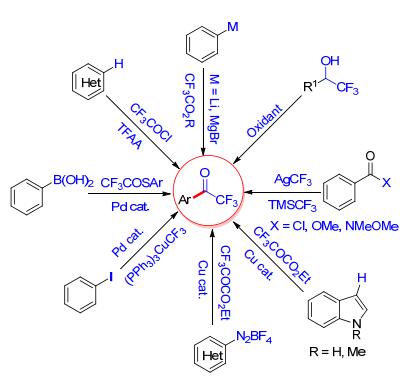
图式 1.
芳基三氟甲基酮化合物的合成
Scheme 1.
Synthesis of aryl trifluoromethyl ketones

有机氟化物因其具有特殊的物理、化学性质及生物活性, 被广泛应用于化工、材料、农药和医药等领域[1~3].随着人们对有机氟化合物的需求量不断增加, 选择性地向有机分子中引入氟原子或者含氟基团, 一直是化学家们十分感兴趣的课题.
芳基三氟甲基酮化合物是一类非常重要的含氟有机化合物, 其三氟甲基的强吸电子作用使羰基的碳具有较强的亲电性, 而羰基的氧具有较弱的亲核性.该类化合物不仅是有机合成中的重要中间体, 可以合成含三氟甲基的杂环化合物、氟醇类化合物等其它含氟化合物; 而且可以作为药物合成的催化剂以及酶的抑制剂.因此, 化学家们一直在不断寻求合成芳基三氟甲基酮化合物的新方法.
目前, 文献报道芳基三氟甲基酮化合物的合成方法有很多(Scheme 1)[4, 5].金属有机试剂与三氟乙酸衍生物的反应, 富电子芳烃的Friedel-Crafts酰基化反应, α-三氟甲基醇的氧化以及与羧酸衍生物的三氟甲基化反应是合成芳基三氟甲基酮化合物的经典的方法.另外, 利用过渡金属催化芳基硼酸、卤化物及重氮盐的三氟乙酰化反应是合成该类化合物的一种新的化学转化.本文将主要介绍近年来芳基三氟甲基酮化合物的合成研究进展, 并对相关反应的机理进行探讨.
α-三氟甲基醇原料廉价易得, 其制备方法也很多, 主要由卤代三氟甲基化合物与醛的还原偶联[6], 以及TMSCF3试剂与醛的亲核-加成反应得到[7].利用不同氧化剂, 可以将α-三氟甲基醇直接、高效地氧化成三氟甲基酮化合物.虽然该方法具有很多优点, 如底物的范围广、反应条件简单, 然而, 该方法也存在一定的问题, 除了需要预先合成相应的醇, 在强氧化条件下, 官能团的耐受性也较差.因此, 化学家们探索了一系列的氧化反应体系(表 1), 以期实现α-三氟甲基醇的高效氧化.
 下载:
导出CSV
下载:
导出CSV
| Entry | Substrate | Oxidant | Conditions and yields | Ref. |
| 1 |  |
 |
3.7 equiv. Dess-Martin reagent, DCM, r.t., 15 min, 75%~95% | [8, 9] |
| 2 |  |
CsSO4F | 1.2 or 2.3 equiv. CsSO4F MeCN, Ar, 30~35 ℃, 44% | [10] |
| 3 |  |
 |
2.5 equiv. AcNH-TEMPO, 2.25 equiv. 2, 6-lutidine, DCM, r.t., 71%~99% | [11] |
| 4 |  |
 |
2.0 equiv. IBX, EtOAc, reflux, 9~30 h, 94%~99% | [12] |
| 5 |  |
Oxone | 5 mol% Organoiodine, 0.9 equiv. oxone, MeCN, 90 ℃, 18 h, 44%~86% | [13] |
| 6 |  |
 |
1.1 equiv. Bi complex, 1.1 equiv. DBU or TMG, CDCl3 or C6D6, r.t., 95% | [14, 15] |
| 7 |  |
O2 or Air | 5 mol% CuCl, 5 mol% Phen, 5 mol% DBADH2, 2. equiv. K2CO3, PhMe, 70~90 ℃, 97% | [16, 17] |
| 8 |  |
NaIO4 and Air |  |
[18] |
| 9 |  |
O2 |  |
[19] |
| 10 |  |
t-BuOOH (TBHP) | 5 mol% MOF, 4.0 equiv. TBHP, PhMe, r.t., 6%~99% | [20] |
| 11 |  |
 |
4c-substituted TEMPO (5 mol%), 1.5~2.0 equiv. Py-HBr3, DCM-NaHCO3, r.t., 70% | [21] |
| 12 |  |
 |
keto-ABNO (5 mol%), NaNO2 (10 mol%), AcOH (0.5 mol/L), O2 (101 kPa), r.t, 18 h, 31%~97% | [22] |
20世纪80年代, Linderman课题组[8, 9]第一次报道了利用Dess-Martin试剂将α-三氟甲基醇直接氧化成三氟甲基酮化合物.一系列含芳基、烷基、苄基、烯基、炔基的α-三氟甲基醇都能得到收率较高的目标产物, 而且官能团的耐受性好.然而, 该反应需要3.7 equiv.的氧化剂, 才能极大地缩短反应时间, 反应仅需15 min, 并能有效地提高反应的转化率.
随后, 氟氧代硫酸铯(CsSO4F)[10]、N-氧铵盐(AcNH-TEMPO)[11]、2-碘酰基苯甲酸(IBX)[12]、有机高价碘[13]、三芳基二氯化铋[14, 15]等化合物也被应用于该反应.尽管这些反应的效率不错, 但也需要加入当量的氧化剂.
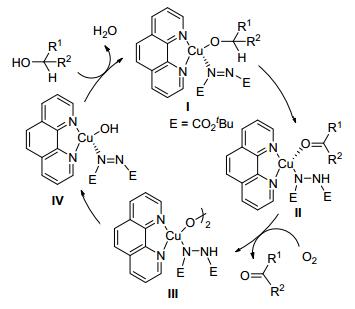
近年来, 过渡金属催化醇的氧化得到快速的发展, 尤其是以空气或氧气为终端氧化剂, 由于具有价廉、清洁等优点而备受人们青睐. 1996年, Markó课题组[16, 17]报道了氯化亚铜/邻菲罗啉的催化反应体系, 以氧气或空气为氧化剂, 能得到产率为97%的目标产物, 水是反应中唯一的副产物.他们发现该反应中加入催化量的偶氮化合物(如偶氮二甲酸二叔丁酯, DBAD)或相应的肼类化合物(DBADH2), 能极大地提高催化剂的转化量和寿命, 以及反应的速率.
该反应可能的机理(Scheme 2)如下:首先铜配合物(Ⅰ)中分子内的氢转移, 可以得到铜-羰基配位的中间体(Ⅱ); 在有氧的条件下, 该中间体(Ⅱ)被氧化得到双核的铜过氧化物(Ⅲ); 紧接着氢原子被攫取, 发生分子内均裂反应得到铜-羟基的物种(Ⅳ), 最后配合物中的羟基与醇分子发生快速的配体交换, 又得到铜配合物(Ⅰ)以及副产物水, 实现了催化循环.
2000年, Bégué课题组[18]报道了以当量的高碘酸钠和空气为氧化剂, 利用钌配合物催化醇的氧化反应, 得到了较高产率的三氟甲基酮化合物.随后, Pedro课题组[19]报道了以氧气为氧化剂, 利用钴配合物催化α-三氟甲基-α-羟基羧酸的氧化脱羧反应.然而, 该反应需要加入当量的特戊醛, 在一定程度上影响产物的分离提纯.最近, Wright课题组[20]报道了金属有机框架物(MOFs)催化的α-三氟甲基醇的氧化反应.而且, 他们发现在过氧叔丁醇氧化剂条件下, MIL-100 (Sc, Fe)催化剂既具有路易斯酸的性能、又具有氧化催化的特点, 能有效地催化杂环芳烃与半缩醛反应得到相应的酮.
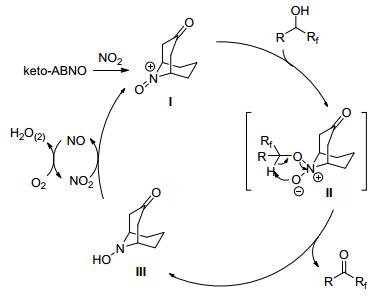
氮氧自由基作为温和的氧化催化剂, 在氧化反应中的应用越来越受到化学家们的重视.该类催化体系具有易操作、反应条件温和、选择性好及转化率高的特点, 其中最常见的是2, 2, 6, 6-四甲基哌啶-1-氧自由基(TEMPO). 2008年, Inokuchi课题组[21]报道了TEMPO衍生物催化α-三氟甲基醇的氧化反应.他们发现该反应利用当量的氢溴酸吡啶过溴化物(Py-HBr3)作为共氧化剂, 能更好地将仲醇氧化为相应的酮.另外, 对于缺电子醇的氧化, C-4位缺电子基团取代的TEMPO衍生物能极大地提高其反应的活性.最近, Kanai课题组[22]报道了以氧气为氧化剂, 利用keto-ABNO催化α-氟烷基醇的氧化反应.该反应具有条件温和、底物范围广、官能团耐受性好以及高原子经济性等优点.
该反应可能的机理(Scheme 3)如下:首先, 氮氧自由基被二氧化氮氧化为具有较高氧化活性的N-氧铵阳离子(Ⅰ); 接下来, α-氟烷基醇与N-氧铵阳离子(Ⅰ)反应, 得到中间体Ⅱ; 醇很快被氧化为酮, 同时产生还原性催化剂Ⅲ; 最后又被二氧化氮氧化, 实现催化循环.
羧酸衍生物的亲核取代反应是基础有机合成中最基本的化学转化. 1979年, Kobayashi课题组[23]首次报道了苯甲酰溴与三氟甲基铜的亲核三氟甲基化反应.虽然该反应能够得到苯基三氟甲基酮, 但产率相对较低, 且伴随苯甲酰氟的副产物. 1991年, Prakash课题组[24]利用四丁基氟化铵(TBAF)促进TMSCF3与苯甲酰氯反应, 可以得到苯基三氟甲基酮产物, 但过量的TMSCF3试剂会进一步与酮反应, 生成硅醇化合物.随后, Naumann课题组[25, 26]探索了三氟甲基金属试剂(如Cd(CF3)2•Glyme和AgCF3)作为亲核试剂与苯甲酰氯反应.尽管也有少量的副产物产生, 但该反应条件温和, 目标产物的产率相对较高, 且底物的适用范围广.然而, 这些三氟甲基金属试剂对空气和水敏感, 要临时或原位反应制备. 2005年, Cai课题组[27]报道了以三氟乙酸钠为三氟甲基化试剂, 以DMF为溶剂, 利用碘化亚铜催化该反应, 能得到产率为80%的苯甲酰氯三氟甲基化产物.值得注意的是, 该反应的后处理非常关键.如果用盐酸水溶液进行处理, 则水解得到三氟甲基醇、双-三氟甲基醇的混合物.
除了酰氯, 羧酸酯也被用来作为底物参与此类反应. 1997年, Mochida课题组[28]报道了三乙基锗钠(Et3- GeNa)促进羧酸酯与三氟甲基苯硫醚(PhSCF3)的反应, 高效地合成芳基三氟甲基酮化合物.有意思的是, 当环己基酯、异丙基酯、叔丁基酯与甲基酯化合物共存时, 只有甲基酯化合物能发生该化学转化.这些结果表明该反应具有很好的化学选择性.随后, Prakash等分别报道了以TMSCF3为三氟甲基化试剂, 利用TBAF[29], CsF[30]以及AcONBu4[31]催化羧酸酯的三氟甲基化反应.这些反应具有反应条件温和、底物适用范围广及产率高等优点.最近, Prakash课题组[32]报道了在双(三甲基硅烷基)氨基钾(KHMDS)的强碱性条件, 以三氟甲烷(CF3H)为亲核性三氟甲基化试剂, 实现了苯甲酸甲酯的三氟甲基化反应.尽管反应产率较低, 但三氟甲烷作为亲核试剂是有机合成中重大突破性进展.
与酰氯、羧酸酯相比, 酰胺的反应活性相对较低. 2012年, Leadbeater课题组[33]首次报道了酰胺的亲核三氟甲基化反应.他们利用氟化铯促进Weinreb酰胺与TMSCF3反应, 合成了一系列产率较高的芳基三氟甲基酮化合物.该反应体系并没有发现双-三氟甲基醇等副产物产生, 只是位阻较大的酰胺不适合反应, 没有得到相应的酮(表 2).其他羧酸衍生物的三氟甲基化也有零星的报道, 如氟化钾促进的N-甲基靛红酸酐脱二氧化碳的亲核三氟甲基化[34]; TBAF促进氧氮杂萘酮亲核开环的亲核三氟甲基化, 得到邻氨基-2, 2, 2-三氟苯乙酮化合物[35].
下载:
导出CSV
| Entry | Carboxylic acid derivative | Trifluoromethylation reagent | Conditions and Yields | Ref. |
| 1 | PhCOBr | CF3Cu | 45 ℃, 24 h, 24% | [23] |
| 2 | PhCOCl | TMSCF3 | TBAF, THF, r.t., mixture products | [24] |
| 3 | ArCOCl | Cd(CF3)2•Glyme | DMAP, DCM, 0 ℃~r.t., 12 h, 57%~85% | [25] |
| 4 | ArCOCl | AgCF3 | Method A: EtCN, -30 ℃~r.t., 12 h, 50%~77% Method B: DMAP, DCM, -30 ℃~r.t., 12 h, 50%~78% |
[26] |
| 5 | PhCOCl | CF3COONa | CuI, DMF, 80% | [27] |
| 6 | RCO2Me (R=Ar, Alkyl) | C6H5SCF3 | 1.1 equiv. Et3GeNa, THF/HMPA, -60 ℃, 1.5 h, 93%~98% | [28] |
| 7 | RCO2Me (R=Ar, Alkyl, Alkenyl, etc.) | TMSCF3 | 2.5 mol% TBAF, PhMe or DCM, or pentane, -78 ℃~r.t., 68%~95% | [29] |
| 8 | RCO2R' (R=Ar, Alkyl, Alkenyl, Alkynyl, etc; R'=Me, Et) | TMSCF3 | 1.0 mol% CsF, THF, r.t., 1~4 h, 84%~90% | [30] |
| 9 | RCO2R' (R=Ar, Alkyl, Alkenyl, etc.; R'=Me, Et, iPr) | TMSCF3 | 5.0 mol% AcONBu4, 0 ℃~r.t., 1 h, 64%~90% | [31] |
| 10 | PhCO2Me | CF3H | KHMDS, THF, 30% | [32] |
| 11 |  |
TMSCF3 | (1) 0.2 equiv. CsF, PhMe, r.t., 24 h (2) TBAF, H2O, 50 ℃, 2h, 48%~99% | [33] |
| 12 |  |
TMSCF3 | 0.3 equiv. KF, 0.3 equiv. TBAB, DMF, 0~r.t., 16 h, 64%~90% | [34] |
| 13 |  |
TMSCF3 | 0.1 equiv. TBAF, DMSO, r.t., 13%~57% | [35] |
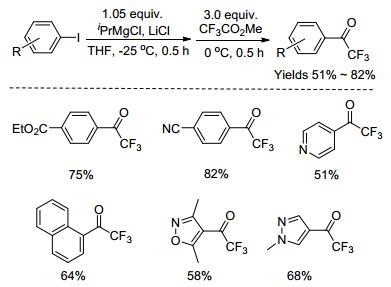
金属有机试剂(芳基锂试剂、芳基格氏试剂)与三氟乙酸及其衍生物(三氟乙酰氯、三氟乙酸酐及三氟乙酸酯)的亲电三氟乙酰化反应是合成芳基三氟甲基酮最经典的方法[36~41].尽管该方法要求严格的无水无氧条件, 但该反应的产率较高, 且原料廉价易得.因此, 该方法无论是在学术还是工业生产上都被经常使用.最近, Funabiki课题组[42]报道了以芳基碘化物为起始原料, 与异丙基氯化镁-氯化锂原位制备芳基格氏试剂, 再与三氟乙酸甲酯“一锅”反应, 得到相应的芳基三氟甲基酮化合物(Scheme 4).该反应具有条件温和、底物范围广、官能团耐受性好及产率高等优点.
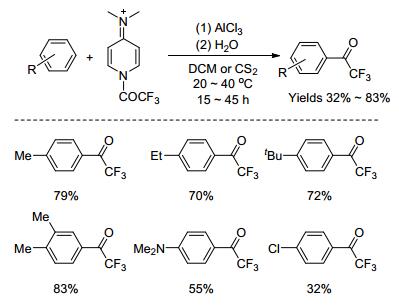
路易斯酸催化富电子芳烃的Friedel-Crafts酰基化反应也是合成芳基三氟甲基酮最常用的方法. 1996年, Simchen课题组[43]报道了三氯化铝催化富电子芳烃三氟乙酰基化反应, 得到一系列芳基三氟甲基酮化合物(Scheme 5).该反应体系用三氟乙酸酐作为酰化试剂, 活性不够.因此, 反应需要由4-N, N-二甲基吡啶(DMAP)与三氟乙酸酐制备活性较高的三氟乙酰化试剂. 2004年, Stalick课题组[44]报道了二乙基氯化铝催化吲哚与三氟乙酸酐的酰基化反应, 得到了区域选择性产率为61%的3-位酰基化产物.
2014年, Ma课题组[45]报道了铜催化吲哚与三氟乙胺、亚硝酸钠的三组分反应, 合成了一系列三氟甲基酮肟化合物, 再经后续的反应可以得到3-(三氟乙酰)吲哚衍生物化合物(Scheme 6).然而, 这种多步反应的条件较为复杂, 官能团的耐受性差, 产率也不高.
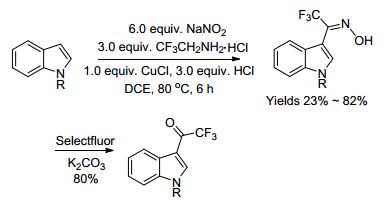
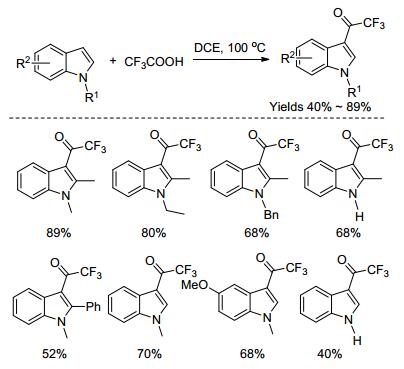
2016年, Guan课题组[46]报道了在无过渡金属催化剂及任何添加剂的条件下, 以三氟乙酸为酰化试剂, 实现了吲哚3-位的三氟乙酰化反应, 合成了各种3-(氟乙酰)吲哚衍生物化合物(Scheme 7).然而, 这些反应都是在强酸条件下进行, 官能团的兼容性不好, 产物的产率也不够高.
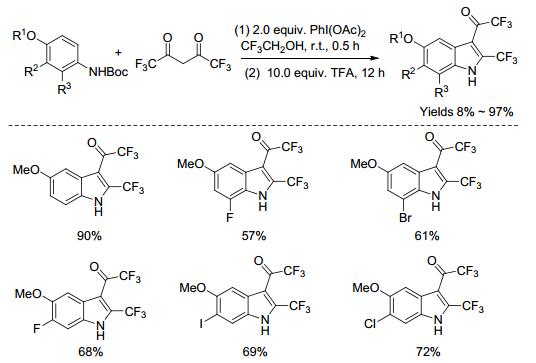
随后, Njardarson课题组[47]通过氧化去芳构化的策略, 利用醋酸碘苯促进苯胺的衍生物与六氟乙酰丙酮的氧化反应, 高效地合成了3-(氟乙酰)吲哚衍生物化合物(Scheme 8).该反应具有条件温和, 官能团耐受性好, 底物范围广, 产率高, 批量合成, 且不需过渡金属催化剂等特点.反应可能的机理(Scheme 9)如下:在氧化剂条件下, 苯胺衍生物去芳构化, 得到中间体Ⅰ, 继而与六氟乙酰丙酮发生加成反应得到中间体Ⅱ, 重新芳构化产生化合物Ⅲ, 再发生分子内亲核加成反应, 形成化合物Ⅳ, 最后消除一分子水, 以及脱去保护基, 得到目标产物.
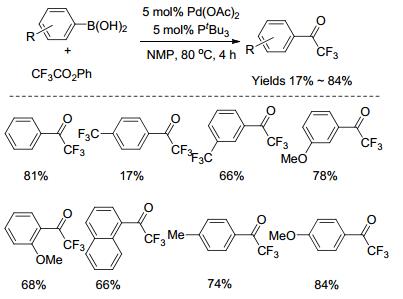
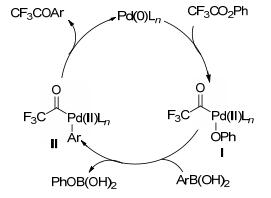
过渡金属催化的交叉偶联反应是现代有机合成的重要工具, 利用此类反应可高效、高选择性地构建碳-碳及碳-杂键等化学键. 2001年, Yamamoto课题组[48]报道了钯催化芳基硼酸与三氟乙酸苯酯的偶联反应, 合成了一系列的芳基三氟甲基酮化合物(Scheme 10).尽管该反应产率较高, 但官能团的耐受性比较差, 只能适用于甲基、三氟甲基、甲氧基等.另外, 底物的适用范围不广, 杂环芳基硼酸都不发生反应.反应可能的机理(Scheme 11)如下:首先Pd(0)与三氟乙酸苯酯发生氧化加成得到Pd(Ⅱ)的中间体Ⅰ, 再与芳基硼酸发生转金属, 形成中间体Ⅱ, 最后还原消除得到目标产物以及Pd(0), 实现催化循环.
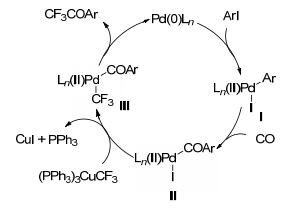
2016年, Wu课题组[49]报道了钯催化芳基碘化物与三氟甲基铜、一氧化碳的羰基化偶联反应, 能高效地合成芳基三氟甲基酮化合物(Scheme 12).该反应具有条件温和, 底物范围广, 官能团耐受性好, 产率高等优点.反应可能的机理(Scheme 13)如下:首先Pd(0)与芳基碘化物发生氧化加成得到ArPd(Ⅱ)的中间体(Ⅰ), 与一氧化碳发生迁移插入, 形成中间体(Ⅱ), 再与三氟甲基铜发生转金属得到中间体(Ⅲ), 最后还原消除得到目标产物以及Pd(0), 实现催化循环.
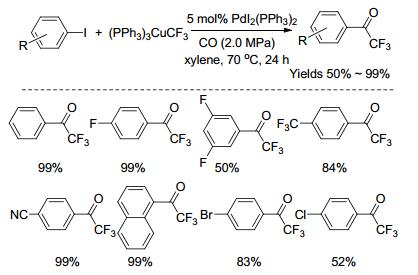
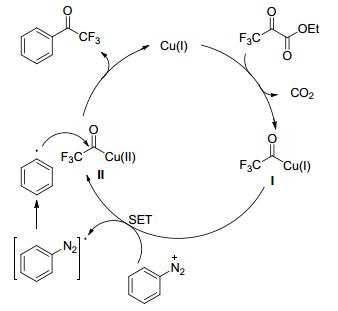
2016年, Weng课题组[50]报道了铜催化芳基重氮盐与三氟丙酮酸乙酯的三氟乙酰化反应(Scheme 14).与三氟乙酰氯、三氟乙酸酐相比, 三氟丙酮酸乙酯相对稳定, 反应体系无需严格无水的条件.该反应的官能团耐受性强, 主要包括酯基、酮羰基、羟基、卤基(氟、氯、溴、碘)、三氟甲基、甲氧基、烷基等.另外, 反应的条件温和, 原料易得, 操作步骤简单, 收率较高, 且对水和空气不敏感, 便于工业化生产.尽管确切的反应机理还不是很清楚, 但他们也提出了可能的机理(Scheme 15).首先, 三氟丙酮酸乙酯与Cu(Ⅰ)催化剂反应, 得到酰基-Cu(Ⅰ)的物种(Ⅰ), 并放出二氧化碳气体; 接下来, 酰基-Cu(Ⅰ)的物种(Ⅰ)与芳基重氮盐之间发生单电子转移, 形成酰基-Cu(Ⅱ)的物种(Ⅱ)以及芳基重氮自由基, 而芳基重氮自由基不稳定, 会释放出氮气, 得到芳基自由基; 最后, 芳基自由基与酰基-Cu(Ⅱ)的物种(Ⅱ)反应, 得到芳基三氟甲基酮化合物, 并重新产生Cu(Ⅰ)催化剂, 实现催化循环.
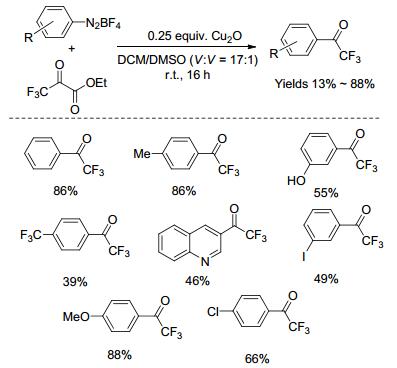
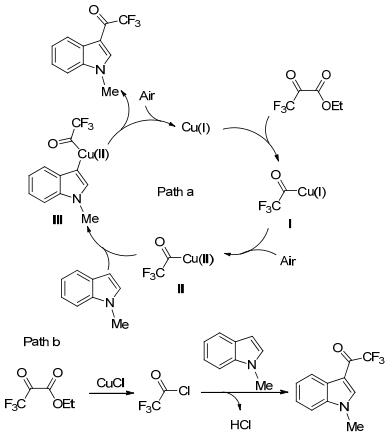
随后, 我们课题组[51]以三氟丙酮酸乙酯为三氟乙酰化试剂, 利用氯化亚铜促进了吲哚衍生物3-位的三氟乙酰化反应(Scheme 16).尽管大部分反应的产率较高, 但对于有硝基、氰基、乙酰基、对甲苯磺酰基等强吸电子官能团, 以及吲哚4-位上有位阻的底物, 几乎得不到目标产物.另外, 对于吲哚氮原子上没有保护基团的底物, 在该反应条件下, 也能得到中等产率的产物.因此, 该方法也是合成3-(氟乙酰)吲哚衍生物的一种重要补充.反应的机理有两种可能(Scheme 17):一方面, 三氟丙酮酸乙酯与Cu(Ⅰ)催化剂反应, 得到酰基-Cu(Ⅰ)的物种(Ⅰ), 在空气条件下, 被氧化成酰基-Cu(Ⅱ)的物种(Ⅱ), 再与吲哚反应, 得到中间体Ⅲ, 最后还原消除得到目标产物; 另一方面, 三氟丙酮酸乙酯与CuCl反应, 得到三氟乙酰氯, 再与吲哚发生Friedel-Crafts酰基化反应, 得到目标产物.

综上所述, 近年来有机化学工作者探索了各种三氟甲基试剂、三氟乙基试剂, 为合成芳基三氟甲基酮提供了新的方法学.在过渡金属催化的研究领域中, 发展稳定的、廉价易得的、高效的三氟甲基或三氟乙基试剂以及探索环境友好的催化剂体系是未来发展的方向.
Prakash, S. G.; Hu, J, B.; Alauddin, M, M.; Conti, P, S.; Olah, G, A. J. Fluorine Chem. 2003, 121, 239. doi: 10.1016/S0022-1139(03)00039-3
Wang, J. J.; Li, F.; Xu, Y.; Wang, J.; Wu, Z. Y.; Yang, C. Y.; Liu, L. T. Chin. J. Org. Chem. 2018, 38, 1155. doi: 10.6023/cjoc201709049
Zeng, Y. W.; Ni, C. F.; Hu, J. B. Chem.-Eur. J. 2016, 22, 3210. doi: 10.1002/chem.201504036
Kelly, C. B.; Mercadante, M. A.; Leadbeater, N. E. Chem. Commun. 2013, 49, 11133. doi: 10.1039/c3cc46266h
Wu, W.; Weng, Z. Q. Synthesis 2018, 50, 1958. doi: 10.1055/s-0036-1591971
Gassman, P. G.; O'Reilly, N. J. J. Org. Chem. 1987, 52, 2481. doi: 10.1021/jo00388a025
Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757. doi: 10.1021/cr9408991
Linderman, R. J.; Graves, D. M. Tetrahedron Lett. 1987, 28, 4259. doi: 10.1016/S0040-4039(00)96479-7
Linderman, R. J.; Graves, D. M. J. Org. Chem. 1989, 54, 661. doi: 10.1021/jo00264a029
Stavber, S.; KoŠir, I.; Zupan, M. J. Org. Chem. 1997, 62, 4916. doi: 10.1021/jo962424a
Kelly, C. B.; Mercadante, M. A.; Hamlin, T. A.; Fletcher, M. H.; Leadbeater, N. E. J. Org. Chem. 2012, 77, 8131. doi: 10.1021/jo301477s
Cheng, H.; Pei, Y.; Leng, F.; Li, J.; Liang, A.; Zou, D.; Wu, Y.; Wu, Y. Tetrahedron Lett. 2013, 54, 4483. doi: 10.1016/j.tetlet.2013.06.045
Tanaka, Y.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2012, 137, 99. doi: 10.1016/j.jfluchem.2012.03.002
Matano, Y.; Hisanaga, T.; Yamada, H.; Kusakabe, S.; Nomura, H.; Imahori, H. J. Org. Chem. 2004, 69, 8676. doi: 10.1021/jo0485740
Matano, Y.; Suzuki, T.; Iwata, T.; Shinokura, T.; Imahori, H. Bull. Chem. Soc. Jpn. 2008, 81, 1621. doi: 10.1246/bcsj.81.1621
Markó, I. E.; Giles, P. R.; Tsukazaki, M.; Brown, S. M.; Urch, C. J. Science 1996, 274, 2044. doi: 10.1126/science.274.5295.2044
Markó, I. E.; Giles, P. R.; Tsukazaki, M.; Chellé-Regnaut, I.; Gautier, A.; Brown, S. M.; Urch, C. J. J. Org. Chem. 1999, 64, 2433. doi: 10.1021/jo982239s
Kesavan, V.; Bonnet-Delpon, D.; Bégué, J.-P.; Srikanth, A.; Chandrasekaran, S. Tetrahedron Lett. 2000, 41, 3327. doi: 10.1016/S0040-4039(00)00378-6
Blay, G.; Fernández, I.; Marco-Aleixandre, A.; Monje, B.; Pedro, J. R.; Ruiz, R. Tetrahedron 2002, 58, 8565. doi: 10.1016/S0040-4020(02)00947-X
Mitchell, L.; Williamson, P.; Ehrlichová, B.; Anderson, A. E.; Seymour, V. R.; Ashbrook, S. E.; Acerbi, N.; Daniels, L. M.; Walton, R. I.; Clarke, M. L.; Wright, P. A. Chem. Eur. J. 2014, 20, 17185. doi: 10.1002/chem.201404377
Mei, Z.-W.; Omote, T.; Mansour, M.; Kawafuchi, H.; Takaguchi, Y.; Jutand, A.; Tsuboi, S.; Inokuchi, T. Tetrahedron 2008, 64, 10761. doi: 10.1016/j.tet.2008.08.051
Kadoh, Y.; Tashiro, M.; Oisaki, K.; Kanai, M. Adv. Synth. Catal. 2015, 357, 2193. doi: 10.1002/adsc.v357.10
Kobayashi, Y.; Yamamoto, K.; Kumadaki, I. Tetrahedron Lett. 1979, 20, 4071. doi: 10.1016/S0040-4039(01)86506-0
Krishnamurti, R.; Bellew, D. R.; Prakash, G. K. S. J. Org. Chem. 1991, 56, 984. doi: 10.1021/jo00003a017
Naumann, D.; Finke, M.; Lange, H.; Dukat, W.; Tyrra, W. J. Fluorine Chem. 1992, 56, 215. doi: 10.1016/S0022-1139(00)81104-5
Kremlev, M. M.; Mushta, A. I.; Tyrra, W.; Naumann, D.; Fischer, H. T. M.; Yagupolskii, Y. L. J. Fluorine Chem. 2007, 128, 1385. doi: 10.1016/j.jfluchem.2007.06.009
Chang, Y.; Cai, C. J. Fluorine Chem. 2005, 126, 937. doi: 10.1016/j.jfluchem.2005.04.012
Yokoyama, Y.; Mochida, K. Synlett 1997, 907.
Wiedemann, J.; Heiner, T.; Mloston, G.; Prakash, G. K. S.; Olah, G. A. Angew. Chem., Int. Ed. 1998, 37, 820. doi: 10.1002/(ISSN)1521-3773
Singh, R. P.; Cao, G.; Kirchmeier, R. L.; Shreeve, J. M. J. Org. Chem. 1999, 64, 2873. doi: 10.1021/jo982494c
Kawano, Y.; Kaneko, N.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2006, 79, 1133. doi: 10.1246/bcsj.79.1133
Prakash, G. K. S.; Jog, P. V.; Batamack, P. T. D.; Olah, G. A. Science 2012, 338, 1324. doi: 10.1126/science.1227859
Rudzinski, D. M.; Kelly, C. B.; Leadbeater, N. E. Chem. Commun. 2012, 48, 9610. doi: 10.1039/c2cc35037h
Shidlovskii, A. F.; Golubev, A. S.; Gusev, D. V.; Suponitsky, K. Y.; Peregudov, A. S.; Chkanikov, N. D. J. Fluorine Chem. 2012, 143, 272. doi: 10.1016/j.jfluchem.2012.07.002
Allendörfer, N.; Es-Sayed, M.; Nieger, M.; Bräse, S. Tetrahedron Lett. 2012, 53, 388. doi: 10.1016/j.tetlet.2011.11.014
McGrath, T. F.; Levine, R. J. Am. Chem. Soc. 1955, 77, 3656. doi: 10.1021/ja01618a075
Chen, L. S.; Chen, G. J.; Tamborski, C. J. Fluorine Chem. 1981, 18, 117. doi: 10.1016/S0022-1139(00)82308-8
Chen, L. S.; Chen, G. J.; Tamborski, C. J. Organomet. Chem. 1983, 251, 139. doi: 10.1016/S0022-328X(00)99496-8
Creary, X. J. Org. Chem. 1987, 52, 5026. doi: 10.1021/jo00231a036
Kerdesky, F. A. J.; Basha, A. Tetrahedron Lett. 1991, 32, 2003. doi: 10.1016/S0040-4039(00)78892-7
Yamazaki, T.; Terajima, T.; Kawasaki-Taskasuka, T. Tetrahedron 2008, 64, 2419. doi: 10.1016/j.tet.2008.01.015
Funabiki, K.; Hayakawa, A.; Inuzuka, T. Org. Biomol. Chem. 2018, 16, 913. doi: 10.1039/C7OB02862H
Simchen, G.; Schmidt, A. Synthesis 1996, 1093.
Wynne, J. H.; Lloyd, C. T.; Jensen, S. D.; Boson, S.; Stalick, W. M. Synthesis 2004, 2277.
Wang, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Org. Lett. 2014, 16, 1606. doi: 10.1021/ol500203m
Yao, S.-J.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. J. Org. Chem. 2016, 81, 4226. doi: 10.1021/acs.joc.6b00580
Vitaku, E.; Smith, D. T.; Njardarson, J. T. Angew. Chem., Int. Ed. 2016, 55, 2243. doi: 10.1002/anie.201511149
Kakino, R.; Shimizu, I.; Yamamoto, A. Bull. Chem. Soc. Jpn. 2001, 74, 371. doi: 10.1246/bcsj.74.371
Zhu, F.; Yang, G.; Zhou, S.; Wu, X.-F. RSC Adv. 2016, 6, 57070. doi: 10.1039/C6RA12287F
Wu, W.; Tian, Q.; Chen, T.; Weng, Z. Chem.-Eur. J. 2016, 22, 16455. doi: 10.1002/chem.201604300
Yan, G.; Cao, X.; Zheng, W.; Ke, Q.; Zhang, J.; Huang, D. Org. Biomol. Chem. 2017, 15, 5904. doi: 10.1039/C7OB01335C

图式 2 氯化亚铜催化醇的氧化反应机理
Scheme 2 Oxidative mechanism of of clcohols catalyzed by cuprous chloride

图式 3 keto-ABNO催化α-氟烷基醇的氧化反应机理
Scheme 3 Oxidative mechanism of alpha-fluoroalkyl alcohol catalyzed by keto-ABNO

图式 5 三氯化铝催化芳烃的三氟乙酰化反应
Scheme 5 Trifluoroacetylation of aromatic hydrocarbons catalyzed by aluminium trichloride

图式 6 铜催化吲哚与三氟乙胺、亚硝酸钠的三组分反应
Scheme 6 Three-component reaction of indole with trifluoroethylamine and sodium nitrite catalyzed by copper

图式 7 吲哚与三氟乙酸的三氟乙酰化反应
Scheme 7 Trifluoroacetylation of indole with trifluoroacetic acid

图式 8 醋酸碘苯促进3-(氟乙酰)吲哚衍生物的合成
Scheme 8 Synthesis of 3-(fluoroacetyl) indole derivatives promoted by iodobenzene acetate

图式 10 钯催化芳基硼酸与三氟乙酸苯酯的偶联反应
Scheme 10 Palladium-catalyzed coupling reaction of aryl boric acid with phenyl trifluoroacetate

图式 12 钯催化芳基碘化物与三氟甲基铜的偶联反应
Scheme 12 Palladium-catalyzed coupling reaction of aryl iodides with trifluoromethyl copper

图式 14 铜催化芳基重氮盐与三氟丙酮酸乙酯的三氟乙酰化反应
Scheme 14 Trifluoroacetylation of aryl diazonium salts with ethyl trifluoropyruvate catalyzed by copper

图式 16 铜催化吲哚衍生物与三氟丙酮酸乙酯的三氟乙酰化反应
Scheme 16 Trifluoroacetylation of indole derivatives with ethyl trifluoropyruvate catalyzed by copper
表 1 不同氧化剂条件下, α-三氟甲基醇的氧化
Table 1. The oxidation of α-trifluoromethyl alcohol under different oxidant conditions
| Entry | Substrate | Oxidant | Conditions and yields | Ref. |
| 1 | |
|
3.7 equiv. Dess-Martin reagent, DCM, r.t., 15 min, 75%~95% | [8, 9] |
| 2 | |
CsSO4F | 1.2 or 2.3 equiv. CsSO4F MeCN, Ar, 30~35 ℃, 44% | [10] |
| 3 | |
|
2.5 equiv. AcNH-TEMPO, 2.25 equiv. 2, 6-lutidine, DCM, r.t., 71%~99% | [11] |
| 4 | |
|
2.0 equiv. IBX, EtOAc, reflux, 9~30 h, 94%~99% | [12] |
| 5 | |
Oxone | 5 mol% Organoiodine, 0.9 equiv. oxone, MeCN, 90 ℃, 18 h, 44%~86% | [13] |
| 6 | |
|
1.1 equiv. Bi complex, 1.1 equiv. DBU or TMG, CDCl3 or C6D6, r.t., 95% | [14, 15] |
| 7 | |
O2 or Air | 5 mol% CuCl, 5 mol% Phen, 5 mol% DBADH2, 2. equiv. K2CO3, PhMe, 70~90 ℃, 97% | [16, 17] |
| 8 | |
NaIO4 and Air | |
[18] |
| 9 | |
O2 | |
[19] |
| 10 | |
t-BuOOH (TBHP) | 5 mol% MOF, 4.0 equiv. TBHP, PhMe, r.t., 6%~99% | [20] |
| 11 | |
|
4c-substituted TEMPO (5 mol%), 1.5~2.0 equiv. Py-HBr3, DCM-NaHCO3, r.t., 70% | [21] |
| 12 | |
|
keto-ABNO (5 mol%), NaNO2 (10 mol%), AcOH (0.5 mol/L), O2 (101 kPa), r.t, 18 h, 31%~97% | [22] |
 下载: 导出CSV
下载: 导出CSV
表 2 羧酸衍生物的亲核三氟甲基化反应
Table 2. Nucleophilic trifluoromethylation of carboxylic acid derivatives
| Entry | Carboxylic acid derivative | Trifluoromethylation reagent | Conditions and Yields | Ref. |
| 1 | PhCOBr | CF3Cu | 45 ℃, 24 h, 24% | [23] |
| 2 | PhCOCl | TMSCF3 | TBAF, THF, r.t., mixture products | [24] |
| 3 | ArCOCl | Cd(CF3)2•Glyme | DMAP, DCM, 0 ℃~r.t., 12 h, 57%~85% | [25] |
| 4 | ArCOCl | AgCF3 | Method A: EtCN, -30 ℃~r.t., 12 h, 50%~77% Method B: DMAP, DCM, -30 ℃~r.t., 12 h, 50%~78% |
[26] |
| 5 | PhCOCl | CF3COONa | CuI, DMF, 80% | [27] |
| 6 | RCO2Me (R=Ar, Alkyl) | C6H5SCF3 | 1.1 equiv. Et3GeNa, THF/HMPA, -60 ℃, 1.5 h, 93%~98% | [28] |
| 7 | RCO2Me (R=Ar, Alkyl, Alkenyl, etc.) | TMSCF3 | 2.5 mol% TBAF, PhMe or DCM, or pentane, -78 ℃~r.t., 68%~95% | [29] |
| 8 | RCO2R' (R=Ar, Alkyl, Alkenyl, Alkynyl, etc; R'=Me, Et) | TMSCF3 | 1.0 mol% CsF, THF, r.t., 1~4 h, 84%~90% | [30] |
| 9 | RCO2R' (R=Ar, Alkyl, Alkenyl, etc.; R'=Me, Et, iPr) | TMSCF3 | 5.0 mol% AcONBu4, 0 ℃~r.t., 1 h, 64%~90% | [31] |
| 10 | PhCO2Me | CF3H | KHMDS, THF, 30% | [32] |
| 11 | |
TMSCF3 | (1) 0.2 equiv. CsF, PhMe, r.t., 24 h (2) TBAF, H2O, 50 ℃, 2h, 48%~99% | [33] |
| 12 | |
TMSCF3 | 0.3 equiv. KF, 0.3 equiv. TBAB, DMF, 0~r.t., 16 h, 64%~90% | [34] |
| 13 | |
TMSCF3 | 0.1 equiv. TBAF, DMSO, r.t., 13%~57% | [35] |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们








 下载:
下载: