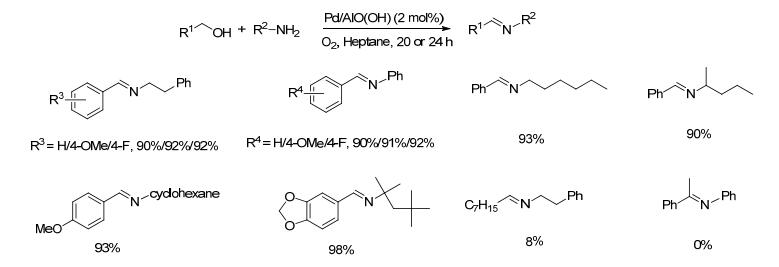
图式 1.
Pd/AlO(OH)催化剂催化醇和胺直接偶联生成亚胺的反应
Scheme 1.
Direct coupling of alcohols and amines catalyzed by Pd/AlO(OH)

亚胺又称席夫碱, 是指醛或酮上氧原子被氮原子取代而形成的一类含碳氮双键的有机化合物, 广泛存在于天然产物、生物活性化合物和药物结构中[1~7]; 同时, 该结构具有较高的反应活性, 可作为氮源应用于不同类型的反应中, 因此在染料、香料、杀菌剂、药品和农用化学品等方面都具有重要的应用[8~18].传统的亚胺制备方法包括酸催化下羰基与伯胺的缩合[19]、次胺的氧化[20, 21]等, 其中, 酸催化下羰基与胺的反应是最常用的亚胺制备方法.相比于醛和酮, 醇类化合物在多数情况下是更稳定和易得的原料; 催化醇和胺直接偶联制备亚胺唯一的副产物为水, 因此具有原子经济性高和环境友好等优点.近年来催化醇胺偶联制备亚胺的方法引起了广泛关注, 并在催化剂尤其是金属催化剂的开发方面取得了一定的进展, 本文系统地总结了金属催化剂催化醇和胺直接偶联制备亚胺的研究进展, 并依据反应机理不同将其分为催化氧化和催化脱氢两大类.
2009年, Park等[22]将钯金属纳米颗粒包埋在勃姆石纳米纤维中, 制备了一种非均相钯催化剂Pd/AlO(OH), 并将其用于催化醇和胺偶联制备亚胺的反应.在2.0 mol%催化剂和氧气存在下, 苄醇和苯乙胺在正庚烷中于90 ℃反应20 h, 亚胺的收率达到90% (Scheme 1).底物拓展发现苄醇类底物中芳环上取代基的电子效应(如4-甲氧基、4-氟)不影响亚胺收率; 苯胺类底物与苄醇的反应则需更多的催化剂(4 mol%)才能获得满意的收率; 脂肪胺分子中烷基的空间体积不影响目标产物的收率, 如2, 4, 4-三甲基戊烷-2-胺与苄醇反应的收率为98%;正辛醇与苯乙胺的反应收率只有8%, 1-苯乙醇与苯胺反应则未得到目标产物.在催化醇和胺偶联的过程中除氧气外不需要其它任何添加剂, 有一定的底物适用范围, 是一种相对绿色且较有效的亚胺制备方法.
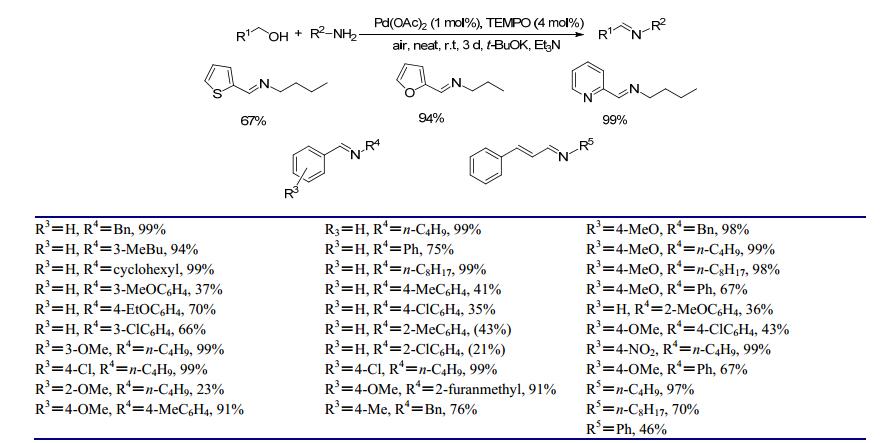
2011年, Xu等[23]研究了钯催化下苄醇和苄胺的直接偶联反应, 在1 mol%醋酸钯(Pd(OAc)2)、15 mol%三乙胺、4 mol%四甲基哌啶氧化物(TEMPO)、20 mol% t-BuOK和空气存在的条件下, 苄醇和苄胺室温反应3 d, 亚胺收率达99%.对于苄醇和脂肪胺类底物, 能以76%~99%的收率得到目标产物; 对于苄醇和取代苯胺类底物, 可采用增加叔丁醇钾用量来提高收率(50 mol%或100 mol%).机理研究表明, 该反应先经历了醇在钯催化下氧化为醛, 随后醛和胺反应生成亚胺的过程, 但催化过程中金属钯以何种活性态存在尚不明确.该方法不需要溶剂且反应条件非常温和, 底物适用范围广, 但反应时间偏长且反应体系需要多种添加物, 对于碱性较弱的苯胺类底物, 收率偏低.
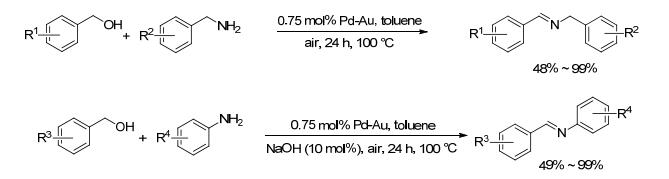
2017年, Dong等[24]以4, 4, 4-三氟-1-(4-(吡啶-4-基)苯基)丁烷-1, 3-二酮和Mn(OAc)2为原料, 在溶液中合成了新的Mn(Ⅱ)金属有机骨架(MOF), 并将该MOF作为Pd-Au双金属合金纳米粒子的载体, 得到了一种高活性含双官能团非均相复合催化体系Pd-Au@Mn(Ⅱ)-MOF.该催化体系在空气条件下, 能有效地促进醇和胺“一锅法”串联反应合成亚胺(Scheme 3).对于19种苄醇和苄胺类底物, 该方法以0.75 mol% Pd-Au为催化剂, 在甲苯中110 ℃反应24 h, 收率在49%~99%之间.当苄胺芳环对位为强的供电性基团(如甲氧基)取代时, 目标产物收率普遍较低.而对于27种苄醇和苯胺类底物, 则需要在反应体系中添加10 mol%的氢氧化钠, 收率在49%~99%之间, 两种底物芳环上取代基的类型和位置对产物收率都有较大影响.
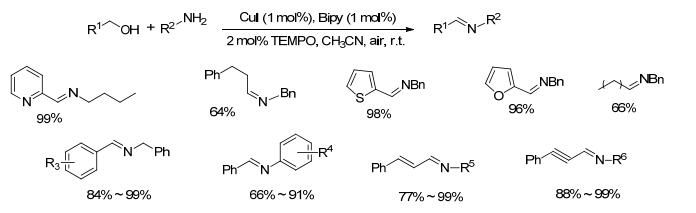
2012年, Xu等[25]以苄醇和苄胺为模型底物, 研究了铜催化剂催化醇和胺直接合成亚胺的方法(Scheme 4).在空气存在下, 以1 mol%的碘化亚铜为催化剂, 1 mol%的2, 2'-联吡啶为配体, 苄醇和苄胺在2 mol%的TEMPO存在下乙腈中室温反应6 h, 以99%的收率得到目标亚胺产物.该方法底物范围较广, 适合苄醇、烯丙醇、炔丙醇、脂肪醇类底物和脂肪胺、芳香胺类底物, 收率在64%~99%之间, 并可以放大到100 mmol的规模.手性胺类底物在反应过程中不发生消旋.在整个反应过程中, 碘化亚铜不仅具有促进醇氧化的作用, 同时还能提高醛和胺的脱水反应速率.该催化方法不需要添加碱和脱水剂, 催化剂廉价易得, 条件温和, 底物适用范围广, 是一种有效的亚胺制备方法.
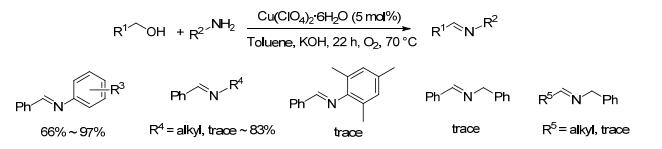
同年, Zhang等[26]研究了Cu(ClO4)2•6H2O催化下醇和胺的亚胺化反应.该方法以5 mol%的Cu(ClO4)2• 6H2O为催化剂、1.5 equiv.氢氧化钾为碱, 氧气存在下甲苯中于70 ℃反应22 h (Scheme 5).除大位阻的2, 4, 6-三甲基苯胺外, 苄醇与芳胺、苄胺、脂肪胺类底物反应能以63%~97%的收率得到目标产物, 而正丁醇、十二醇以及1-苯基-2-丙醇与各种胺的反应效果较差, 均只能得到痕量产物.该方法催化剂较廉价, 但是反应过程需要添加过量的碱.
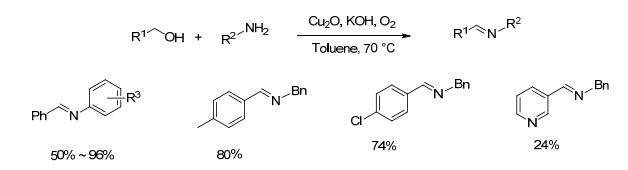
2015年, Bai等[27]以自制的荔枝型氧化亚铜(Cu2O)纳米团聚体替代Cu(ClO4)2•6H2O作催化剂.该催化方法对芳环对位有供电子基团的芳胺类(如甲氧基、甲基苯胺)以及长链脂肪胺的催化效果较好, 收率达到84%~96% (Scheme 6).对于邻位有取代基的苯胺类底物(如2-甲氧基苯胺和2-溴苯胺)与苄醇的反应, 亚胺收率仅为50%和54%, 对于3-吡啶基甲醇与苄胺的反应, 亚胺收率只有24%.该催化体系底物适用范围较窄, 反应体系中需要加入过量的碱; 在反应过程中氧化亚铜可被氧化为氧化铜, 整个过程起催化作用的是氧化亚铜还是氧化铜, 尚不明确.
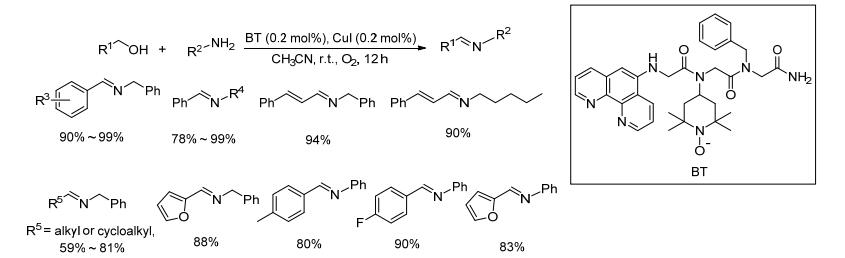
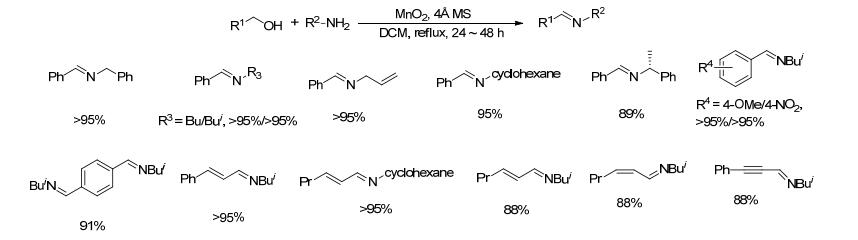
2017年, Galia等[28]将配体与四甲基哌啶氮氧化物结合制备了一类含配体的新型四甲基吡啶氮氧化物的衍生物BT, 并将其替代TEMPO与碘化亚铜组成催化剂.以苄醇和苄胺类化合物为反应底物, 当催化剂(碘化亚铜和BT)的用量为底物的0.1 mol%时, 该催化剂的转化数(TON)较高, 大都达到了900 (Scheme 7).对于苄醇和脂肪胺或芳胺以及脂肪醇和苄胺类底物, 当催化剂的用量为底物的0.1 mol%时, 目标产物的收率一般都<10%;当将催化剂用量提升到底物的0.2 mol%, 同时延长反应时间至24 h, 目标亚胺的收率为59%~99%.该方法催化剂用量少, 反应条件温和, 底物适用范围较广, 但BT不易制备.其催化氧化机理如Scheme 8所示.
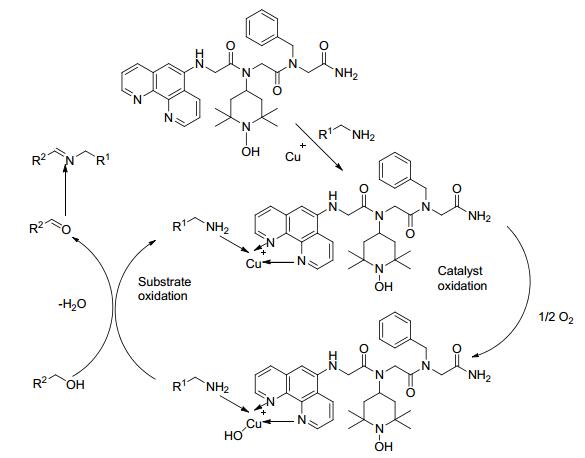
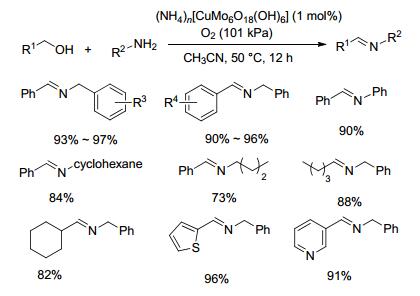
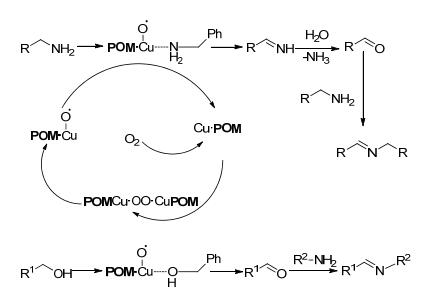
在上述有关铜类催化剂的研究中发现, 通常需要在反应体系中添加碱、TEMPO或有机配体等来提高收率, 但是这些添加物回收困难.针对这一问题, 2017年, Wei等[29]首次制备了无机配体配位的铜催化剂((NH4)n[Cu-Mo6O18(OH)6]), 并将其用于催化醇和胺制备亚胺的反应中(Scheme 9).对于苄醇和苄胺以及脂肪胺类底物, 以乙腈为溶剂, 在1 mol%催化剂和1×105 Pa氧气50 ℃的条件下反应12 h, 目标产物的产率在73%~97%之间.该方法可以避免使用贵重金属和昂贵的、有毒的、对水敏感的有机配体, 减少了对环境的危害, 符合绿色化学发展思路, 为催化剂的开发提供了一种新的思路.其催化机理如Scheme 10所示.
2001年, Taylor等[30]以活性二氧化锰作为原位氧化剂氧化醇和胺制备亚胺(Scheme 11).对于苄醇类底物与胺的反应, 芳环上取代基的电性对反应基本没有影响; 二元醇也可以有效地转化为双亚胺.但是该方法反应时间较长且需要过量的氧化剂, 而且从严格意义来说该方法不属于催化反应.
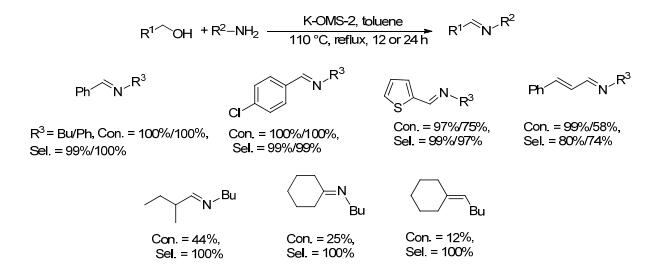
2008年, Steven等[31]制备了锰八面体分子筛催化剂(K-OMS-2), 并将其用于催化醇和胺偶联制备亚胺的反应.在空气存在下, 醇(1 mmol)、胺(2 mmol)和K-OMS-2 (50 mg)在甲苯中110 ℃反应12~24 h, 原料转化率为12%~100%, 亚胺的选择性为39%~100% (Scheme 12).对于环己醇、环戊醇与丁胺的反应, 尽管亚胺选择性为100%, 但醇的转化率只有12%和25%.该催化剂可循环使用4次.该催化方法操作简单, 不需要任何添加剂, 催化剂易回收, 但反应过程所需催化剂用量较大且底物使用范围有限.
2014年, Zhao等[32]将不同形貌的(球形、纳米线以及八面体等)Mn3O4纳米材料嵌入到二乙烯基三烯丙基胺聚合物(DVTA)中, 并将所得的复合材料用于催化空气氧化sp3杂化的C—H键以及醇的选择性氧化, 聚合物中嵌入八面体结构的纳米Mn3O4(Mn@DVTA-3)催化活性最好(Scheme 13).该催化剂催化芳环上有不同取代基的苄醇衍生物与芳香胺或苄胺的反应时, 目标亚胺的收率在80%~88%之间, 而苯丙烯醇与苄胺反应的收率为75%.总体来说催化效率不高, 但是该纳米金属催化剂可以重复使用4次而锰不流失.
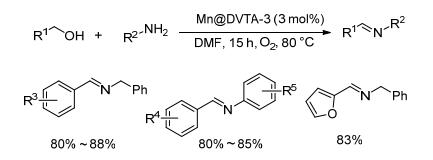
同年, Gao等[33]将MnOx负载于各种不同载体上, 发现以羟磷灰石(HAP)为载体所制备的催化剂(MnOx/ HAP)在空气存在下具有良好的催化醇与胺偶联的性能.对于苄醇类底物和苯胺的反应, 该催化体系能以82%~98%的收率得到相应的亚胺, 而长链的辛醇与苯胺在该条件几乎不反应(Scheme 14).该催化方法不需要添加额外的助剂, 更为重要的是该催化剂易于回收且可重复使用, 以苄醇和己胺为底物时, 催化剂在9次循环使用时收率都达到了98%, 并且可用于克级规模样品的制备.这两类锰催化剂的发现为设计廉价金属以及绿色高效的多相催化剂开辟了一条新途径.
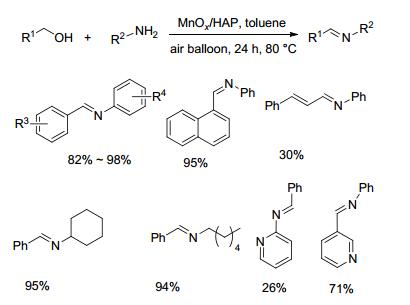
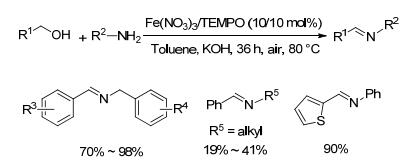
2013年, Xu等[34]报道了以空气为氧化剂, Fe(NO3)3、TEMPO以及氢氧化钾组成的体系催化不同醇胺合成亚胺的方法(Scheme 15).对于苄醇和苯胺类底物, 该方法以70%~98%的收率获得目标产物.然而对于苄醇和脂肪胺类底物, 该方法收率较低(19%~41%), 长链醇与苯胺在该条件下几乎不反应.该方法采用空气作为氧化剂, 催化剂易获得, 反应条件较温和, 但反应体系中需要多种添加物且底物研究范围有限.
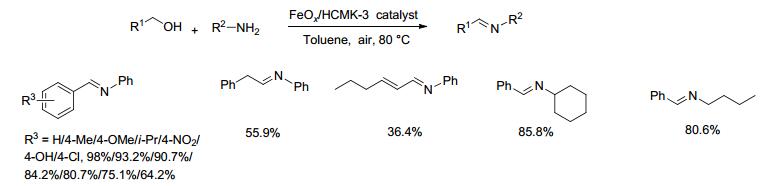
2016年, Liu等[35]研制并报道了一种以介孔碳为载体的FeOx/HCMK-3催化剂, 并将它用于催化醇和胺氧化偶联生成亚胺的反应.在100 kPa空气存在下, 醇(1 mmol)和胺(2 mmol)、0.3 g FeOx/HCMK-3催化剂在甲苯中80 ℃反应8 h, 收率在64.2%~98.0%之间(Scheme 16).芳环上连有供电子基团的苄醇类衍生物(如4-甲基、4-甲氧基和4-异丙基)与苯胺反应时, 分别以93.2%、90.7%和84.2%的收率得到目标产物.脂肪胺(环己胺和正丁胺)与苄醇反应, 能以较好的收率得到亚胺.该催化剂中高分散的FeOx使FeOx/HCMK-3具有较高的还原性和高的催化性能, 亚胺的形成过程经历了一个氧化还原机理, 三价铁在催化氧化醇为醛的同时, 自身被还原为二价铁, 二价铁又被氧氧化为三价, 从而实现了催化剂的再生循环.在亚胺的反应过程中, 醇的氧化脱氢反应是整个反应的速率决定步骤.
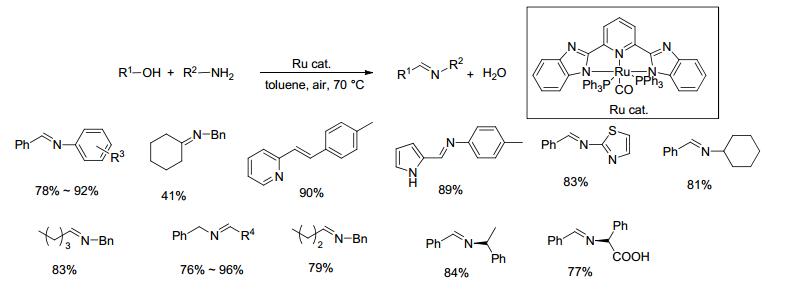
2014年, Ramesh等[36]在研究醇和胺反应制备亚胺时, 以0.002 mol% Ru(Ⅱ)NNN配合物为催化剂, 在空气存在下甲苯中70 ℃反应12~36 h, 收率为41%~96% (Scheme 17).对于不同类型醇和胺类底物的反应, 均能以中等至良好的收率得到目标产物, 催化剂用量低且适用范围较广, 并且可用于手性胺和氨基酸类底物与苄醇的反应.
负载型金纳米粒子作为有机转化催化剂越来越受到人们的关注. 2009年, He等[37]制备了一种以羟基磷灰石(HAP)为载体的金纳米粒子催化剂, 并将其用于催化醇和胺制备亚胺的反应.在1 mol%金(以金的量计算)催化剂和氧气存在下, 醇类和胺类底物在甲苯中60 ℃反应1~16 h, 底物醇的转化率在75%~99%之间, 目标产物亚胺的选择性非常好, 在90%~99%之间, 其中苄醇类底物的转化率为76%~99%之间.强吸电子基团的引入(如硝基)会大大降低其转化率, 4-硝基苄醇的转化率仅为76%, 长链烷基醇如正庚醇的转化率仅为75%.该催化反应经历了一个醇先氧化为醛, 再和胺反应生成亚胺的过程.该催化剂为非均相催化剂, 金在催化过程中不流失, 易回收, 且循环使用5次仍然保持活性.金纳米粒子的引入显著地改变了酸碱分布, 增加了HAP上酸性和碱性位点的总数.
2010年, Riisager等[38]报道了以二氧化钛(TiO2)为载体的金纳米粒子作为多相催化剂, 以纯氧为氧化剂催化醇和胺反应生成亚胺的方法.该反应在室温和常压下进行, 在适当的转化条件下, 具有良好的选择性(>98%), 但是醇类物质的转化率普遍较低(7%~63%), 对于常用作模型底物苄醇的反应, 转化率也只有51%.
2012年, Hensen等[39]制备了一种在碱性水滑石(HT)负载金纳米粒子(AuNP)的非均相催化剂Au/ Mg2Al-HT, 并将其用于催化醇和胺反应制备亚胺.在0.5 mol%催化剂和氧气存在下, 醇和胺类底物在甲苯中于60 ℃反应, 醇类底物的转化率和亚胺的选择性随着反应时间的延长而增加, 反应时间为5~12 h, 转化率在77%~100%之间, 选择性在63%~100%之间.该催化剂明显优于前几种负载金纳米粒子的催化剂, 不仅提高了化学合成的效率, 而且具有稳定性高、产物污染少、催化剂易分离、易再利用的优点, 具有进一步研究和开发的价值.
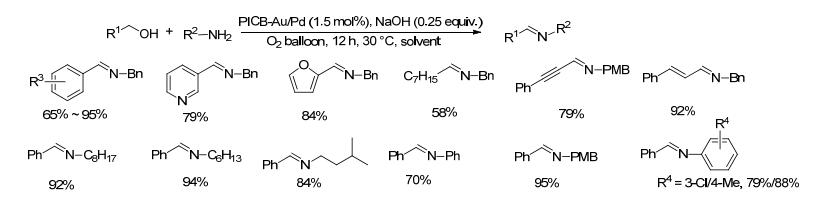
2013年, Kobayashi等[40]报道了一种有效且可重复使用的聚合物嵌顿的金/钯合金纳米粒子的(PICB- Au/Pd)多相催化剂.在1.5 mol% PICB-Au/Pd、25 mol%的氢氧化钠和氧气存在的条件下, 醇类和胺类底物在40 ℃或60 ℃的混合溶剂[V(四氢呋喃)/V(四氟乙烯)=4:1]中反应12 h, 目标产物亚胺的收率在58%~95%之间, 长链的正辛醇底物收率较低只有58%, 其它醇类和胺类底物收率均较好(Scheme 18). PICB-Au/Pd非均相催化剂具有较好的活性和选择性, 且可以容易地多次循环而不损失活性, 但反应需要强碱氢氧化钠的参与.
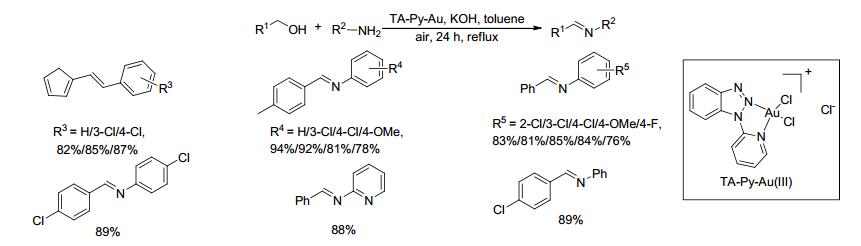
2017年, Wang等[41]将2-吡啶基苯并三唑与金(Ⅲ)配位合成了吡啶基三唑金(Ⅲ)配合物TA-Py-Au(Ⅲ), 并将该配合物作为催化剂用于催化醇和胺合成亚胺的反应.在1 mol%催化剂、120 mol% KOH和空气存在下, 醇和胺在甲苯中回流反应24 h, 亚胺产物的收率为76%~94% (Scheme 19).对于苄醇和芳胺类底物, 当芳环上取代基为F、Cl、OMe和Me时, 亚胺的产率较高; 且具有吡啶基或呋喃基的底物也适用于这一反应.该催化剂中加入了一个配位能力强的配体, 大大提高了催化剂的活性, 有助于该催化剂在高温下分解成金纳米粒子而延迟聚集, 更为重要的是该催化剂对空气和水分不敏感且可催化炔丙醚的3, 3-重排制备丙二烯类化合物.
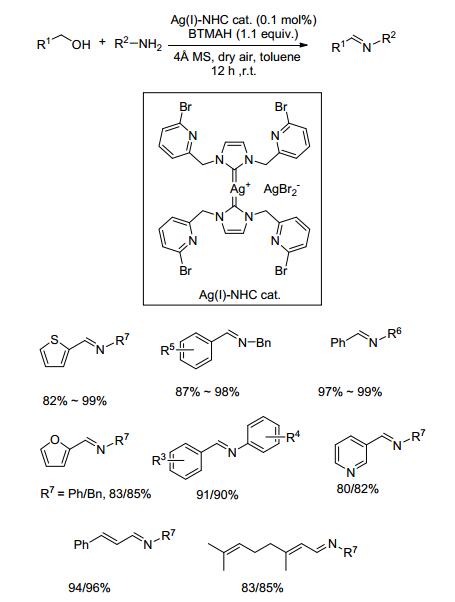
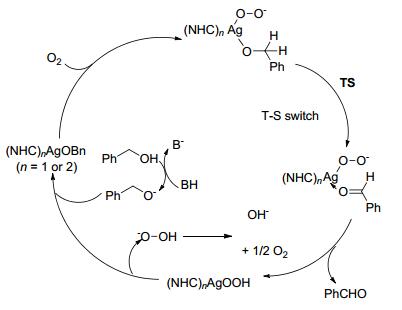
2014年, Jiang课题组[42]在研究醇的选择性氧化时发现, 银-氮杂环卡宾(NHC)复合物具有优异的催化醇胺偶联制备亚胺的性能.在0.1 mol%催化剂、1.1 equiv. BnMe3NOH (BTMAH)、4Å分子筛和干燥的空气存在下, 醇和胺类底物在甲苯中常温反应12 h, 以80%~99%的收率得到目标产物(Scheme 20).对于苄醇类与苄胺类或苯胺类底物, 目标产物的收率在82%~98%之间.苄醇与环己胺或正丙胺的反应收率分别为97%和98%.该催化方法底物应用范围广, 反应条件温和, 产物收率高, 是一种非常有效的亚胺制备方法.催化机理如图(Scheme 21)所示.
2014年, Kegnæs等[43]将银(Ag)负载在氧化铝(Al2O3)上制备了负载的银纳米粒子催化剂(Ag/Al2O3), 并将其用于催化醇和胺制备亚胺的反应.在5 wt%的催化剂和空气条件下, 醇和胺类物质在甲苯中100 ℃反应24 h, 目标亚胺产物的收率在16%~81%之间.对于常用的模板底物苄醇和苄胺的反应, 苄醇的转化率只有10%, 虽然其选择性为99%, 但产物收率只有10%.当采用正己醇为底物时, 基本上不发生转化.总体来说, 该催化剂催化效率较低, 底物适用范围有限.
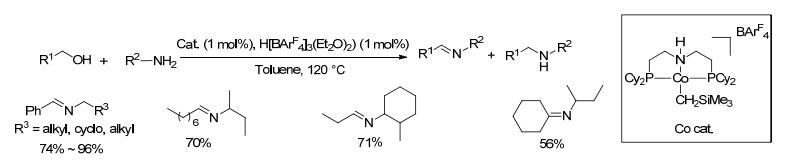
2013年, Hanson等[44]将一种均相的金属钴催化剂用于催化醇和胺偶联合成亚胺(Scheme 22).氘标记研究表明, 反应经历了醇脱氢的过程.对于催化亚胺化, 钴具有与贵金属催化剂相当的活性, 并且对一系列苄醇与脂肪族胺以及脂肪醇与脂肪胺的反应都是有效的, 目标亚胺产物的收率在56%~99%之间.对于部分底物会有少量亚胺双键被还原.该催化方法突出了钴作为贵金属钌、铱和锇催化剂替代品的潜力.
2013年, Xia等[45]采用阴离子交换法将钴离子交换到沸石中制备了钴沸石催化剂, 并首次将其用于醇直接烷基化芳香胺的反应中(Scheme 23).在最佳条件下苄醇对苯胺的烷基化反应中苯胺的转化率为89.8 mol%.钴的负载量、载体、温度和碱等因素对反应均有较大影响, 该催化剂在反应完毕后仍保持多孔结构并具有稳定的催化活性.该催化剂整体活性较低且对底物适应性的研究较少, 无法明确其可能的底物适用范围.
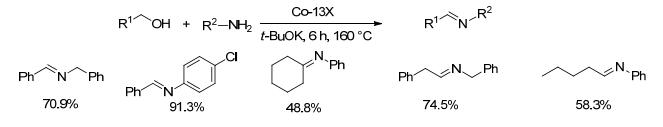
2018年, Balaraman等[46]报道了一种不含膦配体的Co(Ⅱ)-NNN钳子型配合物, 并将其作为催化剂用于催化醇和胺脱氢偶联生成亚胺的反应.在4 Å分子筛、5 mol%催化剂和110 mol%叔丁醇钾存在下, 醇和胺类底物在正辛烷中150 ℃反应32 h, 亚胺收率为72%~84% (Scheme 24).对于各种含供电子和吸电子取代基的苄胺和苄醇, 该方法都能以较好的收率得到亚胺产物.虽然该反应的副产物只有水和氢气, 但需要过量的碱.
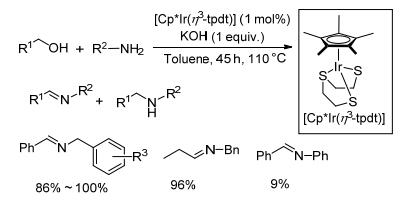
2011年, Pullarkat等[47]制备了一种[Cp*Ir(η3-tpdt)]配合物, 并将其用于催化醇和胺制备亚胺(Scheme 25).该方法以1 mol%铱配合物为催化剂, 1 equiv.氢氧化钾为碱, 醇和胺在甲苯中110 ℃反应45~68 h, 目标产物亚胺的收率与底物的类型相关性较大, 收率在9%~100%之间.对于苄醇、4-甲氧基苄醇与苄胺以及异丙胺的反应, 该催化体系能以几乎定量的产率得到目标产物; 而对于苄醇与苯胺的反应, 目标产物收率较低, 只有9%, 主要副产物是亚胺被还原为苯基苄基胺.该催化剂在催化过程中需要使用1 equiv.的氢氧化钾且研究的反应底物有限, 该方法对底物官能团的耐受性尚不清晰, 总体来说, 该催化体系反应时间偏长, 催化剂催化效率不高.
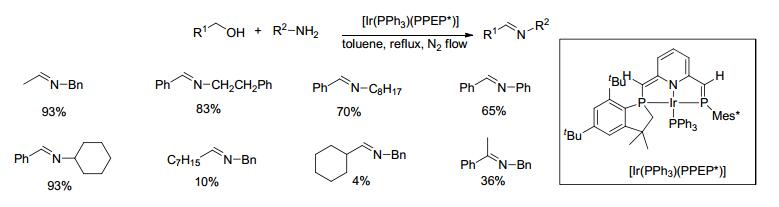
2016年, Ozawa等[48]制备了含去芳香化吡啶环的PNP钳形磷烯配体(PPEP*)的Ir配合物[IrPPh3(PPEP*)], 并研究了其催化醇烷基化胺的反应(Scheme 26).在1 mol%铱配合物和氮气氛围下, 醇和胺类底物在甲苯中回流反应24 h, 产物亚胺的产率在4%~93%之间, 同时会产生部分亚胺双键还原的副产物.
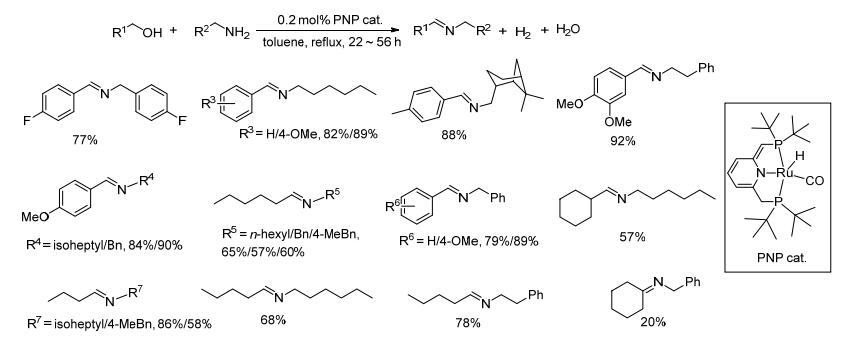
2010年, Milstein等[49]以均相的PNP型钌螯合物催化醇和胺脱氢缩合制备亚胺, 在0.2 mol%催化剂和氩气存在下, 醇和胺在甲苯中回流反应22~56 h, 亚胺收率在20%~92%之间(Scheme 27).苄醇类底物与苄胺或脂肪胺反应能以77%~92%的产率得到亚胺.链状的正己醇、正戊醇或正丁醇与苄胺或脂肪胺反应虽能以57%~86%的收率得到产物, 但体系中会产生一定量的酰胺和酯; 环己醇与苄胺的反应收率只有20%.总体来说, 该方法对苄醇类底物与苄胺和脂肪胺的反应催化效果较好; 对于脂肪醇类底物与胺的反应, 会产生一定量的酰胺和酯等副产物.该催化方法催化剂用量少, 转化数高, 除了氢气和水之外几乎无其它废弃物生成, 且反应过程中亚胺不会被生成的副产物氢气还原; 但该催化剂的制备以及回收相对较为困难, 且亚胺收率仍然有较大的提高空间.
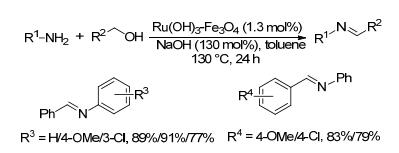
2011年, Ramón等[50]将钌浸渍在磁铁矿上制备了一种新型催化剂(Ru(OH)3-Fe3O4), 并将其用于催化醇对弱亲核性的氨基衍生物(芳胺、磺酰胺、磺胺)的烷基化反应; 当芳胺类底物和醇反应时, 依据所用碱的不同, 会以不同比例得到亚胺和亚胺双键被还原的(二级胺)产物.当采用氢氧化钠为碱时, 在1.3 mol%催化剂和氩气存在下, 醇和芳胺在甲苯中130 ℃反应24 h, 以77%~91%的收率得到目标亚胺产物(Scheme 28).利用其磁性, 该催化剂回收容易; 催化剂回收后可循环使用十次.
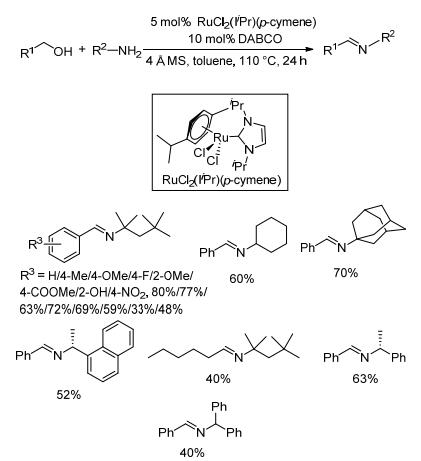
2012年, Madsen等[51]制备了一种新的钌杂环卡宾配合物[RuCl2(IiPr)(p-cymene)]催化剂, 并将其用于催化有位阻的胺和醇直接脱氢偶联制备亚胺.在4Å分子筛、5 mol%催化剂、10 mol% DABCO和氮气存在下, 醇和胺类底物在甲苯中回流反应24 h, 亚胺的收率为33%~80% (Scheme 29).其中, 邻羟基苄醇与叔辛胺反应收率只有33%, 其它苄醇类底物与叔辛胺的收率在48%~80%之间, 其中以苄醇与叔辛胺反应收率为最高(80%), 芳环上取代基位置及类型对收率影响的规律不明显.苄醇与环己胺、1-金刚烷胺反应的收率分别为60%和70%;光学纯(R)-1-苯基乙胺和(R)-1-(1-萘基)乙胺与苄醇反应过程中不发生消旋; 大位阻的三苯甲胺与苄醇反应时, 虽然苄醇的转化率达到了75%, 却没有目标产物生产, 主要产物是苄醇自身缩合形成的苯甲酸苄酯.通过氘标记实验发现, 初始的醇β-位脱氢至醛的过程是个可逆过程, 活性催化组分为二氢钌化物.其反应机理是醇在催化剂作用下脱氢生成醛与催化剂的配合物, 然后胺亲核攻击醛与钌催化剂形成的配合物形成半胺醛, 半胺醛从催化剂脱离后脱水生成亚胺.该催化方法操作简单, 副产物只有水和氢气, 但反应收率不高.由于作者只研究了有位阻的胺类与醇的亚胺化反应, 因此无法推测小位阻胺类与醇类的反应效果.
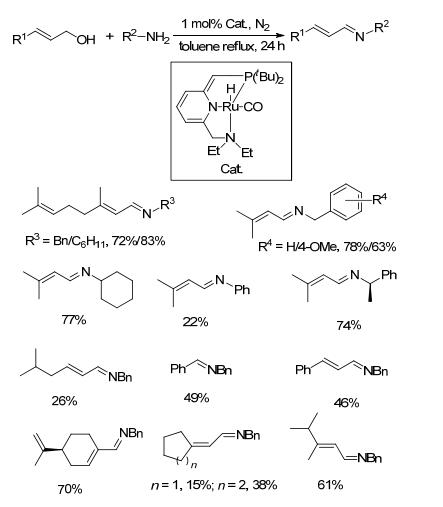
2012年, Schomaker等[52]以PNN型钌螯合物为催化剂, 研究了其催化烯丙基醇类和胺类底物直接脱氢偶联制备α, β-不饱和亚胺的方法.在1 mol%催化剂和氮气氛围下, 烯丙基醇和胺类底物在甲苯中回流反应24 h, α, β-不饱和亚胺产物收率为15%~83% (Scheme 30). 3-甲基- 2-丁烯-烯醇和富电子的伯胺反应时α, β-不饱和亚胺的产率较好; 当与弱亲核性的苯胺反应时, 则收率较低.重要的是烯丙醇类底物结构中其它孤立的双键在反应体系中不会被还原, 且体系中的双键在反应条件下也不会发生明显的迁移.该催化方法针对烯丙醇类与胺类底物直接偶联, 无需任何其它添加剂, 虽然总体来说收率不高, 但属首次且较为系统和深入, 为进一步研究该类底物的反应提供了很好的借鉴.
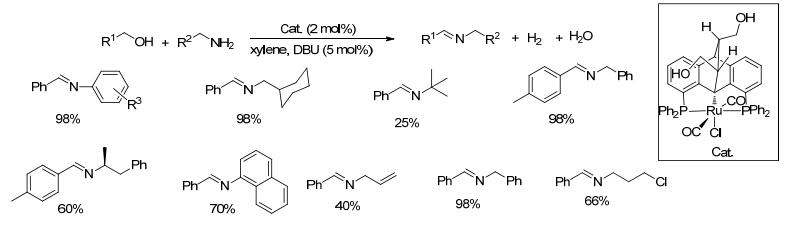
2013年, Gelman等[53]报道了含二苯并苯二烯配体结构片段的双官能化的新型钌配合物, 该配合物在空气中稳定, 具有良好的催化醇无受体脱氢生成酯和醇以及胺无受体脱氢偶联生成亚胺的活性.在2 mol%催化剂和5 mol% DBU存在的条件下, 苄醇类和胺类底物在对二甲苯中回流反应, 以25%~98%的收率得到目标产物(Scheme 31).该催化剂对于大位阻的胺类如叔丁胺收率较低, 只有25%, 对于苄醇和苯胺以及芳胺类底物, 目标产物收率很高, 达到了98%.
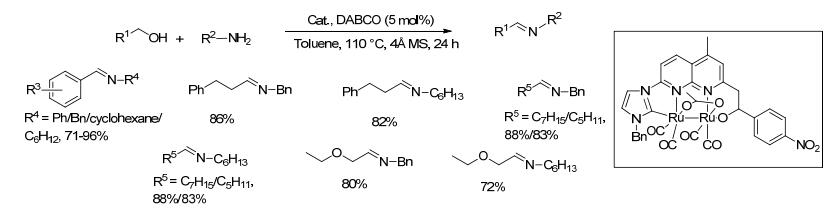
2014年, Bera等[54]报道了一种新型的双钌配合物, 并以其为催化剂研究了其催化醇和胺制备亚胺的性能(Scheme 32).在1 mol%催化剂、5 mol% DABCO以及4Å分子筛存在的条件下, 醇和胺在甲苯中回流反应24 h, 目标产物亚胺的收率在72%~96%之间.该方法底物适用范围较广, 对于苄醇类与芳胺、苄胺以及脂肪胺的反应, 目标亚胺产物的收率均较高, 对于脂肪醇类底物与不同胺的反应, 目标亚胺产物的收率也较高.机理研究表明, 该反应经历了一个醇脱氢生成醛, 随后醛与胺迅速形成亚胺的过程, 但是该催化剂只有在碱性条件下才有较好的催化醇脱氢的能力.
同年, Guan等[55]将Saito课题组开发的Ru-Macho催化剂(用于催化氢化还原酯为醇)转化为可用于脱氢的预催化剂, 并将该预催化剂用于伯醇和胺脱氢制备酰胺的反应中, 该预催化剂在碱存在下转化为具有催化活性的Ru-Macho催化剂, 当底物醇为仲醇时, 则生成了亚胺类产物, 该催化剂对于催化醇和胺脱氢制备酰胺具有重要的意义(Scheme 33).
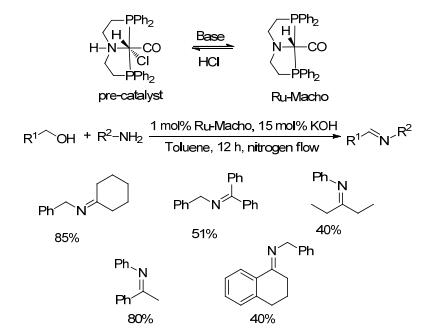
2017年, Mashima等[56]将RuCl2(dppea)2与Zn(OCOCF3)2和叔丁醇钾组成复合催化体系, 研究了其催化醇和胺脱氢偶联制备亚胺的性能(Scheme 34).以0.5 mol%钌配合物、1 mol% Zn(OCOCF3)2和20 mol%的叔丁醇钾为催化剂, 醇和胺类底物在二氧六环中回流反应18 h, 目标亚胺的收率在25%~83%之间.对于苄醇类底物与正己胺的反应, 收率在43%~95%之间, 而正己胺与苯丙醇反应的收率为76%, 说明芳环上取代基的类型和位置对收率影响较大.此外, 对催化剂进行结构变换得到的新钌配合物([Ru(OCOCF3)2{(S)-dppmp}2]), 可用于催化醇和胺直接偶联制备酰胺的反应, 这一发现对于催化醇和胺制备酰胺同样具有十分重要的意义.
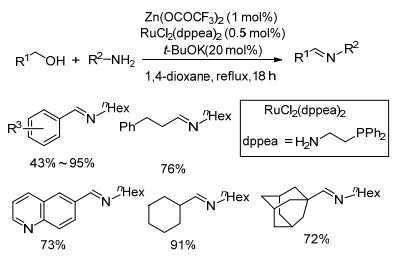
2018年, Li等[57]开发了氮膦功能化卡宾配体, 并将其和Ru(CON)Cl2组成催化体系用于催化醇烷基化胺的反应.发现在该催化体系中, 所得产物是亚胺和胺的混合物, 且配体的类型和反应时间对产物(亚胺和胺)的比例有重要影响.
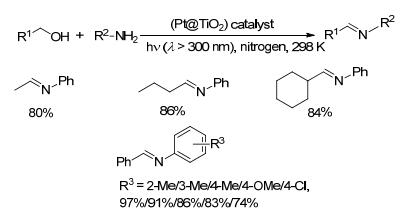
2011年, Shiraishi等[58]将Pt纳米颗粒负载在二氧化钛(TiO2)上制备了一种Pt@TiO2催化剂, 并研究了其在紫外光条件下催化醇和芳胺偶联制备亚胺的性能.在紫外光照射(λ>300 nm)和氮气氛围下, 过量的醇和50 μmol胺在5 mg催化剂作用下室温反应, 亚胺的收率为74%~97% (Scheme 35).苄醇与甲基苯氨反应时, 亚胺的收率为2-Me>3-Me>4-Me; 环己醇与苯胺反应的收率为84%.该反应在室温进行, 除紫外光外无需其它添加剂, 无其它副产物生成.该催化方法虽然底物类型研究相对较少, 但收率较好且较为绿色, 值得进一步研究.
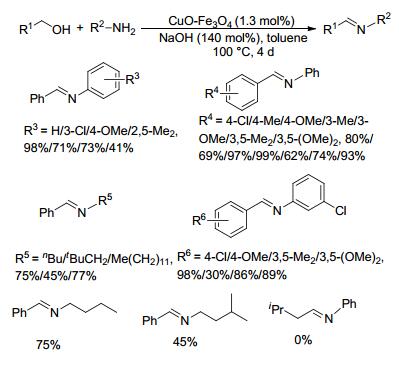
2012年, Ramón等[59]将氧化亚铜浸渍在在磁铁矿上制备了一种CuO-Fe3O4催化剂, 并研究了其催化醇和芳胺类底物直接脱氢偶联制备亚胺的性能.在1.3 mol%催化剂和140 mol%氢氧化钠存在下, 苄醇和芳胺类底物在甲苯中100 ℃反应4 d, 亚胺产物的收率为0%~99% (Scheme 36).大部分苄醇类与芳胺类底物的反应收率都较高(62%~99%), 3-氯苯胺与苄醇的反应收率较低只有30%, 位阻较大的2, 5-二甲基苯胺和苄醇反应收率也只有41%;异丙醇与苯胺在该反应条件下无目标产物生成; 当苄醇芳环上为单甲基取代时, 亚胺的收率为间位>对位; 而取代基为甲氧基则恰好相反.当芳香醇和芳香胺上的取代基都为Cl时, 亚胺的收率为98%.该催化剂廉价且易制备, 利用载体磁性回收容易, 但是该催化反应所需的时间较长且反应体系里需要加入过量的碱.
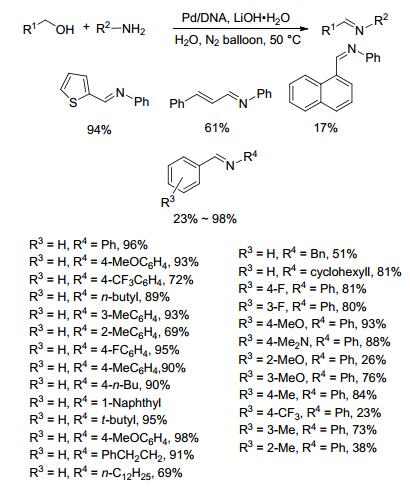
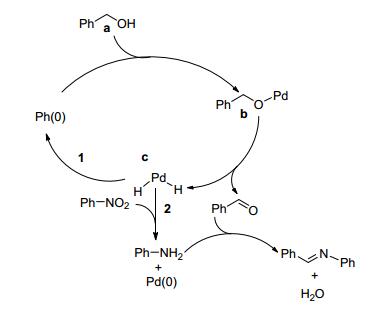
2012年, Wang等[60]以苄醇和苯胺为模型反应, 研究了Pd/DNA、Au/DNA、Pt/DNA、Ag/DNA、Pd/PVP、Pd/starch、Pd/Arabic gum、Pd/diatomite、Pd/MMT、Pd/C、PdCl2等催化剂催化的直接亚胺化反应, 发现相同条件下Pd/DNA的催化效果最好.该方法以1.5 equiv.氢氧化锂为碱、Pd/DNA (2.9 mol%钯)为催化剂, 水为溶剂在50 ℃下反应, 以96%的产率得到目标产物亚胺(Scheme 37).底物拓展发现苄醇类底物中芳环取代基的类型和位置对收率有较大影响, 当底物为2-取代的苄醇或1-萘甲醇时, 目标产物亚胺的收率均较低.当苄醇上的取代基为强供电子基团, 如甲基、甲氧基时, 产物亚胺的收率与芳环上取代基位置之间总体呈现如下规律:对位>间位>邻位.初步的机理研究(Scheme 38)表明, 该反应为一串联反应, 醇先在催化剂作用下脱氢生成醛, 随后醛和胺反应生成亚胺.该催化剂循环使用5次而保持催化活性.该方法以水为溶剂, 条件较温和, 底物适用范围较广, 催化剂回收容易且能循环使用, 该催化方法唯一的不足是需要添加过量的碱氢氧化锂.
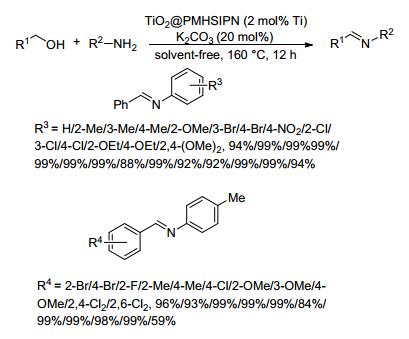
2014年, Yang和Xu等[61]将二氧化钛(TiO2)负载在有机硅上制备了二氧化钛TiO2@PMHSIPN催化剂, 并将其用于催化醇与苯胺类底物直接脱氢偶联制备亚胺的反应.在无溶剂、2 mol%催化剂、20 mol%碳酸钾和氩气氛围下, 苄醇类和芳胺类底物在160 ℃反应12 h, 亚胺的收率为59%~99% (Scheme 39).对于苄醇与苯胺类底物的反应, 无论芳环连有吸电子或供电子取代基, 都能以优异的收率得到目标亚胺; 对于4-甲基苯胺与苄醇类底物的反应, 除2, 6-二氯苄醇与4-甲基苯胺的收率较低外(59%), 也都能以优异的产率得到目标产物, 而且芳环上取代基位置对亚胺收率几乎没有影响.该催化剂以环境友好的有机硅材料和商业上可用的钛前驱体为原料, 避免了过渡金属的使用.该反应体系不需要使用氧化剂或其它添加剂, 且副产物仅为水或氢气, 但该反应温度太高且反应体系中需要碳酸钾的参与.
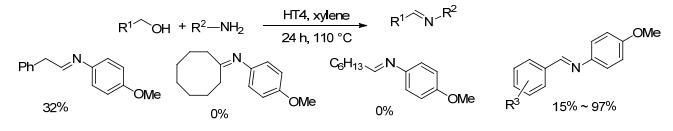
2015年, Voutchkova-Kostal等[62]研究水滑石(HT)类非均相催化剂时, 发现Fe:Mg:Al HT (HT4)能催化醇类物质的脱氢反应以及醇和胺的亚胺化反应, 该催化体系催化4-甲氧基苄醇和4-甲氧基苯胺反应的收率达到了97% (Scheme 40).底物拓展研究发现, 对于苄醇类底物与芳胺类底物的反应, 收率在15%~97%之间.而环辛醇、正辛醇与4-甲氧基苯胺在该条件下不发生反应.作者研究的底物类型较少, 且从有限的反应结果来看, 该催化剂活性较低、底物适用范围有限且总体收率均不高.
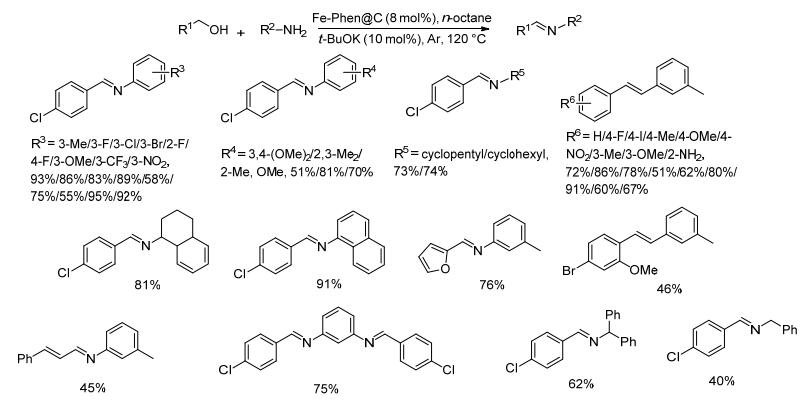
2016年, Balaraman等[63]制备了Fe-Phen@C催化剂, 并将该非均相催化剂用于催化醇与胺的无受体脱氢直接偶联生成亚胺的反应.在8 mol%催化剂、10 mol% t-BuOK和氩气存在下, 醇和胺在正辛烷中120 ℃反应24 h, 目标亚胺的收率良好(Scheme 41).对底物研究发现, 对氯苄醇与不同取代基的苯胺反应时, 当苯胺3, 4位连有二个甲氧基时, 亚胺的收率较低只有51%.对苯胺上取代基的位置研究发现, 亚胺的产率为:间位>对位>邻位.该催化剂对对氯苄醇与正已胺以及己醇与3-甲基苯胺的反应几乎没有效果, 均以痕量收率得到目标产物.该催化剂回收容易可以循环使用六次, 可以放大到克级以上的规模.该催化方法的副产物只有氢气和水, 对环境没有污染, 符合绿色化学的发展理念, 唯一不足是反应体系中需要加入强碱叔丁醇钾.
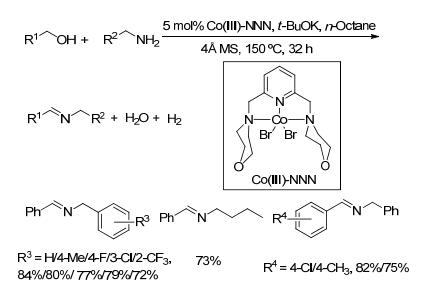
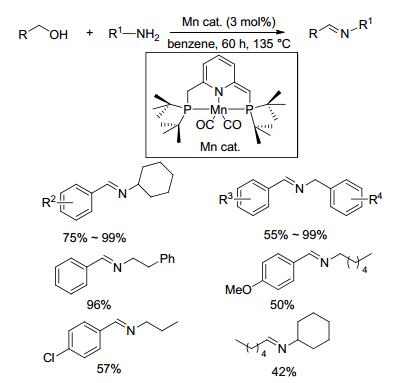
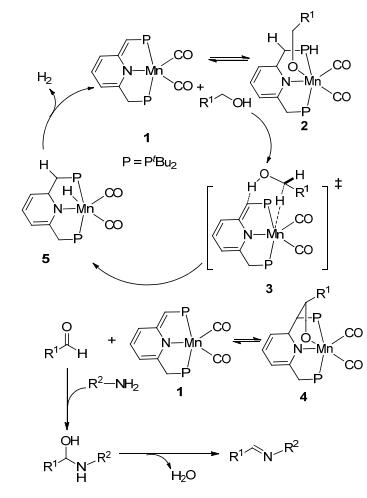
2016年, Milstein等[64]制备了Mn-PNP双齿配合物催化剂, 首次将其用于催化醇与胺脱氢偶联制备亚胺的反应(Scheme 42).该方法以苯为溶剂, 氮气氛围下将物质的量比为1:1的底物醇和胺在135 ℃反应60 h, 以42%~99%的收率生成目标产物亚胺.其可能的催化机理如Scheme 43所示.
同年, Kirchner等[65]也报道了MnI-PNP双齿配合物催化醇与胺脱氢偶联制备亚胺的反应.以甲苯替代上述方法的苯为溶剂, 且在反应体系中加入分子筛以去除反应产生的水, 以3 mol% MnI-PNP为催化剂, 将苄醇和芳胺在140 ℃下反应16 h, 所得亚胺产物的产率为60%~92%.当醇为正丁醇时, 亚胺的收率只有17%, 该催化体系可能不适合长链的醇类底物.该反应体系无需氧化剂参与, 所采用的金属廉价易得, 催化剂易制备且具有绿色环保等特点, 但催化剂的回收和循环使用不易.
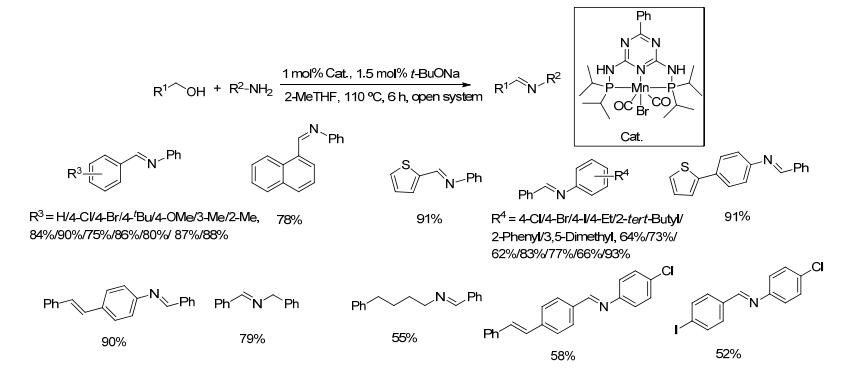
2018年, Kempe等[66]将PN5P型锰催化剂用于催化醇和胺的偶联反应, 并惊奇地发现, 醇和胺的偶联产物类型取决于反应体系中的碱.当采用叔丁醇钾为碱时, 得到的产物为胺; 而采用叔丁醇钠为碱时, 则得到亚胺(Scheme 44).对于苄醇类底物与苯胺的反应, 在催化剂和叔丁醇钠作用下, 亚胺的收率在75%~91%之间, 长链的醇与苯胺则无亚胺产物生成; 对于苄醇类和芳胺类底物的反应, 亚胺的收率在52%~93%之间; 4-苯基丁胺与苄醇反应的收率只为55%.该方法催化剂用量少且适用于碱性较弱的芳胺类底物与醇的反应.锰作为廉价金属, 展现了替代贵重金属铱和钌的巨大潜力.
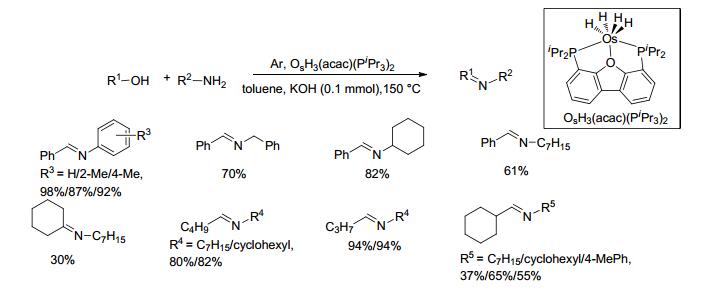
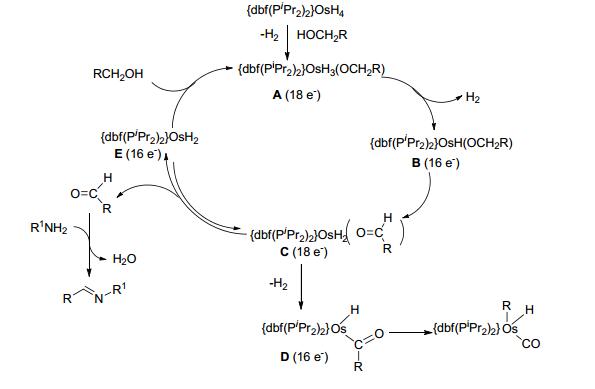
2011年, Honczek等[67]制备了一种磷氧磷(POP)型锇复合物, 并研究了其在醇胺无受体脱氢制备亚胺反应中的催化活性.在0.2 mol%的催化剂和10 mol%的氢氧化钾存在条件下, 醇和胺类底物在150 ℃的甲苯中带压反应3~24 h, 收率在30%~98%之间.对于苄醇和苯胺类底物的反应, 收率在87%~98%之间(Scheme 45).对于链状脂肪醇和脂肪胺或环己胺的反应, 收率在80%~94%之间; 环己基甲醇与正庚胺反应的收率仅为37%, 环己醇与正庚胺的收率在30%.该催化方法反应温度较高, 有一定的底物适用范围, 但总体收率不高.由于作者对每一类底物的类型研究较少, 所以底物结构类型对收率的影响规律尚不易得出.然而, 金属锇是非常有希望作为金属钌的替代物应用于催化醇胺偶联制备亚胺的反应.催化机理如Scheme 46所示.
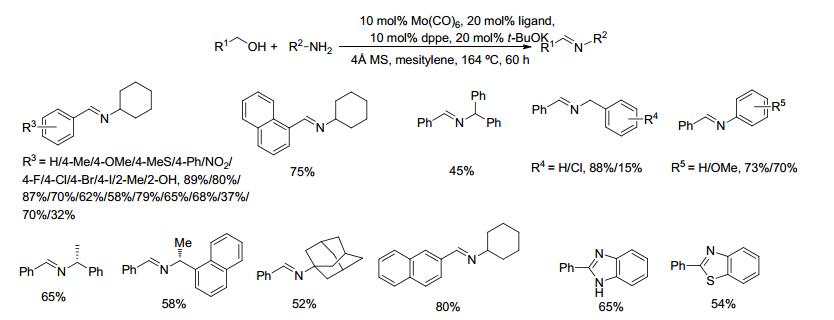
2018年, Madsen等[68]开发了一种基于氮杂环卡宾、dppe和金属钼组成的催化体系, 并将其应用于催化醇与胺脱氢偶联生成亚胺的反应.在10 mol% Mo(Co)6、20 mol%的1, 3-双环己基咪唑氯化铵、10 mol% dppe、20 mol%叔丁醇钾和分子筛存在下, 醇和胺在1, 3, 5-三甲苯中回流反应60 h, 目标亚胺产物的收率在15%~89%之间(Scheme 47).对于苄醇类底物和环己胺的反应来说, 亚胺的收率在32%~89%之间, 芳环的4位上含有供电子取代基时, 亚胺产物收率较高; 4-硝基苄醇与环己胺的反应, 由于硝基发生竞争性还原使得其亚胺收率仅为58%.对碘苄醇则由于脱碘或亚胺还原等副反应的影响, 亚胺产率只有37%. 1-或2-萘基甲醇与环己胺反应的收率分别为75%和80%.不同的胺类底物与苯甲醇反应时, 对氯苄胺由于脱卤等反应的影响, 产率仅为15%.空间位阻大的1-金刚烷基胺与苄醇反应的亚胺收率为52%.该催化方法也适用于苯胺和对甲氧及苯胺与苄醇的反应, 产率分别为73%和70%.该方法催化剂相对廉价, 底物适用范围较广.催化体系虽然需要添加多种配体和碱, 但属首次使用钼类金属作为脱氢催化剂, 具有开创性.
从现有研究成果来看, 金属催化剂催化醇胺偶联制备亚胺, 基本可以分为两类:一类是在氧气或空气存在下, 选择性氧化醇为醛, 随后醛与胺反应生成亚胺, 其中铜、锰、金三类金属的研究较为深入, 而金属钯和钌催化的氧化偶联则具有非常重要的引领意义.另外一类是催化醇和胺脱氢偶联生成亚胺, 相比催化氧化偶联方法, 这类方法更加绿色环保, 除贵金属钌研究较为深入外, 其它如铱、铂、铜、钯、钛、锇、钼、钴、铁、锰等金属的催化醇胺偶联脱氢也相继展开, 虽然不是很成熟, 但都是开创性的工作, 且具有重要的引领作用.金属催化剂的发展为催化合成亚胺提供了新的途径, 虽然已经获得了重要进展, 但是仍然存在不足.催化醇胺氧化偶联方面, 虽然所研究的金属类型已经较多, 但是大都处于初期阶段, 大部分催化体系中仍然需要多种添加物且其底物适用范围不明确; 催化醇胺脱氢偶联方面, 除均相钌类催化剂外, 其它金属类型的研究虽较多但同样处于初期阶段, 也存在催化体系需要多种添加物; 反应温度高时间长, 催化剂回收困难, 底物适用范围有限; 产物收率不高且易生成亚胺被还原的副产物等不足.因此开发新型催化剂时, 应尽可能减少或避免催化体系中的添加物, 在提高催化剂活性和选择性的同时, 还应考虑催化剂的回收循环; 且更要注重廉价金属催化剂的开发.
Marques, C. S.; Burke, A. J. ChemInform 2011, 3, 635.
Bayrak, H.; Demirbas, A.; Karaoglu, S. A.; Demirbas, N. Eur. J. Med. Chem. 2009, 44, 1057. doi: 10.1016/j.ejmech.2008.06.019
Gawronski, J.; Wascinska, N.; Gajewy, J. Chem. Rev. 2008, 108, 5227. doi: 10.1021/cr800421c
Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626. doi: 10.1021/cr100204f
And, S. K.; Ishitani, H. Chem. Rev. 1999, 99, 1069. doi: 10.1021/cr980414z
Nielsen, M.; Worgull, D.; Zweifel, T.; Gschwend, B.; Bertelsen, S.; Jørgensen, K. A. Chem. Commun. 2011, 47, 632. doi: 10.1039/C0CC02417A
Marques, C. S.; Burke, A. J. ChemCatChem 2011, 3, 635. doi: 10.1002/cctc.201000369
Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1996, 118, 4916. doi: 10.1021/ja960364k
Thalji, R. K.; Ahrendt, K. A.; Bergman, R. G.; Ellman, J. A. ChemInform 2010, 33, 9692.
Nieto, S.; Dragna, J. M.; Anslyn, E. V. Chem.-Eur. J. 2010, 16, 227. doi: 10.1002/chem.v16:1
Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. ChemCatChem 2010, 2, 1438. doi: 10.1002/cctc.201000175
Nakajima, R.; Ogino, T.; Yokoshima, S.; Fukuyama, T. J. Am. Chem. Soc. 2010, 132, 1236. doi: 10.1021/ja9103233
Hadjipavlou-Litina, D. J.; Geronikaki, A. A. Drug Des. Discovery 1998, 15, 199.
Akhmetova, V. R.; Khabibullina, G. R.; Rakhimova, E. B. Mol. Diversity 2010, 14, 463. doi: 10.1007/s11030-010-9248-3
Nielsen, M.; Worgull, D.; Zweifel, T.; Gschwend, B.; Bertelsen, Jørgensen, K. A. Chem. Commun. 2011, 47, 632. doi: 10.1039/C0CC02417A
Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626. doi: 10.1021/cr100204f
Xie, J. H.; Zhu, S. F.; Zhou, Q. L. Chem. Rev. 2011, 111, 1713. doi: 10.1021/cr100218m
Marques, C. S.; Burke, A. J. ChemCatChem. 2011, 3, 635. doi: 10.1002/cctc.201000369
Nieto, S.; Dragna, J. M.; Anslyn, E. V. Chem.-Eur. J. 2010, 16, 227. doi: 10.1002/chem.v16:1
Dhakshinamoorthy, A.; Alvaro, M. ChemCatChem 2010, 2, 1438. doi: 10.1002/cctc.201000175
Aschwanden, L.; Mallat, T.; Maciejewski, M.; Krumeich, F.; Baiker, A. ChemCatChem 2010, 2, 666. doi: 10.1002/cctc.201000092
Min, S. K.; Kim, S.; Park, S.; Park, S.; Bosco, W.; Chidrala, R. K.; Park, J. J. Org. Chem. 2009, 74, 2877. doi: 10.1021/jo8026609
Jiang, L.; Jin, L. L.; Tian, H. W.; Yuan, X. Q.; Yu, X. C.; Xu, Q. Chem. Commun. 2011, 47, 10833. doi: 10.1039/c1cc14242a
Chen, G. J.; Ma, H. C.; Xin, W. L.; Li, X. B.; Jin, F. Z.; Wang, J. S.; Liu, M. Y.; Dong, Y. B. Inorg. Chem. 2017, 56, 654. doi: 10.1021/acs.inorgchem.6b02592
Tian, H. W.; Yu, X. C.; Li, Q.; Wang, J. X.; Xu, Q. Adv. Synth. Catal. 2012, 354, 2671. doi: 10.1002/adsc.v354.14/15
Kang, Q.; Zhang, Y. G. Green Chem. 2012, 43, 1016.
Bai, L.; Dang, Z. RSC Adv. 2015, 5, 10341. doi: 10.1039/C4RA14890H
Darapanani, C. M.; Arghya, S.; Galia, M. J. Catal. 2017, 355, 139. doi: 10.1016/j.jcat.2017.09.018
Wei, Y. G.; Yu, H.; Zhai, Y. Y.; Dai, G. Y.; Ru, S.; Han, S. Chem.-Eur. J. 2017, 23, 13883. doi: 10.1002/chem.201703185
And, L. B.; Taylor R, J. K. Org. Lett. 2001, 4, 1637.
Sithambaram, S.; Kumar, R.; Son, Y. C.; Steven, L. J. Catal. 2008, 253, 269. doi: 10.1016/j.jcat.2007.11.006
Mondal, J.; Borah, P.; Sreejith, S.; Nguyen, K. T.; Han, X. G.; Ma, X.; Zhao, Y. L. ChemCatChem 2014, 6, 3518. doi: 10.1002/cctc.201402512
Chen, B.; Li, J.; Dai, W.; Wang, L. Y.; Gao, S. Green Chem. 2014, 16, 3328. doi: 10.1039/c4gc00336e
Zhang, E, L.; Tian, H. W.; Xu, S. D.; Yu, X. C.; Xu, Q. Org. Lett. 2013, 15, 2704. doi: 10.1021/ol4010118
Geng, L. L.; Song, J. L.; Zheng, B.; Wu, S. J.; Zhang, W. X.; Jia, M. J.; Liu, G. Chin. J. Catal. 2016, 37, 1451. doi: 10.1016/S1872-2067(16)62506-8
Sindhuja, E.; Ramesh, R. Tetrahedron Lett. 2014, 55, 5504. doi: 10.1016/j.tetlet.2014.08.035
Sun, H.; Su, F. Z.; Ni, J.; Cao, Y.; He, H. Y.; Fan, K. N. Angew. Chem., Int. Ed. 2009, 48, 4390. doi: 10.1002/(ISSN)1521-3773
Kegnæs, S.; Mielby, J.; Mentzel, U. V.; Christensen, C. H.; Riisager, A. Green Chem. 2010, 12, 1437. doi: 10.1039/c0gc00126k
Liu, P.; Li, C.; Hensen, E. J. Chem.-Eur. J. 2012, 18, 12122. doi: 10.1002/chem.201202077
Soulé, J. F.; Miyamura, H.; Kobayashi, S. Chem. Commun. 2013, 49, 355. doi: 10.1039/C2CC36213A
Huang, R.; Yang, Y.; Wang, D. S.; Zhang, L.; Wang, D. W. Org. Chem. Front. 2017, 5, 203.
Han, L.; Xing, P.; Jiang, B. Org. Lett. 2014, 16, 3428. doi: 10.1021/ol501353q
Mielby, J.; Poreddy, R.; Engelbrekt, C.; Kegnæs, S. Chin. J. Catal. 2014, 35, 670. doi: 10.1016/S1872-2067(14)60033-4
Zhang, G. Q.; Hanson, S. K. Org. Lett. 2013, 15, 650. doi: 10.1021/ol303479f
Sun, Y. W.; Lu, X. H.; Wei, X. L.; Zhou, D.; Xia, Q. H. Catal. Commun. 2014, 43, 213. doi: 10.1016/j.catcom.2013.10.008
Midya, S. P.; Pitchaimani, J.; Landge, V. G.; Madhu, V.; Ekam-baram, B. Catal. Sci. Technol. 2018, 8, 3469. doi: 10.1039/C8CY00859K
Xu, C.; Lai, Goh, L. Y.; Pullarkat, S. A. Organometallics. 2011, 30, 6499. doi: 10.1021/om200883e
Chang, Y. H.; Tanigawa, I.; Takeuchi, K.; Taguchi, H.; Ozawa, F. Eur. J. Inorg. Chem. 2016, 5, 754.
Gnanaprakasam, B.; Zhang, J.; Milstein, D. Angew. Chem., Int. Ed. 2010, 49, 1468. doi: 10.1002/anie.200907018
Cano, R.; Ramón, D. J.; Yus, M. J. Org. Chem. 2011, 76, 5547. doi: 10.1021/jo200559h
Maggi, A.; Madsen, R. Organometallics 2012, 31, 451. doi: 10.1021/om201095m
Jared, W. R.; Sara, A. M.; Simon, D. P.; Jennifer, M. S. Org. Biomol. Chem. 2012, 10, 1746. doi: 10.1039/c2ob06921k
Musa, S.; Fronton, S.; Vaccaro, L.; Gelman, D. Organometallics 2013, 32, 3069. doi: 10.1021/om400285r
Saha, B.; Daw, P.; Sengupta, G.; Rahaman, S. M.; Bera, J. K. Chem.-Eur. J. 2014, 20, 6542. doi: 10.1002/chem.201304403
Oldenhuis, N. J.; Dong, V. M.; Guan. Z. B. Tetrahedron 2014, 70, 4213. doi: 10.1016/j.tet.2014.03.085
Higuchi, T.; Tagawa, R.; Iimuro, A.; Akiyama, S.; Nagae, H.; Mashima, K. Chem.-Eur. J. 2017, 23, 12795. doi: 10.1002/chem.v23.52
Yu, X. J.; Li, Y. Q.; Fu, H. Y.; Zheng, X. L.; Chen, H.; Li, R. X. Appl. Organomet. Chem. 2018, 32, 4277. doi: 10.1002/aoc.v32.4
Shiraishi, Y.; Ikeda, M.; Tsukamoto, D.; Tanaka, S.; Hirai, T. Chem. Commun. 2011, 47, 4811. doi: 10.1039/c0cc05615d
Pérez, J. M.; Cano, R.; Yus, M.; Ramón, D. J. Eur. J. Org. Chem. 2012, 24, 4548.
Tang, L.; Sun, H. Y.; Li, Y. F.; Zha, Z. G.; Wang, Z. Y. Green Chem. 2012, 14, 3423. doi: 10.1039/c2gc36312g
Wang, H.; Zhang, J.; Cui, Y. M.; Yang, K. F.; Zheng, Z. J.; Xu, L. W. RSC. Adv. 2014, 4, 34681. doi: 10.1039/C4RA06685E
Bain, J.; Cho, P.; Voutchkova-Kostal, A. Green Chem. 2015, 17, 2271. doi: 10.1039/C5GC00312A
Jaiswal, G.; Landge, V. G.; Jagadeesan, D.; Balaraman, E. Green Chem. 2016, 47, 3232.
Mukherjee, A.; Nerush, A.; Leitus, G.; Shimon L, J. W.; David, Y. B.; Jalapa N, A. E.; Milstein, D. J. Am. Chem. Soc. 2016, 138, 4298. doi: 10.1021/jacs.5b13519
Mastalir, M.; Glatz, M.; Gorgas, N.; Stçger, B.; Pittenauer, E.; Allmaier, G.; Veiros, L. F.; Kirchner, K. Chem.-Eur. J. 2016, 22, 12316. doi: 10.1002/chem.v22.35
Fertig, R.; Irrgang, T.; Freitag, F.; Zander, J.; Kempe, R. ACS Catal. 2018, 9, 8525.
Esteruelas, M. A.; Honczek, N.; Valencia, M.; Oñate, E.; Oliván, M. Organometallics 2011, 30, 2468. doi: 10.1021/om200290u
Madsen, R.; Azizi, K. ChemCatChem 2018, 53, 1.

图式 1 Pd/AlO(OH)催化剂催化醇和胺直接偶联生成亚胺的反应
Scheme 1 Direct coupling of alcohols and amines catalyzed by Pd/AlO(OH)

图式 2 醋酸钯催化醇和胺直接偶联生成亚胺的反应
Scheme 2 Direct coupling of alcohols and amines catalyzed by Pd(OAc)2

图式 3 含双官能团的非均相复合催化体系Pd-Au@Mn(Ⅱ)-MOF催化醇和胺一锅合成亚胺的反应
Scheme 3 Direct coupling of alcohols and amines catalyzed by Pd-Au@Mn(Ⅱ)-MOF

图式 4 铜催化剂催化醇和胺直接合成亚胺的反应
Scheme 4 Direct coupling of alcohols and amines catalyzed by copper catalysts

图式 5 碱性条件下Cu(ClO4)2•6H2O催化醇和胺合成亚胺的反应
Scheme 5 Direct coupling of alcohols and amines catalyzed by Cu(ClO4)2•6H2O

图式 6 荔枝型氧化亚铜(Cu2O)纳米团聚体催化醇和胺合成亚胺的反应
Scheme 6 Direct coupling of alcohols and amines catalyzed by uniform litchi shaped Cu2O nanoaggregates

图式 7 BT与碘化亚铜组成的催化剂催化醇和胺生成亚胺的反应
Scheme 7 Direct coupling of alcohols and amines catalyzed by BT and CuI

图式 8 BT和CuI催化醇和胺合成亚胺的反应机理
Scheme 8 Proposed mechanism for the catalytic oxidative synthesis of imines catalyzed by BT and CuI

图式 9 无机配体配位的铜催化剂催化醇和胺制备亚胺的反应
Scheme 9 Direct coupling of alcohols and amines catalyzed by inorganic ligand coordinated copper catalysts

图式 10 无机配体配位铜催化醇和胺合成亚胺的反应机理
Scheme 10 Proposed mechanism for the direct coupling of alcohol and amine catalyzed by Cu complex

图式 11 MnO2催化醇和胺合成亚胺的反应
Scheme 11 Direct coupling of alcohols and amines catalyzed by MnO2

图式 12 八面体分子筛催化剂催化醇和胺合成亚胺的反应
Scheme 12 Direct coupling of alcohols and amines catalyzed by K-OMS-2

图式 13 Mn@DVTA-3催化醇和胺制备亚胺的反应
Scheme 13 Direct coupling of alcohols and amines catalyzed by Mn@DVTA-3

图式 14 MnOx/HAP催化醇和胺偶联制备亚胺的反应
Scheme 14 Direct coupling of alcohols and amines catalyzed by MnOx/HAP

图式 15 Fe(NO3)3/TEMPO催化醇和胺制备亚胺的反应
Scheme 15 Direct coupling of alcohols and amines catalyzed by Fe(NO3)3/TEMPO

图式 16 FeOx/HCMK-3催化醇和胺制备亚胺的反应
Scheme 16 Direct coupling of alcohols and amines catalyzed by FeOx/HCMK-3

图式 17 Ru(Ⅱ)NNN配合物催化醇与胺偶联生成亚胺的反应
Scheme 17 Direct coupling of alcohols and amines catalyzed by Ru(Ⅱ)NNN complex

图式 18 PICB-Au/Pd多相催化剂催化醇与胺偶联生成亚胺的反应
Scheme 18 Direct coupling of alcohols and amines catalyzed by PICB-Au/Pd

图式 19 TA-Py-Au(Ⅲ)催化剂催化醇与胺偶联生成亚胺的反应
Scheme 19 Direct coupling of alcohols and amines catalyzed by TA-Py-Au(Ⅲ)

图式 20 Ag-NHC催化剂催化醇与胺偶联生成亚胺的反应
Scheme 20 Direct coupling of alcohols and amines catalyzed by Ag-NHC catalyst

图式 21 Ag-NHC催化剂催化醇与胺偶联生成亚胺可能的反应机理
Scheme 21 Possible mechanism of imine formation catalyzed by Ag-NHC catalyst

图式 22 富土金属催化剂钴催化醇和胺偶联制备亚胺的反应
Scheme 22 Direct coupling of alcohols and amines catalyzed by Co complex

图式 23 钴沸石催化剂催化醇和胺偶联制备亚胺的反应
Scheme 23 Direct coupling of alcohols and amines catalyzed by cobalt zeolite catalyst

图式 24 Co(Ⅱ)-NNN型配合物催化醇与胺偶联制备亚胺
Scheme 24 Direct coupling of alcohols and amines catalyzed by Co(Ⅱ)-NNN complex

图式 25 [Cp*Ir(η3-tpdt)]配合物催化醇与胺偶联生成亚胺的反应
Scheme 25 Direct coupling of alcohols and amines catalyzed by [Cp*Ir(η3-tpdt)] complex

图式 26 含去芳化PNP钳型磷烯配体(PPEP*)的Ir配合物催化醇与胺偶联生成亚胺的反应
Scheme 26 Direct coupling of alcohols and amines catalyzed by Ir complexes

图式 27 均相钌螯合物催化醇与胺偶联生成亚胺的反应
Scheme 27 Direct coupling of alcohols and amines catalyzed by PNP-type ruthenium pincer complex

图式 28 (Ru(OH)3-Fe3O4)催化醇和胺偶联制备亚胺的反应
Scheme 28 Direct coupling of alcohols and amines catalyzed by Ru(OH)3-Fe3O4

图式 29 [RuCl2(IiPr)(p-cymene)]催化醇与胺偶联生成亚胺的反应
Scheme 29 Direct coupling of alcohols and amines catalyzed by RuCl2(IiPr)(p-cymene)

图式 30 钌催化剂催化醇与胺偶联生成亚胺的反应
Scheme 30 Direct coupling of alcohols and amines catalyzed by ruthenium complex

图式 31 新型钌配合物催化醇与胺无受体脱氢偶联生成亚胺的反应
Scheme 31 Direct coupling of alcohols and amines catalyzed by ruthenium complex

图式 32 新型的二钌配合物催化醇与胺偶联生成亚胺的反应
Scheme 32 Direct coupling of alcohols and amines catalyzed by double ruthenium complex

图式 33 Ru-Macho催化醇与胺偶联生成亚胺的反应
Scheme 33 Direct coupling of alcohols and amines catalyzed by Ru-Macho

图式 34 钌配合物[Ru(OCOCF3)2{(S)-dppea}2]催化醇与胺偶联生成亚胺的反应
Scheme 34 Direct coupling of alcohols and amines catalyzed by [Ru(OCOCF3)2{(S)-dppea}2]

图式 35 Pt@TiO2催化醇与胺偶联生成亚胺的反应
Scheme 35 Direct coupling of alcohols and amines catalyzed by Pt@TiO2

图式 36 CuO-Fe3O4催化醇与胺偶联生成亚胺的反应
Scheme 36 Direct coupling of alcohols and amines catalyzed by CuO-Fe3O4

图式 37 Pd/DNA催化醇和胺的直接亚胺化反应
Scheme 37 Direct coupling of alcohols and amines catalyzed by Pd/DNA

图式 38 钯纳米粒子(Pd/DNA)催化醇和胺合成亚胺的反应机理
Scheme 38 Proposed mechanism for the catalytic oxidative synthesis of imines catalyzed by Pd/DNA

图式 39 TiO2@PMHSIPN催化剂催化醇与胺偶联生成亚胺的反应
Scheme 39 Coupling reaction of alcohol and amine to produce imines catalyzed by TiO2@PMHSIPN

图式 40 水滑石(HT4)类非均相催化剂催化醇和胺的亚胺化反应
Scheme 40 Direct coupling of alcohols and amines catalyzed by hydroimide (HT4)

图式 41 Fe-Phen@C催化醇和胺制备亚胺的反应
Scheme 41 Direct coupling of alcohols and amines catalyzed by Fe-Phen@C

图式 42 双齿配合物催化剂Mn-PNP催化醇和胺制备亚胺的反应
Scheme 42 Direct coupling of alcohols and amines catalyzed by Mn-PNP

图式 43 双齿配合物催化剂Mn-PNP催化醇和胺制备亚胺的反应机理
Scheme 43 Proposed mechanism of preparation of imines from alcohols and amines catalyzed by Mn-PNP complex

图式 44 Mn催化剂催化醇与胺偶联生成亚胺的合成的反应
Scheme 44 Direct coupling of alcohols and amines catalyzed by Mn complex

图式 45 OsH3(acac)(PiPr3)2催化醇与胺偶联生成亚胺的合成的反应
Scheme 45 Direct coupling of alcohols and amines catalyzed by OsH3(acac)(PiPr3)2

图式 46 三氢化物OsH3(acac)(PiPr3)2催化的亚胺的合成的反应机理
Scheme 46 Proposed mechanism for the synthesis of imines catalyzed by OsH3(acac)(PiPr3)2

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

 下载:
下载: