得到应用.
2013年, Yan等[31 以2-氨基吡啶、苯甲醛、硝基甲烷在铜盐催化下合成了2-苯基-3-硝基咪唑并[1, 2-a ]吡啶衍生物(Eq. 23).首先2-氨基吡啶和醛生成了亚胺, 然后亚胺和硝基甲烷通过迈克尔加成得到仲胺中间体, 该中间体在二价酮的催化下失去一个电子, 形成自由基离子, 接下来在氧化剂作用下失去氢自由基得到氮鎓离子, 通过质子消除形成亚胺, 亚胺迅速平衡成更稳定的烯胺结构, 再经过一轮重复得到最终产物(Scheme 9
图式 9
2015年, Kamal等[32 以取代吡啶、α -溴代酮和苯甲酰叠氮化合物为原料, 以独特的区域选择性合成咪唑并[1, 2-a ]吡啶(Eq. 24).其中取代吡啶和α -溴代酮生成吡啶鎓盐.叠氮化物在Cu(OAC)2 (2 mol%)和Et3 N体系下分解产生螯合的亚胺.螯合的亚胺被吡啶鎓叶立德攻击, 产生的中间体迅速异构化成另一种N正离子中间体.由于螯合作用, 该中间体的胺基(NH)表现为弱碱, 并且进攻吡啶亚胺(弱酸)发生[3+2]偶极环加成得到另一种咪唑并[1, 2-a ]吡啶基本骨架中间体.在氧气的作用下, 该中间体迅速芳香化成咪唑并[1, 2-a ]吡啶(Scheme 10
图式 10
2017年, Rao等[33 在氧气氛围中, 以2-氨基吡啶为原料, 以乙基叔胺为碳源, Cu催化高效合成3-甲酰基咪唑并[1, 2-a ]吡啶 (Eq. 25), 即用分子氧作为乙基氧化剂, 同时选择性地断裂乙基C—C键和C—N键.该反应具有广泛的底物范围、良好的功能基团耐受性等特点.
2017年, Dheer等[34 报道了由CuI/β -CD在水中催化2-氨基吡啶和末端炔烃合成2-碘-咪唑并[1, 2-a ]吡啶的方法(Eq. 26).该方法的显著特征是, 在温和的反应条件下原位形成1-碘炔和烷基/芳基叠氮化物, 具有高区域选择性和使用水作为溶剂的优点.
2018年, Ghosh等[35 报道了2-氨基吡啶和取代苯乙酮在碘催化剂的作用下快速有效地构建咪唑并[1, 2-a ]吡啶(Eq. 27).该方法具有反应时间短、底物范围广泛和所收率良好等优点.另外咪唑并[1, 2-a ]吡啶的后期官能化可以通过C—H键活化和C—C交叉偶联反应进行.
2.
微波反应
微波常用来加热材料, 被应用于有机化学领域的时间是20世纪80年代, 但由于缺乏控制性和安全性, 发展缓慢.近年来, 随着科学技术的不断发展, 应用微波反应器进行有机合成, 因其可以大大缩短时间且改善收率, 受到广泛的关注.
早期, 微波反应器辅助合成多应用于异腈多组分反应, 最早报道于1999年, 此后不断被应用于此类反应中.例如2009年, Larhed课题组[36 使用2-氨基吡啶、醛、环戊基异腈在微波反应器的辅助下进行多组分反应, 合成了多取代的咪唑并[1, 2-a ]吡啶(Eq. 28), 以MgCl2 作催化剂于160 ℃微波辐射20~30 min完成.
2012年, Beifuss课题组[37 在甲苯溶液中, 在以蒙脱石为催化剂催化2-氨基吡啶、醛和异腈酸酯之间的三组分反应的研究基础上, 用离子溶液代替甲苯, 利用微波辅助反应使胍盐在无任何催化剂的情况下在几分钟内制备3-氨基取代的咪唑并[1, 2-a ]吡啶(Eq. 29), 产率高达98%.
2013年, Maiti等[38 报道了一种新的以三维方式快速产生复杂的分子框架的合成方法, 实现了一种新的一锅三组分反应.在无溶剂条件下由三氟甲磺酸钪(Ⅲ)催化各种取代的苯并咪唑连接的氨基吡啶、醛和异腈合成苯并咪唑-咪唑并[1, 2-a ]吡啶(Eq. 30).该方法绿色环保, 且允许引入三个结构多样性点以扩展化学空间, 具有高纯度和高产率的优点.
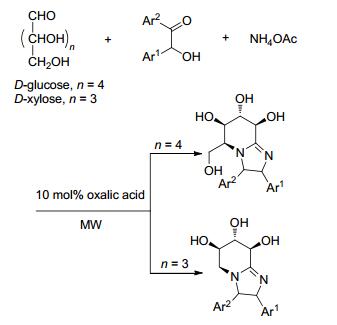
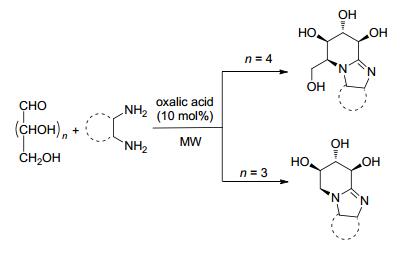
微波辅助合成除了应用于异腈多组分反应, 近些年来也报道了一些其他反应. 2010年, Yadav和Awasthi[39 开发了两种之前未见报道过的一锅法高产合成方法, 用于从碳水化合物中提取咪唑并[1, 2-a ]吡啶.第一种方法是微波辅助10 mol%草酸催化的无溶剂D -葡萄糖/D -木糖与乙酸铵和苯偶姻的串联反应, 得到带有多羟基亚氨基糖的四氢咪唑并[1, 2-a ]吡啶(Scheme 11 D -葡萄糖/D -木糖和1, 2-二胺作为起始底物, 在10 mol%的草酸存在下通过胺驱动串联反应, 以极好的产率成功合成了多羟基四氢苯并咪唑并[1, 2-a ]吡啶(Scheme 12 D -葡萄糖作为生物可再生资源, 为获得一系列与生物、化学和药学相关的手性产品提供新的方法学路线.
图式 11
图式 12
2015年, Zhang等[40 在无溶剂的条件下, 微波辅助TsOH催化2-氨基吡啶、3-苯基丙醛和醇三组分反应(Eq. 31), 首先, 通过TsOH催化的2-氨基吡啶和3-苯基丙醛的脱水反应形成亚胺产物.随后, 向炔烃中加入醇, 得到(1Z , 2E )-3-甲氧基-3-苯基-N -(吡啶-2-基)丙-2-烯-1-亚胺, 然后亲核加成得到吡啶六元环并吡咯五元环中间体, 该中间体在TsOH的帮助下进行顺序质子转移生成N正离子中间体, 得到最终产物取代3-(烷氧基(苯基)甲基)咪唑并[1, 2-a ]吡啶(Scheme 13 a ]吡啶.
图式 13
2015年, Karamthulla等[41 报道了在微波辐射和分子碘存在下, 2-氨基吡啶、芳基乙二醛和环状1, 3-二羰基的快速一锅三组分反应(Eq. 32).使用该方法可以合成多种2, 3-二取代的咪唑并[1, 2-a ]吡啶衍生物.该方法的显着特点是无金属催化剂、反应时间短、产率高使用微波加热及无有害副产物.
2016年, Wagare等[42 以2-氨基吡啶、苯乙酮、N -溴代琥珀酰亚胺(NBS)为原料, 在聚乙二醇(PEG-400)和水(体积比为1:2)的溶剂中借助微波反应加热生成2位取代的咪唑并[1, 2-a ]吡啶(Eq. 33).此方法用苯乙酮和NBS代替α -卤代酮, 反应过程简便, 安全经济绿色, 减少了反应时间, 提高了反应收率.
2018年, Rao等[43 开发了在无催化剂微波辐射下在绿色溶剂中合成咪唑并[1, 2-a ]吡啶的杂环化反应.反应使用H2 O-IPA作为反应介质, 各种取代的2-氨基吡啶作为原料, 在微波辐射下与α -溴代苯乙酮进行环化反应, 以极好的产率得到相应的咪唑并[1, 2-a ]吡啶(Eq. 34).
3.
固相合成
固相合成是将反应物连接在一个不溶性的固体载体上的合成方法, 一般选用树脂材料作为固体载体, 此反应可直接选用简单反应器进行反应, 避免了中间体的分离和纯化, 最终产物直接从固体载体上分离下来.与微波合成具有共同的优势, 如反应条件比较温和、反应时间短和副产物少, 产率越来越高.
2001年, Chen课题组[44 应用固相试剂合成了咪唑并[1, 2-a ]吡啶.将Rink树脂用二异丙基碳二亚胺甲酰化(DIC)/HCOOH、POCl3 /二异丙胺(DIPEA)脱水处理转化成异腈树脂, 并用该树脂、醛和2-氨基吡啶在TsOH作用下合成树脂键合的咪唑并[1, 2-a ]吡啶, 有效处理后生成3-酰胺基咪唑并[1, 2-a ]吡啶衍生物(Scheme 14
图式 14
2002年, Lam小组[45 应用α -卤代酮树脂实现了咪唑并[1, 2-a ]吡啶的固相合成.先将树脂材料用亚磺酸盐砜烷基化得到α -卤代酮树脂, 再用该树脂处理2-氨基吡啶得到咪唑[1, 2-a ]吡啶树脂键合产物, 用环氧化物对砜阴离子进行烷基化得到醇, 然后在琼斯试剂中氧化成酮, 最后水解释放出产物(Scheme 15
图式 15
2004年, Lyon和Kercher[46 将乙醛酸在甲醇溶液中与大孔聚苯乙烯碳酸酯树脂(MP-CO3 )结合得到乙醛酸树脂, 乙醛酸树脂与2-氨基吡啶和异腈反应合成了2-未取代的-3-氨基咪唑杂环(Scheme 16
图式 16
4.
总结与展望
总结了近几年有关咪唑并[1, 2-a ]吡啶的绿色合成方法.一锅“多组分”反应可以直接从简单易得的原料出发, 不经中间体的分离, 直接获得我们所要合成的化合物, 而且具有高效、高选择、高收率、反应条件温和及操作简洁方便等优点; 微波合成和固相合成属于有机化学新时代的新技术, 这种新型的合成方法具有反应条件温和、时间短、副产物少和高转化率等优势.
经过有机化学家们的不断探索, 新催化剂(纳米粒子负载金属催化剂)和“新”溶剂(共熔溶剂类似的绿色溶剂)的开发及新技术的应用近年来不断被报道, 应扩大其在化学合成上的应用, 改变传统有机合成的污染重及消耗大等缺点.利用绿色实验原料及绿色实验技术, 不断创新从源头上减少或消除化学试剂对环境的污染, 实现更大的原子经济性.但从目前的研究进展来看, 有关的新技术应用到化学合成仍不普遍, 大多数报道的新催化剂的合成与应用都是以2-氨基吡啶为原料, 而以其他原料构建咪唑并[1, 2-a ]吡啶的绿色合成鲜见报道.




 下载:
下载:










































 下载:
下载:

















 下载:
下载:
