
图 1.
环糊精的分子结构(a)和三维结构示意图(b)
Figure 1.
Molecular structure (a) and three-dimentional structure diagram (b) of cyclodextrin

超分子化学(Supramolecular chemistry)[1]是一门新型的前沿学科, 与诸多学科如配位化学、材料科学和生命科学等相互交织.超分子的形成主要是基于多个分子相互识别的过程, 该过程不仅是主体分子与客体分子的包合过程, 更重要的是主体分子对于底物的选择过程.超分子包合是一种有目的、有选择的作用机制.
超分子化学研究的内容主要包括分子识别、自组装和主客体化学等, 而主客体化学便是对超分子主体化合物与其他小分子客体之间的作用进行探索, 例如冠醚对金属阳离子的选择性包合、环糊精对芳香醛的包合催化反应等[2].环糊精(缩写为CD)是在冠醚后超分子化学研究最多的一类大环主体分子, 是一种通过α-1, 4-糖苷键将D-吡喃型葡萄糖单元连接在一起而形成的环状低聚糖, 主要包括α-、β-和γ-环糊精(如图 1a)[3].通过X射线分析结果可以观察到环糊精具有一个特殊的刚性锥形空腔结构(如图 1b).环糊精外侧含有大量的羟基, 使得环糊精具有较好的亲水性, 可以溶解在水里, 而内部不具有羟基, 所以环糊精具有独特的疏水空腔.正是由于这个独特的“内疏水、外亲水”空间结构, 使得环糊精从被发现以来, 越来越受到科学工作者的关注.
在立体空间允许的情况下, 环糊精的空腔能够包含各种疏水性的客体分子, 于是便形成一种独特的主客体包合物, 借此便能更好地将其应用于催化有机反应; 另外, 环糊精因其无毒无害、价格低廉, 又具有良好的客体识别性、优良的生物相容性与适当的空腔形状等优点, 且存在大量氢键作为活化基团, 使得环糊精催化有机反应可以工业化、产业化.
然而天然环糊精的催化活性是有限的, 且在有机溶剂中溶解性也较差, 极大地限制了其应用范围, 通常会经过对其表面上的羟基进行修饰来完善其性能.金属催化剂作为重要的工业催化剂, 与环糊精体系结合可同时发挥金属的催化性能和环糊精的分子识别和相转移等功能, 极大地改善其催化性能.近年来, 发表了基于不同反应类型的环糊精催化反应方面的综述, 例如环糊精衍生物在氧化、水解、还原、偶联等催化反应中的应用进展综述[4].而本文则主要综述了环糊精参与的过渡金属催化有机反应, 并以金属价态分类对0~4价常见过渡金属参与催化的有机反应进行介绍.
纳米金(AuNPs)由于具有独特的电学和光学特性, 在纳米科学和纳米技术领域是一项非常热门的研究, 并具有广泛的应用潜力.纳米金和大环超分子的复合更是提供了一种新型的混合纳米材料, 并且有望带来新的特性、新的功能和新的应用. Zhao等[5]合成了一种由环糊精修饰的纳米金粒子(CD@AuNPs), 可用于传感、自组装和级联催化.单分散的纳米金粒径为15~20 nm, 环糊精作为还原剂和稳定剂, 以一种生态友好的一步胶体合成法合成了混合的纳米材料.首先, 纳米金粒子可以作为基于客体置换反应产生的荧光传感的支架和能量接收器.其次, 大环超分子功能化的纳米金粒子可以有效地进行组装, 并利用四(4-羧基苯基)卟啉作为介体, 形成良好的一维、二维结构.最后, 除了常规的主客体交互作用之外, CD@AuNPs还展现出了令人意想不到的催化活性——同时具有葡萄糖氧化酶和辣根过氧化物酶(HRP)这两种常见酶的模拟特性, 在级联反应(首先催化氧化葡萄糖生成葡萄糖酸和H2O2, 然后H2O2和四甲基联苯胺被分别催化转化为H2O和相应的氧化物)中被用作唯一的催化剂.结合实验和密度泛函理论(DFT)计算, Zhao等推测CD@AuNPs的独特催化活性可能是由环糊精分子的特殊拓扑结构以及由此产生的电子转移效应引起.该纳米材料的出现启示人们纳米金属材料仍有许多“隐藏”的特性等待去发现, 同时设计配体结构也可能是一种新型的策略用来操纵纳米金属材料的物理化学性质, 例如颗粒大小、晶面和催化特性.
Menuel等[6]则研究了环糊精和其它糖类添加剂在机械合成纳米金粒子中的作用, 以及它们催化硝基苯转化为相应的苯胺产物中的作用.在这里, 环糊精不仅可以使纳米金粒子更加稳定, 还可以通过超分子作用在固体混合物中扩散基质, 使化学反应定向到选择性生成苯胺衍生物. Menuel等仔细研究了影响纳米金的形成和苯胺衍生物合成的因素, 研究表明, 糖类添加剂的性质和苯取代基的位置均起到了很大的决定性作用, 同时水也在硝基的还原过程和超分子与底物的相互作用中起着至关重要的作用.此外, 该催化系统可以在连续三次使用后回收利用, 且催化活性不会造成明显的损失, 这便突出了机械化学、超分子化学和催化反应三者结合的特别之处.
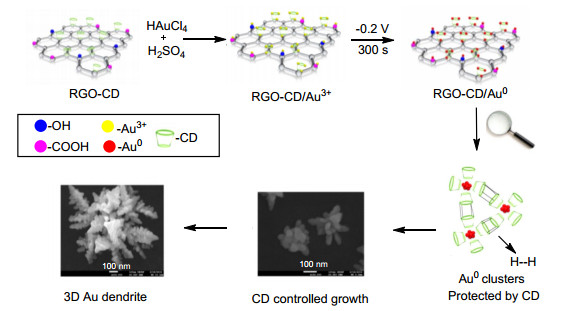
纳米金的化学和物理性质主要是取决于它们的大小和形状, 而这些性质是可以通过改变形态和结构来调节的.在纳米金的不同形态中, 树枝状的纳米金由于其在多个领域中的潜在应用受到了极大的关注[7, 8]. Shanmugam等[9]在环糊精的辅助下将纳米金负载在氧化石墨烯上, 并利用电子显微镜观察了其生长机制, 研究表明环糊精修饰的氧化石墨烯(RGO)可通过电沉积技术由最初的Au3+最终形成树枝状纳米金(Scheme 1), 且CD修饰的RGO对于纳米金的成核和成长都起着至关重要的作用. Shanmugam等紧接着分析了纳米金的结构和电化学性质, 结果显示纳米金同时也对亚硝酸盐和葡萄糖的电解氧化反应具有协同催化作用, 经分析其催化活性正是来源于其典型的树枝状纳米金结构, 由于其具有很多的活性位点, 且在RGO-CD的支持下可充当微小的纳米电极, 因此具有很高的电催化活性.该合成方法简单、有效、低成本, 同样也适用于制备其他种类的纳米金属, 由此可见, 该方法未来将在催化、生物传感和纳米器件方面均具有很好的应用前景.
科学家们一直致力于寻找成本效益高和新的激活技术以高效催化C—C偶合反应和点击反应, 尽管目前的催化剂通过各种回收方式可以达到很高的周转率, 但仍存在诸如由金属催化剂引起的产品污染和高成本等缺点. Cravotto等[10]为了解决这一问题, 将CuⅠ阳离子紧密地嵌入到β-CD的腔体内, 并通过微波辐射进行物理激活, 从而合成了一种新型的β-CD/CuⅠ催化剂.这种催化剂具有独特的极性结构, 本身对电介质加热就十分敏感的环糊精更是由于嵌入的阳离子而增强了这一效果, 因此它特别适用于微波辅助反应, 并且可以在短时间内达到最高产量. Cravotto等将此催化剂成功地应用于最常见的点击反应中, 即苄基叠氮和苯乙炔的叠氮炔烃环加成反应(Eq. 1), 反应温度为70 ℃时在100 W的微波辐射下10 min便可使反应达到终点.由于反应后金属的损失可忽略不计, 因此该催化剂可循环使用, 且制备过程简单, 符合绿色化学的要求.
|
|
(1) |
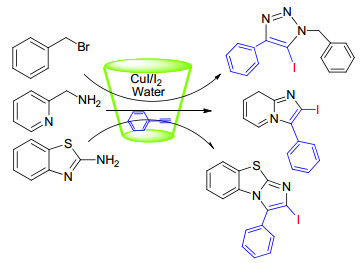
碘代三唑已知的合成方法是在亲电碘化试剂的存在下由CuⅠ催化有机叠氮物和末端炔烃发生环加成作用而生成[11], 而绿色化学的一项基本原则是提倡使用水作为一种更安全、更环保的溶剂, 但是使用水作为反应介质对有机合成化学家来说是一个巨大的挑战, 因为有机化合物在水中的溶解度很低, 然而环糊精可以通过包合有机材料提高其在水中的溶解性.基于这些研究, Dheer等[12]研究了一种新型的CuI(碘化亚铜)/β-CD催化剂, 并首次用于水中区域选择性合成5-碘-1, 2, 3-苯并三唑、2-碘咪唑吡嗪和2-碘苯咪唑噻唑(Scheme 2), 该方法的优点是条件温和、操作简单、产量高、底物通用性高以及选择水作为绿色溶剂.
Patil等[13]开发了一种常温下在水溶液中铜环糊精催化醇的氧化工艺, 首次报道了在氧化过程中形成了Cu2O-CD纳米结构.研究证明了不同铜盐、CD和碱基对纳米颗粒的形态具有影响, 该催化剂对一锅化三组分的丙炔氨化反应显示出了良好的催化效率, 收率可高达85%, 且反应后通过添加乙酸乙酯回收粗产物即可实现催化剂的再利用.他们还对使用后的催化剂进行了XRD分析, 以探测其成分和结构的变化, XRD图谱分析表明, 峰值没有变化即没有新的晶相物种产生, 从而证明催化剂没有发生任何变化, 这些研究解释了该催化过程可能是一种表面介导催化作用.
在分析金属与环糊精相互作用的文献资料时, 可以看到大部分都是涉及到二价金属, CuⅡ更是其中的“佼佼者”.早在1927年, Messmer[14]便首次在碱性溶液中将CuⅡ配位在β-环糊精上.
在过去的20年中, “点击化学”在学术界以及工业上都引起了相当多的关注, 因为它能从简单的基质中高效地制备更复杂的产品[15~17].其中, 铜催化的叠氮炔烃环加成反应(CuAAC)便是最原始也是最著名的一个例子[18, 19]. CuAAC法的特点是区域选择性高和生物相容性良好, 能在缓和的条件下高效地合成1, 2, 3-苯并三唑衍生物, CuⅡ在该反应中起到了至关重要的作用.同样CuAAC法也可用于其他的有机反应[20~24], 经Sharpless和Medal研究发现, 不同的铜离子化合物, 例如CuI[25]、Cu(OAc)2[26]、CuOTf·C6H6[27, 28]和[Cu(phen)(PPh3)2]-NO3[29], 都曾用于催化此类反应.然而, 尽管这些均相催化剂具有如此优良的特性, 它们的应用仍然受到限制, 因为它们具有不易分离回收、含有细胞毒性和在反应过程中易被破坏等缺点.为了克服这些问题, 研究者们将更多的关注放在了非均相催化剂上, 因为它们相比于同类型的均相催化剂更容易分离回收, 且在自然界中是低毒的[30~32].通过将铜离子固载于不溶性的载体之上, 便可以取两者之所长.如今, 人们越来越热衷于将对环境无害的物质作为非均相催化剂[33~35]. Nie等[36]首次将CuⅡ修饰的γ-CD负载在氮化硼上(h-BN@γ-CD@- Cu(OAc)2)(图 2), 这种可回收再重复利用的非均相催化剂可用于合成1, 2, 3-苯并三唑衍生物.且该催化剂具有良好的官能团耐受性, 可以高收率催化水相中的反应.此外, 催化剂还可以直接通过离心回收, 循环使用7次以后收率依旧很高.
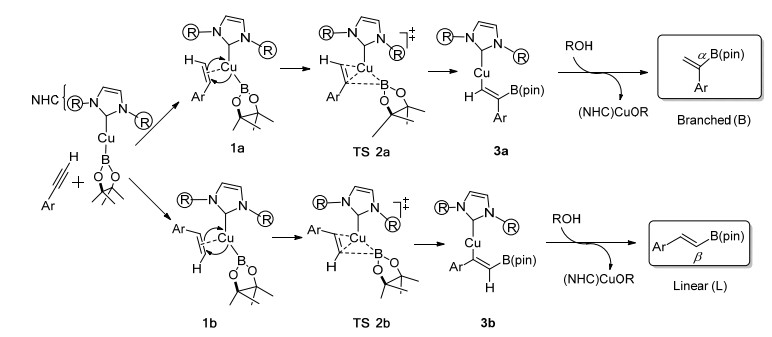
环糊精是一种很好的能够包合客体分子的主体化合物, 大范围地应用于水相反应中.由于其能够识别不同的小分子和改善有机小分子在水中的溶解性, 环糊精经常被用作水相催化剂.近些年以来, 硼氢化反应引起了学术界极大的关注.在大量文献报道中, 铜催化的硼氢化反应更是尤为重要.一方面, 研究发现使用过渡金属催化剂可以显著加快硼氢化反应的速率, 甚至能够实现立体选择性或反应区域的反转; 另一方面, 与贵金属如Pd、Pt、Rh、Ir等相比, 铜具有价格低、毒性小等优点. Yao等[37]报道了一种在水介质中条件温和、环境友好型的铜催化的末端炔烃硼氢化反应, 在环糊精-双吡啶配体(CD-1) (Eq. 2)的存在下, 可进行区域选择性控制.该反应可成功应用于保护失活的末端炔烃, 且该配体至少可以回收重复利用5次后仍具有催化活性.这是首次报道的环糊精衍生物能够催化炔烃进行区域选择性硼氢化反应, 成为从简单的末端炔烃中获得乙烯基硼烷的有效途径.
|
|
(2) |
Zhang等[38]研究合成了一种新型环糊精衍生物, 是由N-杂环化合物包裹而形成的环糊精复合物(ICyD), 分别由α-CD、β-CD得到了α-ICyD、β-ICyD, 在铜催化的硼氢化反应中发生了相反的区域选择性.反应的结果主要取决于环糊精的性质, α-CD衍生物提供的是末端炔烃的线性产物, 而β-CD衍生物提供的则是支链产物(Scheme 3).这两种催化剂的区别在于其机制有所差异:传统的并行机制和新型的正交机制, 腔体形状的差异性不仅能引起一个区域选择性开关, 同时还能产生一个机械开关. ICyD中的金属表面是由环糊精支架包裹着的, 腔体内附加金属的位置迫使外部的配体受其形状影响而与金属中心发生相互作用, 因此, 这种特殊的结构导致在催化环异构化反应时诱发了受形状依赖的立体选择性和区域选择性. α-ICyD和β-ICyD的结构分析表明, 这两种环糊精衍生物在形状上有着明显的差异, 尤其是在金属存在的情况下.
Kaboudin等[39]将环糊精和金属铜有效结合, 合成了一种高效的纳米催化剂CuⅡ-β-CD, 可用于在均相条件下通过偶联反应一锅法制得1, 2, 3-苯并三唑化合物. CuⅡ-β-CD是一种具有良好稳定性的双核复合材料, 可以通过将硫酸铜溶液加入到β-环糊精和NaOH的溶液中获得.尽管该种催化剂具有很多优点, 例如良好的活性和选择性以及较高的转化率, 广泛应用于各行各业, 但是将这种可溶性的催化剂与产品和反应物分离开来仍然很困难.因此, 在此基础上, Kaboudin等[40]制备了一种高效的、易于回收、可重复利用的Fe3O4磁性纳米粒子支撑的CuⅡ-β-CD复合物催化体系(Fe3O4-β- CD-Cu2), 可通过催化芳基硼酸发生自身偶联反应, 从而高效地合成联苯化合物, 还可以直接在室温下以水为介质催化芳基硼酸、叠氮化钠与炔烃通过环化反应合成1, 2, 3-苯并三唑.经检验, 催化剂通过磁铁回收循环使用4次后催化活性仅略有损失.
PdⅡ可催化交叉耦合反应形成C—C键, 此反应广泛应用于合成和医疗化学中以制备具有巨大治疗潜力的新型药物[41, 42].一般来说, 磷配体会用来激活钯元素, 但是, 它们对空气和湿度都很敏感, 限制了催化剂的重复使用, 而且在水介质中反应也会生成许多副产物, 极大地限制了其应用[43, 44].因此, 钯催化系统中避免使用磷化氢被认为是有机合成领域中最具有挑战性的研究之一. Raihana等[45]将Pd(Ⅱ)离子负载在水溶性的吡啶修饰的β-CD上(PdⅡ@Pyr:β-CD), 研究表明此复合物可高效催化水介质中的Suzuki-Miyaura、Heck C—C耦合反应, 利用少量的PdⅡ@Pyr:β-CD便可以高收率催化各种卤代芳烃参与的耦合反应, 包括氯化物和苯硼酸、氯化物和苯乙烯发生的反应.这种均相催化剂可在保证其催化活性的基础上至少再循环使用6次以上.
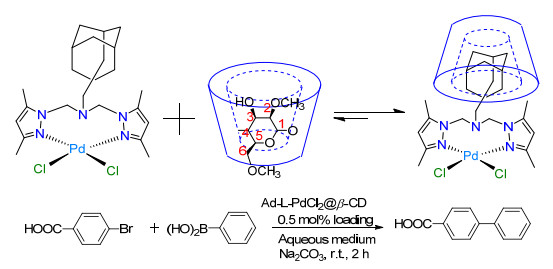
Qi等[46]设计了一种利用非共价键结合来设计和制备水溶性金属催化剂的新方法, 是将疏水性的钯二吡唑复合物负载于金刚烷上和相互补的亲水性反应形成了配合物(Ad-L-PdCl2 @β-CD).这一催化剂对于亲水性芳基硼酸和芳基溴代物在有机溶剂中发生的Suzuki-Miyaura耦合反应具有超高的催化活性(Scheme 4).相比较之下, 在相同的反应条件下, 单纯的钯复合物(Ad-L-PdCl2)的催化效率大大降低.反应完毕后, 可以n-Bu4NBr为稳定剂对其进行离心分离回收再利用, 但该催化剂负载量较高.为降低贵金属的使用量, Zhou等[47]最近又新合成了一种低钯负载量(仅0.00084 wt%)的催化剂——PdCl2(Ln@β-CD), 显示出了更良好的催化性能, 可在水中高效催化芳基硼酸和芳基卤化物的Suzuki耦合反应.实验表明, 当K3PO4·7H2O存在时, 以PdCl2(Ln@β-CD)为催化剂催化芳基溴化物, 4 h后分离得到的联芳烃衍生物的收率便可以达到80%~100%, 而控制实验表明没有PdCl2(Ln@β-CD)存在的条件下只能获得微量的产品.为了进一步研究该催化剂中三种配体的影响, 又进行了一系列比较实验, 发现只有微量β-CD、PdCl2、邻溴甲苯和苯基硼酸作为反应底物时, 只能获得6%的收率, 该结果更清楚地表明了该复合型催化剂的重要性.
Guo等[48]也合成了一种金属钯修饰形成的环糊精复合物(DACH-Pd-β-CD), 并对其进行了表征.结果表明, 此催化系统在常温下、水溶液中存在硼氢化钠的情况下, 对不同的硝基苯类化合物还原成相应的苯胺具有较高的催化性能, 所需产品的产量最高可达99% (Eq. 3).此外, 催化剂可以很方便地进行分离回收, 并且循环使用5次之后仍然可以维持较高的催化活性.
|
|
(3) |
近年来, 许多传统催化剂被报道用于不同条件下的转移氢化反应, 这些报道大多使用高温、过量的不可回收催化剂和有毒试剂.在此背景下, Imran等[49]合成了一种水溶性的PdⅡ@PyPDA:β-CD复合物, 可作为绿色环保的均相催化剂, 在温和的反应条件下以异丙醇为还原剂, 将芳香族酮加氢转化为仲醇.该催化剂重复使用5次后, 其催化活性仅略有降低.作者提出了一种可行的氢化机理.首先, 异丙氧基和PdⅡ@PyPDA:β-CD结合, 紧接着发生β位的氢消除生成了一个瞬态的氢化物, 然后芳香族酮插入到β-CD腔体中生成了一个中间产物, 最后迅速与异丙醇发生转移氢化反应, 从而触发第二个催化循环.该催化剂还有许多显著的优点, 如制备简单、稳定性好、底物选择性强、易回收、可重复利用, 因此在其他反应和工业应用中也有极大的潜在用途.
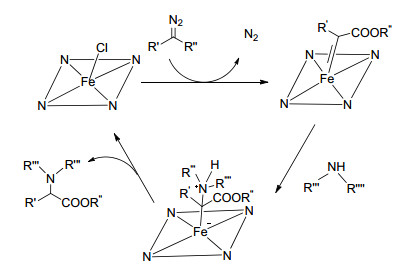
血红素不仅是最丰富、使用最广的金属卟啉催化剂之一, 也是多种金属酶的重要辅因子.近年来, 许多含血红素的金属酶在生物合成和生物分析化学中都得到了相当大的关注[50].金属催化的有机胺和α-重氮酯的N—H插入反应为含有C—N键活性分子的合成提供了简单易行的方法, 然而这种反应大多数使用有机溶剂, 并且需要严格的反应条件, 另一方面, 由于水溶液中的O—H键和底物的水不溶性, 酶催化的卡宾体转换反应仍然很难发生于水介质中[51].同样的问题也阻碍了血红素催化卡宾体嵌入有机胺中, 并且由于卟啉环的疏水性, 血红素在水介质中的催化活性通常也会有所降低.此种情况, “内疏水, 外亲水”的环糊精便可以为血红素提供有利的微环境以提高有机基质的溶解度. Xu等[52]利用环糊精协助血红素在水介质中成功催化了α-重氮酯与芳香胺的N—H键插入反应(Eq. 4), 产率可高达96%, 且反应体系对不同的芳香胺均展现出了良好的兼容性.同时也在设计控制实验的基础上初步提出了反应机理(Scheme 5), 首先, 重氮化合物经过亲电加成, 通过失去一个N2分子来协调血红素的不饱和Fe3+中心, 形成了一个血红素卡宾配合物; 然后, 通过胺基的亲核进攻产生了N—H键嵌入物.这种以水为介质的催化体系在卡宾体转化反应中占有很大的优势, 血红素与环糊精的协同作用也为血红素酶应用于重氮酯的转化反应提供了一种初步的可能性.
|
|
(4) |
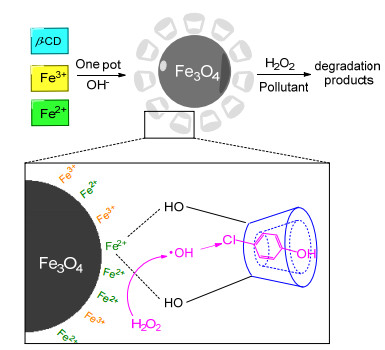
Wang等[53]也以一锅法制备了Fe3O4@β-CD磁性纳米复合材料, 并利用其催化污染物的降解(Scheme 6).在这里, 4-氯苯酚和氯苯被选中作为模型污染物, 它们通常被添加到生活中常见的除草剂、杀菌剂、杀虫剂和防腐剂中. Wang等对Fe3O4@β-CD复合材料的理化性能进行了表征, 并根据pH值、H2O2浓度和催化剂用量等关键变量的影响, 对其催化性能进行了评价, 同时也对反应动力学、材料稳定性和降解机理进行了评估.结果表明, Fe3O4@β-CD比Fe3O4表现出更高的催化活性, 对于4-对氯苯酚的降解速率分别为0.0373和0.0162 min-1, 这可能是由于形成了三元复合物, 其允许所产生的羟基自由基直接攻击4-对氯苯酚, 同时也增加了有机污染物的溶解度. Fe3O4@β-CD对于催化氯苯的降解也表现出了较高的催化活性, 其降解速率为0.0392 min-1 (Fe3O4催化的降解速率为0.0099 min-1), 可能由于Fe3O4@β-CD复合材料所具有的协同效应.此外, Fe3O4@β-CD还表现出稳定的机械强度和较好的重复利用性.仅含有Fe3+或无定形氧化铁的矿物质在催化剂表面的稳定性较差、溶解度较高, 因此, 稳定性是多相催化剂的一个重要特性.为此, Wang等对催化剂的稳定性做了测试, 为了验证溶液中溶解的铁离子对反应的影响, 在相同反应条件下, 以最大浸出铁离子量为基础进行了分批实验.最后的研究表明, H2O2的活化即·OH的生成, 主要还是归因于Fe3O4@β-CD的催化作用, ·OH的生成主要发生在固体催化剂的表面, 这样, 吸附在催化剂表面的有机物就可以直接被·OH攻击降解, 便不再需要经过浸出的Fe离子催化降解.因此, 该复合材料可以有效地回收, 不会在溶液中损失大量的铁离子, 且催化剂的结构和组成也不会发生明显变化.同时, 通过对4-对氯苯酚降解的中间产物和氯离子含量的分析, 提出了污染物可能的降解机理, 并根据密度泛函理论(DFT)探究了污染物和环糊精之间的主-客体相互作用, 阐述了由于环糊精的特定空间选择性而导致了降解中间产物的单一性.
TiO2是一种储量丰富、价格低廉、环境友好的材料, 结构稳定、有较大的能带隙(3.2 eV), 然而其光响应范围过于狭窄, 此时由TiO2和石墨烯制成的纳米复合材料便可以解决这一问题.石墨烯在没有功能化的情况下维持其稳定的分散状态是十分困难的, 这是由于其自身会发生聚集和重叠, 而在π-π相互作用和范德华力作用下其表面积将会减小, 这便限制了氧化石墨烯的实际应用.因此, 石墨烯的表面功能化更显得尤为重要.据报道, 在极性溶剂中, 石墨烯和环糊精的超分子体系具有很高的分散性[54].该超分子体系可稳定分散于水溶液中, 可为金属盐提供原位还原以形成超分子金属纳米复合材料. Sharavath等[55]首次在90 ℃低温下用一种简单的水相法合成了TiO2-β-CD-石墨烯(TiO2-CD@GNS)纳米复合材料(Scheme 7), 利用β-CD作为稳定剂将石墨烯固定于水相中, 防止其发生聚集, 从而促进了TiO2纳米粒子在石墨烯纳米薄片上的原位自组装过程.这种新型材料比原来的TiO2-氧化石墨烯材料具有更好的能量储存能力和更高的光催化活性, 在可见光下使用TiO2-CD@GNS, 可在25 min内实现亚甲基蓝的100%光降解, 由此可见其高效的光催化活性.研究表明这可能是由于增加了可见光的吸收度和通过Ti—O—C键在Ti和C中进行电子转移, 从而极大地阻碍了由光产生的电子空穴对的重组.
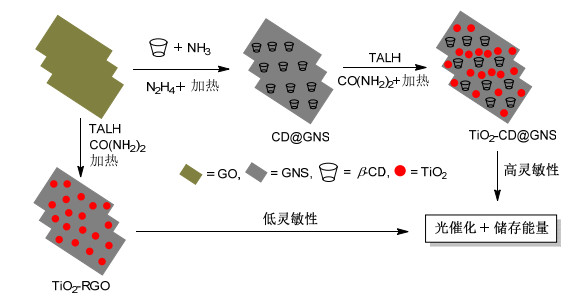
Mohamed等[56]也合成了一种TiO2-CD-GN纳米复合材料, TiO2纳米颗粒通过光照和环糊精的协助固定在石墨烯纳米薄片上.研究表明, 该复合材料也可用于可见光下4-氯苯酚的光降解, 且催化活性远高于TiO2. Subramanian等[57]则制备了一种TiO2-CD复合材料用于光降解4-硝基苯酚, 环糊精同样也大大增强了TiO2的光催化活性, 通过UV-DRS、FE-SEM和PXRD分析, 主要可归结为两个关键因素: (1) TiO2-CD的带隙能量比相对应的TiO2要低; (2) TiO2-CD中TiO2的相结构和表面形态都保持不变.此外, β-CD还可以作为4-硝基苯酚和TiO2之间的一个桥梁, 从而促进了它们之间的相互作用.
Sun等[58]采用光诱导装配法合成了新型光催化复合材料WF/β-CD/TiO2 (WF为一种天然纤维素的合成物), 并评估了其对甲基橙(MO)的光催化降解效率.在该材料中, β-CD用来捕获光催化底物, 并将其转移到TiO2表面, 以此来提高降解效率(Scheme 8).他们利用酚酞探针技术和反向滴定法, 测得WF/β-CD/TiO2中分别含有3.5%的β-CD和0.0581 g/0.1 g的TiO2, 它可以在21 min内完全降解100 mL浓度为0.1 mmol·L-1的甲基橙溶液. WF的加入虽然对β-CD/TiO2的光催化活性影响不大, 但为进一步将TiO2制备为高效的光催化剂提供了理想的生物质载体.另外, 在进行了5次循环试验后, 降解效率仍高达86%, 明显高于同样条件下的β-CD/ TiO2, 表现出良好的再生性能.上述所提到的几种环糊精-TiO2复合型催化剂均经过热重分析, 结果表明均具有较好的热稳定性.

本文系统介绍了环糊精参与的过渡金属催化的有机化学反应, 重点探讨了环糊精参与的常见0至4价过渡金属催化的有机反应.由于环糊精腔体外表面上存在许多可作为活化基团的羟基, 因此可以对其进行修饰, 从而获取到具有更加优良性能的环糊精衍生物.环糊精及其衍生物与金属催化剂的结合能够更加高效地催化有机反应在水相中完成, 有效地避免了使用成本高或具有污染性的有机溶剂, 同时, 环糊精本身无毒无害、价格低廉, 绿色环保, 更契合当今“绿色化学”的理念.环糊精参与的过渡金属催化体系中环糊精最重要的作用便是将底物包结于其“内疏水、外亲水”的空腔中, 这便可以做到在为底物提供一个疏水环境的同时, 让此包合物又溶于水中, 借此该包合物便可以顺利地在水相中实现有机合成转化.综上所述, 环糊精参与的过渡金属催化体系优点如下: (1)催化体系的反应速率和区域选择性有了显著提升; (2)可设计制备出新型的水溶性催化剂, 绿色环保; (3)环糊精与底物形成包合物, 有利于加强金属催化剂和底物之间的接触, 使底物更加易于反应.但由于环糊精本身是低聚糖类物质, 高温下易分解, 因此, 如何对环糊精进行合理的修饰才能使其更加稳定也是本领域面临的一大难题.今后该领域的关键是进一步研究环糊精参与的过渡金属催化有机反应的机理, 以便改进或调控催化性能, 加快环糊精参与的过渡金属催化体系的大规模工业化应用.因此希望在不久的将来能开发出更加活跃和更有选择性的催化系统.虽然仍有很长的路要走, 但探索基于环糊精参与的过渡金属催化剂合成的新方法、新技术以及催化新反应将前景光明.
莱恩, Jean, M., 沈兴海, 超分子化学:概念和展望, 北京大学出版社, 北京, 2002.Lai, E.; Jean, M.; Shen, X. H. M. Supramolecular Chemistry:Concepts and Prospects, Peking University Press, Beijing, 2002 (in Chinese).
Xia, D. H.; Jiang, S. J.; Li, L.-L.; Xiang, Y. Z.; Zhu, L. J. Chin. J. Chem. Eng. 2016, 24, 146. doi: 10.1016/j.cjche.2015.06.008
童林荟, 环糊精化学-基础与应用, 科学出版社, 北京, 2001.Tong, L. H. M. Cyclodextrin Chemistry-Basics and Applications, Science Press, Beijing, 2001 (in Chinese).
沈海民, 纪红兵, 有机化学, 2011, 32, 791. http://sioc-journal.cn/Jwk_yjhx//CN/abstract/abstract340916.shtmlShen, H. M.; Ji, H. B. Chin. J. Org. Chem. 2011, 32, 791(in Chinese), http://sioc-journal.cn/Jwk_yjhx//CN/abstract/abstract340916.shtml
Zhao, Y.; Huang, Y.; Zhu, H.; Zhu, Q.; Xia, Y. J. Am. Chem. Soc. 2016, 138, 16645. doi: 10.1021/jacs.6b07590
Menuel, S.; Léger, B.; Addad, A.; Monflier, E.; Hapiot, F. Green Chem. 2016, 18, 5500. doi: 10.1039/C6GC00770H
Stewart, M. E.; Anderton, C. R.; Thompson, L. B.; Maria, J.; Gray, S. K.; Rogers, J.A.; Nuzzo, R. G. Chem. Rev. 2008, 108, 494. doi: 10.1021/cr068126n
Xiao, J.; Qi, L. Nanoscale 2011, 3, 1383. doi: 10.1039/c0nr00814a
Shanmugam, M.; Kim, K. J. Electroanal. Chem. 2016, 776, 82. doi: 10.1016/j.jelechem.2016.06.009
Cravotto, G.; Gaudino, E. C.; Tagliapietra, S.; Carnaroglio, D.; Procopio, A. Green Proc. Synth. 2012, 1, 269.
Hein, J. E.; Tripp, J. C.; Krasnova, L. B.; Sharpless, K. B.; Fokin, V. V. Angew. Chem., Int. Ed. 2009, 48, 8018. doi: 10.1002/anie.v48:43
Dheer, D.; Rawal, R. K.; Singh, V.; Sangwan, P. L.; Das, P.; Shankar, R. Tetrahedron 2017, 73, 4295. doi: 10.1016/j.tet.2017.05.081
Patil, R. N.; Vijay Kumar, A. ACS Omega 2017, 2, 6405. doi: 10.1021/acsomega.7b00898
Messmer, E. Z. Phys. Chem. 1927, 126, 369.
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 113, 2056. doi: 10.1002/(ISSN)1521-3757
Krasinski, A.; Radic, Z.; Manetsch, R.; Raushel, J.; Taylor, P.; Sharpless, K. B.; Kolb, H. C. J. Am. Chem. Soc. 2004, 126, 12809. doi: 10.1021/ja046382g
Hein, J. E.; Tripp, J. P.; Krasnova, L. B.; Sharpless, K. B.; Fokin, V. V. Angew. Chem., Int. Ed. 2009, 48, 1. doi: 10.1002/anie.200890275
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596. doi: 10.1002/(ISSN)1521-3773
Tornoe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057. doi: 10.1021/jo011148j
Aprahamian, I.; Dichtel, W. R.; Ikeda, T.; Heath, J. R.; Stoddart, J. F. Org. Lett. 2007, 9, 1287. doi: 10.1021/ol070052u
Yigit, S.; Sanyal, R.; Sanyal, A. Chem. Asian J. 2011, 6, 2648. doi: 10.1002/asia.v6.10
Yamada, Y. M. A.; Sarkar, S. M.; Uozumi, Y. J. Am. Chem. Soc. 2012, 134, 9285. doi: 10.1021/ja3036543
Collinson, J.-M.; Wilton-Ely, J. D. E. T.; Diez-Gonzalez, S. Chem. Commun. 2013, 49, 11358. doi: 10.1039/c3cc44371j
Xiong, X.; Chen, H.; Tang, Z.; Jiang, Y. RSC Adv. 2014, 4, 9830. doi: 10.1039/c3ra45994b
White, J. R.; Price, G. J.; Schiffers, S.; Raithby, P. R.; Plucinski, P. K.; Frost, C. G. Tetrahedron Lett. 2010, 51, 3913. doi: 10.1016/j.tetlet.2010.05.104
Brotherton, W. S.; Michaels, H. A.; Simmons, J. T.; Clark, R. J.; Dalal, N. S.; Zhu, L. Org. Lett. 2009, 11, 4954. doi: 10.1021/ol9021113
Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302. doi: 10.1039/b904091a
Zhu, L.; Lynch, V. M.; Ansly, E. V. Tetrahedron 2004, 60, 7267. doi: 10.1016/j.tet.2004.06.079
Zhang, H.; Tanimoto, H.; Morimoto, T.; Nishiyama, Y.; Kakiuchi, K. Tetrahedron 2014, 70, 9828. doi: 10.1016/j.tet.2014.10.076
Ramesh, C.; Banerjee, J.; Pal, R.; Das, B. Adv. Synth. Catal. 2003, 345, 557. doi: 10.1002/adsc.200303022
Sheng, S. R.; Wang, Q. Y.; Ding, Y.; Liu, X. L.; Cai, M. Z. Catal Lett. 2009, 128, 418. doi: 10.1007/s10562-008-9767-z
Reddi, M. N. K.; Satheesh, K. B.; Anil, K. M.; Arulselvan, P.; Ibrahim, K. S.; Lasekan, O. Molecules 2012, 17, 7543. doi: 10.3390/molecules17067543
Ramesh, C.; Banerjee, J.; Pal, R.; Das, B. ChemInform 2010, 345, 557. https://www.researchgate.net/publication/230007928_Silica_Supported_Sodium_Hydrogen_Sulfate_and_Amberlyst-15_Two_Efficient_Heterogeneous_Catalysts_for_Facile_Synthesis_of_Bis-_and_Tris1H-indol-3-ylmethanes_from_Indoles_and_Carbonyl_Compounds1
Dabbawala, A. A.; Sudheesh, N.; Bajaj, H. C. Dalton. Trans. 2012, 41, 2910. doi: 10.1039/c2dt11924b
Datta, K. K. R.; Srinivasan, B.; Balaram, H.; Eswaramoorthy, M. J Chem. Sci. 2008, 120, 579. doi: 10.1007/s12039-008-0088-y
Nie, R.; Sang, R.; Ma, X.; Zheng, Y.; Cheng, X.; Li, W.; Wu, Y. J. Catal. 2016, 344, 286. doi: 10.1016/j.jcat.2016.09.022
Yao, Z.; Hong, S.; Zhang, W.; Liu, M.; Deng, W. Tetrahedron Lett. 2016, 57, 910. doi: 10.1016/j.tetlet.2016.01.049
Zhang, P.; Meijide, S. J.; Driant, T.; Derat, E.; Zhang, Y.; Ménand, M. Angew. Chem. 2017, 129, 10961. doi: 10.1002/ange.201705303
(a) Kaboudin, B.; Abedi, Y.; Yokomatsu, T. Eur. J. Org. Chem. 2011, 6656.
(b) Kaboudin, B.; Abedi, Y.; Yokomatsu, T. Org. Biomol. Chem. 2012, 10, 4543.
Kaboudin, B.; Mostafalu, R.; Yokomatsu, T. ChemInform 2013, 44, 2262.
Perez, A. L.; Moseguer, J. O.; Marques, P. R.; Corma, A. Angew. Chem., Int. Ed. 2013, 125, 11768. doi: 10.1002/ange.201303188
Hoffmann, I.; Blumenröder, B.; Thumann, S. O. N.; Dommer, S.; Schatz, J. Green Chem. 2015, 17, 3844. doi: 10.1039/C5GC00794A
Saito, N.; Taniguchi, T.; Hoshiya, N.; Shuto, S.; Arisawa, M.; Sato, Y. Green Chem. 2015, 17, 2358. doi: 10.1039/C4GC02469A
(a) Zhong, R.; Pöthig, A.; Feng, Y.; Riener, K.; Herrmann, W. A.; Kühn, F. E. Green Chem. 2014, 16, 4955.
(b) Billingsley, K.; Buchwald, S. L. J. Am. Chem. Soc. 2007, 38, 3358.
(c) Old, D. W.; Wolfe, J. P.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 30, 4369.
(d) Martin R.; Buchwald, S. L. Acc. Chem. Res. 2008, 41, 1461.
(e) Vellakkaran, M.; Andappan, M. M. S.; Kommu, N. Green Chem. 2014, 16, 2788.
Raihana, I. K.; Kasi, P. Green Chem. 2016, 18, 4791. doi: 10.1039/C6GC90091G
Qi, M.; Tan, P. Z. Xue, F.; Malhi, H. S.; Zhang, Z. X.; Young, D. J. Rsc. Adv. 2014, 5, 3590.
Zhou, X.; Guo, X.; Jian, F.; Wei, G. ACS Omega 2018, 3, 4418. doi: 10.1021/acsomega.8b00469
Guo, Y.; Li, J.; Zhao, F.; Lan, G.; Li, L.; Liu, Y.; Yang, R. RSC Adv. 2016, 6, 7950. doi: 10.1039/C5RA23271F
Imran, K. R.; Pitchumani, K. ACS Sustainable Chem. Eng, 2018.
Poulos, T. L. Chem. Rev. 2014, 114, 3919. doi: 10.1021/cr400415k
Sreenilayam, G.; Fasan, R. Chem. Commun. 2015, 51, 1532. doi: 10.1039/C4CC08753D
Xu, X.; Li, C.; Tao, Z.; Pan, Y. Adv. Synth. Catal. 2015, 357, 3341. doi: 10.1002/adsc.201500418
Wang, M. L.; Fang, G. D; Liu, P. Appl. Catal., B 2016, 188, 113. doi: 10.1016/j.apcatb.2016.01.071
Guo, Y.; Guo, S.; Ren, J.; Zhai, Y.; Dong, S.; Wang, E. ACS Nano. 2010, 4, 4001. doi: 10.1021/nn100939n
Sharavath, V.; Sarkar, S.; Gandla, D.; Ghosh, S. Electrochim. Acta 2016, 210, 385. doi: 10.1016/j.electacta.2016.05.177
Mohamed, M. A.; Shukla, A.; Sandhya, K. Y. Environ. Prog. Sustainable 2016, 35, 1283. doi: 10.1002/ep.v35.5
Subramanian, R.; Ponnusamy, V. J. Mater. Sci.: Mater. Electron. 2016, 28, 1.
Sun, N.; Wang, T.; Liu, C. Wood. Sci. Tenol. 2016, 50, 1. doi: 10.1007/s00226-015-0797-6

图 1 环糊精的分子结构(a)和三维结构示意图(b)
Figure 1 Molecular structure (a) and three-dimentional structure diagram (b) of cyclodextrin

图式 1 树枝状纳米金的形成原理图
Scheme 1 Schematic representation of the mechanism for the formation of 3D Au-NDs

图式 2 CuI(碘化亚铜)/β-CD催化合成5-碘-1, 2, 3-苯并三唑、2-碘咪唑吡嗪和2-碘苯咪唑噻唑
Scheme 2 CuI/β-CD-catalyzed regioselective synthesis of 5-iodo-1, 2, 3-triazoles, 2-iodoimidazopyridines, and 2-iodobenzo- imidazothiazoles

图式 4 Ad-L-PdCl2@β-CD催化的Suzuki-Miyaura耦合反应
Scheme 4 The Suzuki-Miyaura coupling reaction catalyzed by Ad-L-PdCl2@β-CD

图式 5 环糊精协助血红素催化的α-重氮酯与芳香胺的N—H键插入反应机理
Scheme 5 Mechanism of hemin-catalyzed, cyclodextrin-as- sisted N—H insertion reaction of α-diazo ester into aromatic amines

图式 6 Fe3O4@β-CD的制备过程和降解过程原理图
Scheme 6 Preparation process of Fe3O4@β-CD and schematic diagram of degradation process

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





 下载:
下载:




 下载:
下载: