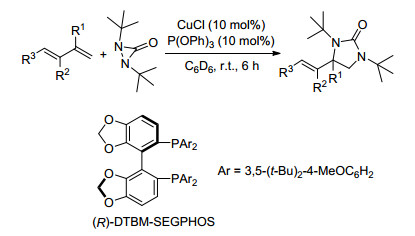
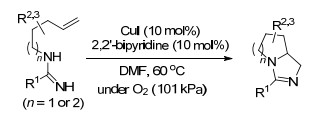
图式 1.
铜(Ⅰ)催化共轭烯烃的二氨化反应
Scheme 1.
Cu(Ⅰ)-catalyzed diamination of conjugated dienes

在有机合成领域, 追求“Ideal Synthesis”是一个经久不衰的话题[1].因此, 有机串联反应和多组分反应, 以其在单步操作中可实现高效的多个化学转化的特点, 引起了化学界的广泛关注.随后, 大量与之相联系的方法出现, 而且还不断地被发现着, 这些发现更充分发挥了相关起始原料的多功能性, 其中烯烃的双官能团化反应一直是个热门话题[2].特别是在C—H功能化领域快速发展的推动下, 过渡金属催化的烯烃的双官能团化反应在近几年取得了巨大的进展[3].
烯烃类化合物由于其便宜易得的特点, 在有机合成的发展中占有极为重要的地位.二十世纪中期, Wacker反应[4]被发现:水溶液中, 在氯化铜及氯化钯的催化作用下, 用空气(氧气)直接把烯烃氧化成醛或酮.这一反应成功地应用到了工业生产中, 同时也广泛地应用于有机合成中, 此后, 对于烯烃的官能团化研究更加受到了研究者们的重视.烯烃是非常普遍的前手性基团, 化学家常通过一步法使那些非手性的化合物转化为手性化合物, 且可以合成高度复杂的化合物, 因此烯烃的双官能团化反应非常重要.
烯烃的双官能团化反应作为一类重要的有机合成反应, 不仅可以经济有效地一步合成多位点反应产物, 而且可以将起始原料转化为多种含有生物活性或药物活性的化合物.近年来, 烯烃的双官能团化反应迅速发展, 主要报道了一些过渡金属如: Rh、Pd、Fe、Cu、Ag、Au等催化的烯烃的双官能团化反应以及一些应用有机小分子催化的反应.由于非金属试剂毒性低、便宜易得、一般符合绿色化学的要求, 因此发展非金属条件下发生的双官能团化反应, 已成为此类反应的一个重要研究方向.
邻二胺作为一种非常重要的官能团, 广泛地存在于生物活性化合物和天然产物中, 是医药制剂和农用化学品中的关键结构元素, 同时也为手性配体、有机催化剂和辅助剂的开发提供了极好的平台[5].因此, 从现有前体化合物快速合成邻二胺一直是有机合成中的一个重要目标, 而人们也为开发高效的合成方法投入了很大的努力.鉴于邻二胺的重要用途, 烯烃的二氨化反应激发了许多合成方法的发展.
2007年, Shi课题组[6]报道了以CuCl作为催化剂, 二叔丁基二氮杂环丙酮为氮源的共轭烯烃的二氨化反应, 具有很广的底物范围, 一些共轭三烯也可以以较好的产率得到二氨化产物, 反应均在端烯位置进行, 且推测该反应可能为自由基反应历程.随后, 2008年该课题组[7]又对此反应进行了不对称方面的研究, 筛选出(R)- DTBM-SEGPHOS为手性配体, 但是只得到了中等的ee值(Scheme 1).
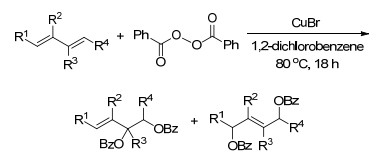
2010年, Shi课题组[8]又报道了铜催化的共轭烯烃与二叔丁基二氮杂环丙酮区域选择性的二氨化反应.与之前不同的是, 这一反应以CuBr为催化剂, 发生在共轭烯烃的非末端位置, 也可以得到较高产率的底物. 2011年, Shi课题组[9]又深入研究了该反应的机理, 一价铜催化剂与底物二烯上的取代基都对该反应的区域选择性有高度的影响, 且反应涉及自由基历程(Scheme 2).
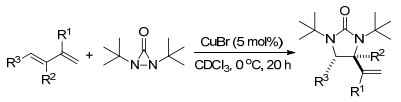
2014年, Shi课题组[10]再次报道了CuBr催化的端烯与二叔丁基二氮杂环丙酮的二氨化反应, 生成咪唑啉- 2-酮, 这是生物活性分子中的一种重要结构, 且此反应产物在此条件下会接着发生脱氢反应(Scheme 3).
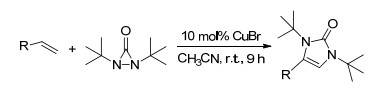
2010年, Chemler课题组[11]首次报道了铜催化剂诱导的烯烃分子内二氨化的不对称反应, 它无论对于分子内还是分子外的氨源都有很好的容忍性, 可以生成各种不同功能化的氮杂环化合物, 产率高达97% (Scheme 4).
2012年, Chiba课题组[12]报道了N-烯丙基脒类化合物以铜(Ⅰ)作为催化剂, 增氧条件下发生[3+2]成环反应(Scheme 5).
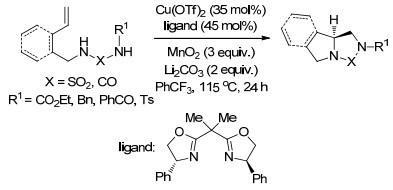
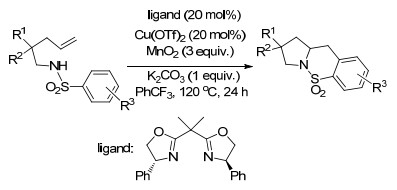
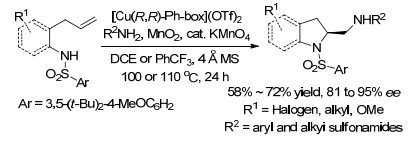
2014年, Chemler课题组[13]再次报道了铜催化的烯烃的二氨基化反应, 且无论是分子内还是分子间反应的底物范围, 都是迄今为止报道的最为广泛的.该氧化反应以便宜易得的MnO2促进, 以很好的产率和极好的对映选择性生成了2-氨基甲基二氢吲哚类化合物和四氢吡咯类化合物(Scheme 6).
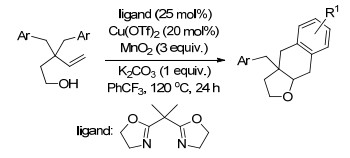
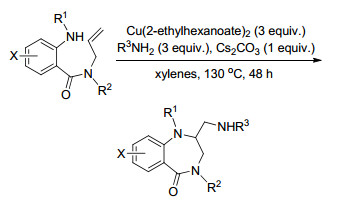
2015年, Chemler课题组[14]又通过烯烃的二氨化反应合成2-氨甲基取代的1, 4-苯并二氮杂卓-5-酮类化合物, 都得到了很好的产率(Scheme 7).
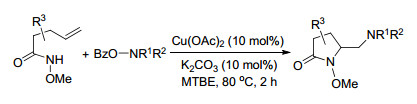
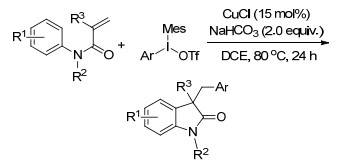
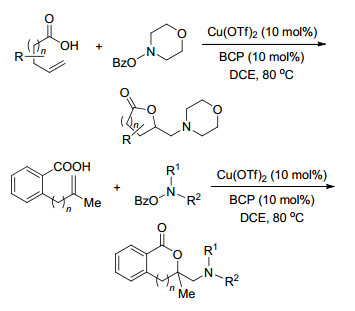
同年, Wang课题组[15]也对铜催化烯烃的二氨化反应做了报道.这是首例铜催化下直接将富电子氨基加成到烯烃上的报道, 以吗啉-4-基苯甲酸酯作为亲电子的氮源和氧化剂, Cu(OAc)2为催化剂, 在80 ℃下, 2 h就可以得到二氨化目标产物.这是一个快速有效地合成二氨化产物的方法(Scheme 8).
2017年, Chemler课题组[16]采用之前未使用过的不饱和氨基甲酸酯作为铜催化二氨化反应的底物, 磺酰胺类、苯胺类、苯甲酰胺、吗啉以及哌啶作为分子外部氨源, 在温和的反应条件下以中等及以上的产率和非对映选择性生成3-氨甲基取代的异噁唑烷类化合物.实验发现, 与2-烯丙基苯胺、4-戊烯基胺相比, 底物羟胺的高反应活性很可能是由于羟胺的强亲核性、强自旋能力或过渡态下较低的跨环张力, 高的反应活性确保了低的反应温度, 反过来低的反应温度又使底物胺的范围更广泛.所以, 富电子胺与氧化条件下发生的铜催化烯烃的二氨化反应并不是本质上不相容的, 相反, 温和的反应条件, 特别是反应温度方面, 有利于其成功利用(Scheme 9).
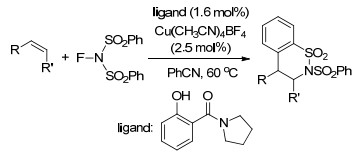
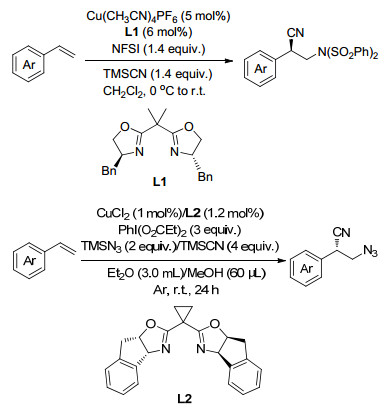
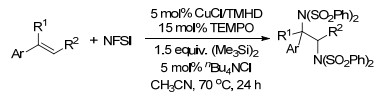
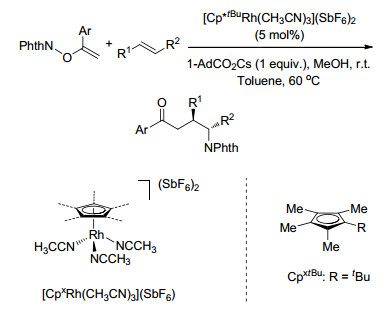
同年, Weng课题组[17]报道了Cu-TEMPO催化的苯乙烯的二氨化反应, 以N-氟代双苯磺酰胺(NFSI)为唯一的氮源, 反应条件温和, 涉及自由基反应历程(Scheme 10).
随后, Liu课题组[18]报道了在Cu(Ⅰ)/磷酸双催化作用下烯烃的不对称自由基二氨化(Scheme 11)和叠氮胺化反应(Scheme 12), 得到了高效率、显著的对映选择性、优良的官能团相容性和广泛的底物范围的对映体富集的α-三取代的吡咯烷类化合物.此外, 得到的吡咯烷类化合物可以显著地促进不对称Michael加成反应的对映选择性, 进一步展示了这一新反应方法的潜力(Scheme 13).
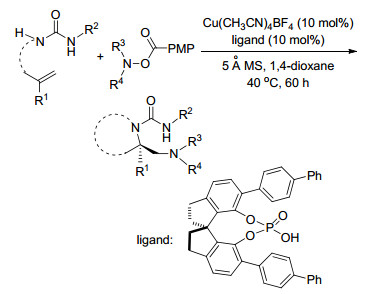
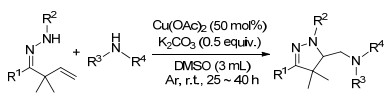
2018年, Wang课题组[19]报道了铜催化下β, γ-不饱和腙的二氨化反应, 以简单的胺作为外部氮源, 二甲基亚砜(DMSO)作为氧化剂, 在温和的反应条件下生成吡唑啉类化合物, 这一类产物在有机合成及药物中都是重要的组成部分(Scheme 14).
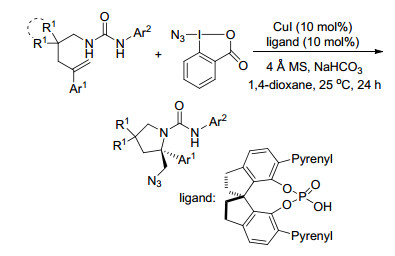
2015年, Zeng课题组[20]首次报道了Cu(Ⅱ)催化的N-烯丙基脲的不对称分子内环化反应, 为手性邻二胺双环杂环化合物的快速合成提供了一种有效的方法, 其产率为中等及以上, ee可高达98%.其中N, N-螯合双齿噁唑啉配体与碱金属碳酸锂一起对获得高对映选择性起了至关重要的作用(Scheme 15).
环邻氯胺酮广泛地存在于天然产物和生物活性化合物中, 是一种多用途的合成中间体[21].
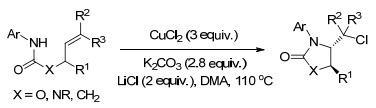
2016年, Xu课题组[22]报道了Cu(Ⅱ)催化的烯烃的氨氯化反应, CuCl2作为氧化剂和氯源, 烯烃类、炔烃类、丙二烯类各种碳-碳多重键化合物均适用于此反应.反应操作简单、底物拓展性强, 为生成邻氯氨化合物提供了有效的途径(Scheme 16).
碳-碳叁键是有机化学中最有价值的官能团之一, 它作为有机合成中的合成中间体, 具有多种反应活性, 在生物化学和材料科学中具有广泛的应用前景.
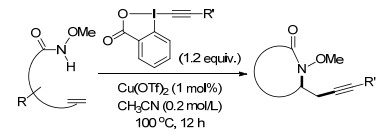
2017年, Wang课题组[23]报道了温和条件下, 以乙炔基苯碘酰酮(EBX)为试剂, 铜催化(催化剂用量仅为1 mol%)的烯烃的氨基炔化反应.该反应不仅可以快速地与炔基成键一步生成氮杂环化合物, 而且对端烯和内烯以及不同的炔基类化合物都具有很好的普适性.可广泛地应用于合成、生物共轭和分子成像技术(Scheme 17).
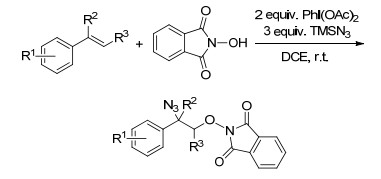
同年, Wang课题组[24]又报道了CuOAc催化的未活化烯烃的氨基叠氮化反应, 反应将酰胺和叠氮从两个不同的氨基前体连接到端烯和内烯上, 具有显著的区域选择性和立体选择性.机理研究表明, 这一二氨化反应是先通过亲核氨基环化反应, 然后利用亲电的叠氮碘形成分子间C—N键.这一反应途径与以往的叠氮碘烷引发的烯烃功能化反应不同, 表明叠碘碘烷具有新的反应活性.此反应不仅可以快速引进各种功能化的二氨分子, 还能在复杂的含氮骨架上引入叠氮化合物, 从而促进其在生物医学和材料研究中的研究和应用(Scheme 18).
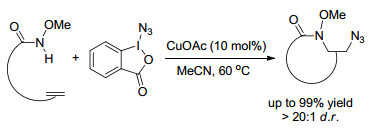
2017, Xie课题组[25]报道了一种新的铜催化烯烃与α-溴代烷基酯和伯胺的串联环化反应, 合成了多取代的吡咯烷-2-酮.该反应一次形成三个新的化学键, 一个C—C键和两个C—N键, 是首个三组分的[2+2+1]成环的烯烃的双官能团化反应, 包括氨基烷基化反应和酰胺化反应, 且研究表明该反应历经自由基历程(Scheme 19).
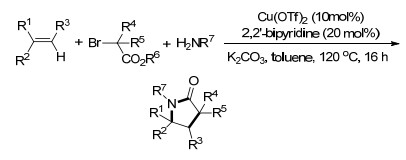
2017年, Liao课题组[26]报道了以手性亚砜磷配体, CuCl为催化剂的苯乙烯的不对称硼胺化反应, 以亲电性胺试剂和联硼酸频哪醇酯分别作为氮源和硼源, 合成的手性β-氨基烷基硼酯可转化为有用的手性β-羟胺类化合物(Scheme 20).
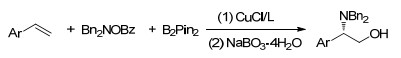
2018年, Hull课题组[27]通过对α-卤代羰基化合物和胺类化合物进行研究, 报道了铜催化的三组分的碳胺化反应.它可以将易得的烯烃、功能化的烷基卤代物与胺偶联在一起, 且三组分的反应范围都很广, 产率由一般到很好.初步的机理研究发现反应经历碳自由基历程(Scheme 21).
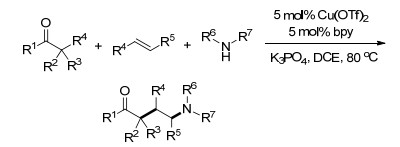
2007年, Chemler课题组[28]报道了一种新颖的铜催化烯烃的不对称芳氨化反应, 反应为芳基磺酰胺对分子内末端烯烃的加成, 从而得到手性磺内酰胺. MnO2作为氧化剂使催化循环顺利地实现, 并以较高的产率得到了目标产物.用手性双噁唑啉配体, 得到了高达94%的ee值(Scheme 22).
烯烃的分子内碳氨化反应是合成氮杂环的有效方法. 2010年, 该课题组[29]又报道了铜催化的烯烃的分子内对映选择性芳基氨化反应, 使用同样手性双噁唑啉配体, 得到的产物同时具有两个手性中心, 且得到了高达20:1的dr值与97%的ee值(Scheme 23).

2013年, Kanai课题组[30]报道了铜催化的分子间的芳氨化反应, 以末端烯烃与NFSI反应, NFSI同时提供芳源和氮源, 得到了产率达91%的磺内酰胺类目标产物, 且反应有较好的底物范围(Scheme 24).
光学纯的2, 2-二芳基乙胺骨架作为药理学中多巴胺受体激动剂的重要构造, 广泛地存在于生物活性分子及药物中.因此, 对映选择性的2, 2-二芳基乙胺骨架是高度需求的, 但是, 关于这一物质的有效的合成方法报道太少.
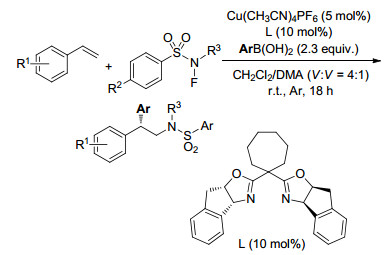
2017年, Lin课题组[31]报道了烯烃的分子间不对称芳基氨基化反应, 以一价铜为催化剂, N-氟代-N-芳基磺酰胺为氨源, 与Cu(Ⅰ)反应释放出氨自由基, 加成到烯烃后产生的苄基自由基与L*CuⅡAr偶联得到对映选择性的芳基化产物(Scheme 25).反应具有很好的对映选择性和底物普适性, 生成了可用于合成生物活性分子的2, 2-二芳基乙胺类化合物.
2012年, Chemler课题组[32]进一步报道了铜催化的烯烃分子内芳基醚化反应生成稠环和桥环两种四氢呋喃类化合物, 该反应具有很好的官能团容忍性, 但手性控制无法实现(Scheme 26).
2013年, Duan课题组[33]报道了铜催化丙烯酰胺类化合物的芳基苄基化反应, 生成氧化吲哚衍生物, 得到了高达96%的产率, 且很好地体现了原子经济性(Scheme 27).
同年, Li[34]、Fu[35]、Zhao[36]课题组均报道了铜催化的烯烃的双芳基化反应, 以高价碘试剂为芳基源生成3, 3-二取代的氧化吲哚衍生物(Scheme 28).
含氟类化合物因其独特的生物活性, 在材料化学、药物化学等多个领域得到了极其广泛的应用, 而三氟甲基具有较低的生物毒性及较强的亲脂性, 因此研究者们对其有了越来越多的兴趣[37].
硫氰类化合物是一类重要的反应中间体, 它可以用于合成具有生物活性的分子, 也可以合成橡胶硫化的加速剂.
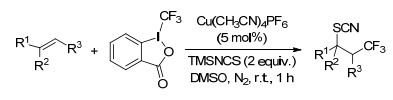
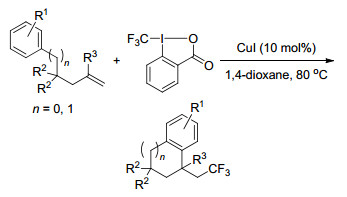
2015年, Liu课题组[38]以三甲硅烷基异氰酸酯(TMSNCS)作为硫氰源, 以Togni试剂作为三氟甲基源, 在Cu(Ⅰ)的催化作用下, 同时将硫氰基和三氟甲基引入分子中.这一反应化学选择性高、反应条件温和、底物普适性及官能团兼容性都比较好.反应历经自由基反应历程, 先生成三氟甲基自由基, 之后对烯烃加成生成β-CF3的苄基自由基, 最后与TMSNCS反应形成C—S键(Scheme 29).
同年, Sodeoka课题组[39]报道了CuI/Togni试剂催化的烯烃的芳基三氟甲基化反应, 以较高的产率得到了三氟甲基化的碳环化合物和杂环化合物(Scheme 30).
2014年, Liang课题组[40]也报道了铜催化的分子内的芳基三氟甲基化反应, 以N-芳基丙烯酰胺类化合物为原料, 以便宜易得的Langlois试剂为三氟甲基源, 室温条件下在水相中发生自由基反应历程生成目标产物(Scheme 31).
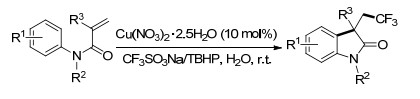
2013年, Sodeoka课题组[41]报道了铜催化的烯基胺的三氟甲基化反应, 包括烯丙基苯胺衍生物的N-迁移氧三氟甲基化反应、氨基三氟甲基化反应及三组分一锅法偶联反应.这些高效的反应将为含有β-三氟甲生物活性化合物的合成开辟一条新的途径(Scheme 32).
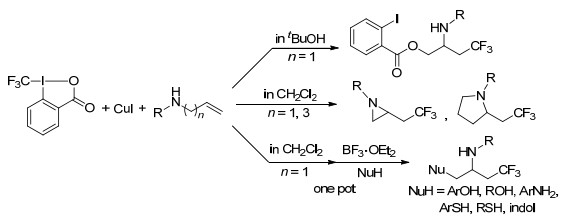
2014年, Tan和Liu课题组[42]首次报道了以(TMS)CF3为CF3源的铜催化分子内未活化烯烃的氨基三氟甲基化反应.该反应与以往的反应相比, 不仅扩大了底物范围, 避免了使用昂贵的亲电CF3试剂和光氧化还原策略, 而且提高了官能团的容忍性, 体现了其在医药化学及材料科学方面的应用性(Scheme 33).
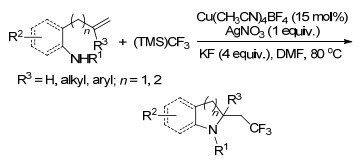
同年, Liu课题组[43]又报道了以铜作为催化剂, Togni试剂为三氟甲基源, 未活化烯烃与多种氨基亲核试剂的氨基三氟甲基化反应, 以优良的产率得到了吡咯烷类及二氢吲哚类化合物(Scheme 34).
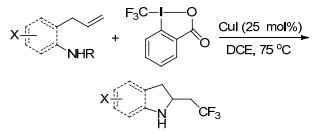
2016年Liu课题组[44]首次报道了CuCl催化下烯烃的不对称自由基氨基三氟甲基化反应.该反应以新奇、简单、高效的方法直接获得了各种含CF3取代基的吡咯烷骨架化合物, 且该反应具有极好的效率、显著的对映选择性及官能团容忍性.而且这一反应的成功不仅仅归功于Cu(Ⅰ)/磷酸催化体系, 含两个N—H的尿素作为亲核试剂和导向基(Scheme 35).
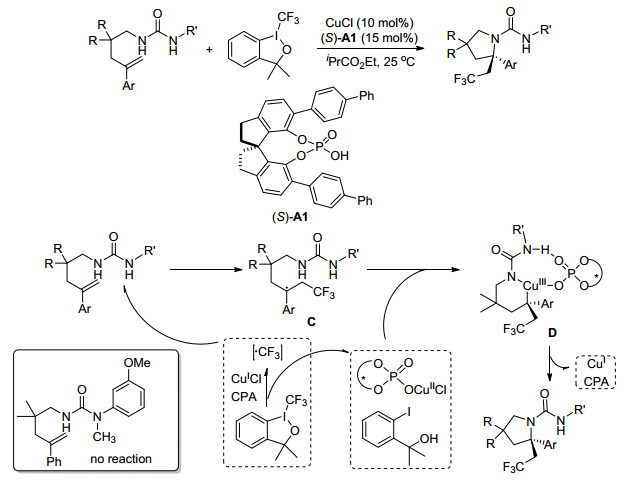
2016年, Wang课题组[45]报道了一种以酰胺为亲核试剂的铜催化烯烃的氨基三氟甲基化反应.该方法可以以较好的产率生成各种含CF3的内酰胺, 反应条件温和, 具有很广的底物范围.为制备含CF3的内酰胺提供了一种有价值的方法, 在医药和农用化学品方面具有很大的应用价值(Scheme 36).
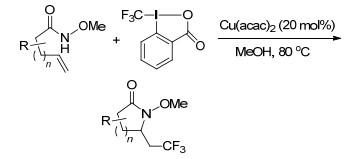
2017年, Liu课题组[46]再次报道了铜催化的不对称自由基氨基氟烷基化反应和氨基二氟甲基化反应.该反应以商业可用的氟烷基磺酰氯为自由基源, 与烯烃反应, 均以极好的产率和对映选择性获得四种类型(β-全氟丁基、β-三氟甲基、β-二氟乙酰基及β-二氟甲基)α-叔碳吡咯烷骨架化合物.该反应成功的关键不仅在于引入了CuBr/手性磷酸双催化体系, 而且还在于使用碳酸银抑制由原位生成的HCl引起的强背景反应和侧氢化胺反应.广泛的底物范围、极好的官能团容忍性、产品多功能化使该方法更加实用(Scheme 37).
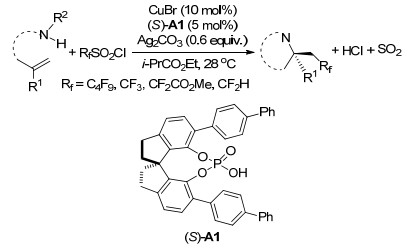
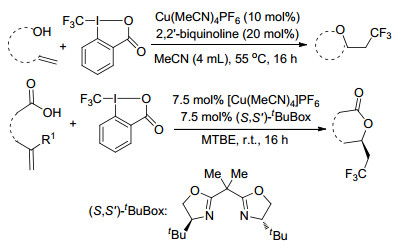
2012年, Buchwald课题组[47]报道了Cu(Ⅰ)/2, 2'-联喹啉催化体系下发生的一种温和、通用、简便的未活化烯烃的氧三氟甲基化反应, 这一方法从简单的起始原料来合成有用的含CF3骨架的化合物.其中, Togni试剂作为三氟甲基源, 羧酸、醇类、酚类均可作为此反应条件下的亲核试剂.随后, 该课题组[48]利用手性双噁唑啉配体研究了以上反映的对映选择性, 并获得了较高的ee值(Scheme 38).
随后, 2013年, Qing课题组[49]报道了铜催化的烯烃与三氟甲基磺酸钠和羟肟酸的三组分氧三氟甲基化反应, 该反应以CuSO4•5H2O为催化剂, 采用稳定廉价的NaSO2CF3作为三氟甲基源, 反应条件温和, 具有良好的官能团容忍性(Scheme 39).
2016年, Yu课题组[50]首次利用无毒易得的温室气体CO2与烯丙胺在铜催化作用下, 和Togni’s试剂发生氧三氟甲基化反应, 高效高选择性地合成具有生理活性的含三氟甲基的2-噁唑烷酮类化合物.特别值得注意的是, 这是在氧化还原中性及温和的条件下发生的高化学选择性、高区域选择性及高非对映选择性的反应.此外, 该反应具有良好的官能团兼容性及底物拓展性, 易实现产品的多样化(Scheme 40).
2017年, Liu课题组[51]以非手性吡啶作为辅助配体, 在CuI/磷酸双催化作用下, 首次实现了烯烃与醇的不对称自由基氧三氟甲基化反应.这一反应条件温和, 底物范围广, 具有极好的官能团容忍性, 为获得对映选择性的三氟甲基取代的四氢呋喃类化合物提供了一种独特的、直接的途径(Scheme 41).
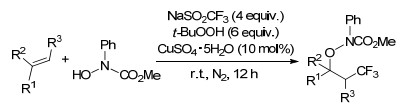
2018年, Liu课题组[52]通过铜催化烯烃肟化合物与Togni’s试剂的不对称自由基氧三氟甲基化反应, 得到了对映选择性的α-叔碳立体中心的含CF3的异噁唑啉类化合物.该反应成功的关键在于合理设计以金鸡纳-生物碱基磺胺为中性/阴离子混合配体, 有效控制包含游离烷基自由基的铜催化反应的立体化学.将产物转化为含CF3基团的手性1, 3-氨基醇, 进一步说明该方法的实用性(Scheme 42).

2016年, Liu课题组[53]首次成功研发了烯烃的自由基1, 2-双官能团化型甲酰化反应.该反应由未活化烯烃的三氟甲基化、叠氮化、磺酰化、全氟烷基化或二氟甲基化引发1, 2-、1, 4-和1, 5-甲酰自由基迁移而发生.这是一种新颖的、可持续的自由基反应, 且能以良好的效率、显著的选择性和良好的官能团容忍性生成β-官能团化醛类化合物.这一方法可用于生成苯并九、十、十一元环化合物以及多功能化的桥环化合物(Scheme 43).
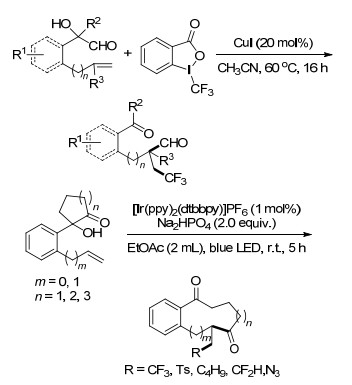
2017年, Zhu课题组[54]首次报道了铜催化烯烃的酰基三氟甲基化反应, 以优良的产率和非对映选择性得到了三氟甲基茚酮类化合物及相关的环酮类化合物, 为生物活性化合物的合成提供了有效的方法(Scheme 44).
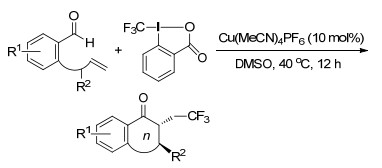
2018年, Liu课题组[55]报道了一种新颖的铜对映选择性催化的苯乙烯的三氟甲基炔基化反应, 反应在非常温和的条件下通过自由基传递过程以高产率和极好的对映选择性获得了结构多样的含CF3基团的炔基化合物.该反应底物范围广, 官能团容忍性好.此外获得的三氟甲基炔基化产物可以转化为具有综合用途的手性末端炔烃、丙二烯、Z-烯烃及CF3修饰的非甾体抗炎药(Scheme 45).
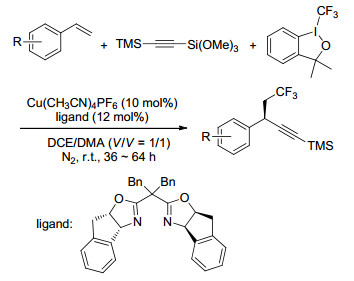
有机叠氮化合物是一类重要的结构单元和有机活性中间体[56], 可以在有机合成中进行多样性转换, 例如转化为异氰、胺或者其它杂环化合物, 所以叠氮化合物可以作为天然产物和生物活性化合物的前体, 合成药物、分子配体及生物活性化合物.此外, 有机叠氮化合物在多肽化学、聚合物合成及材料科学中都有非常广泛的应用[57].
2014年, Jiao课题组[58]报道了铜催化3-取代吲哚的氧叠氮化反应和烷氧化叠氮化反应, 这一反应条件温和, 铜催化下高价碘叠氮试剂产生叠氮自由基, 然后迅速地经过脱芳构化作用以优良的产率得到目标产物, 反应不需要加入任何的碱或配体(Scheme 46).
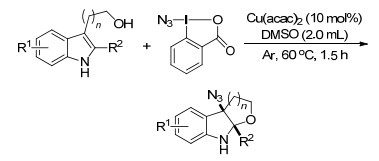
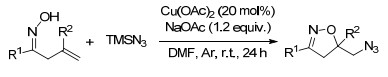
随后, Xu课题组[59]报道了烯烃的分子内自由基氧叠氮化反应, 以便宜易得的TMSN3作为叠氮试剂, 有机碱(如NaOAc、Na2CO3)可有效地促进肟自由基的形成, 在温和的反应条件下, 以优良的产率生成叠氮取代的异噁唑啉类化合物(Scheme 47).
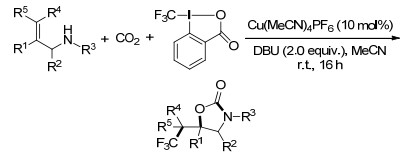
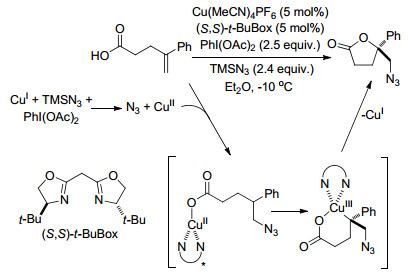
2015年, Buchwald课题组[60]报道了通过铜催化的烯烃的自由基氧化官能团化作用, 对映选择性地合成功能化内酯的反应, 其中含有链烯烃羧酸的分子内氧叠氮化反应.反应用了催化量的Cu(MeCN)4PF6、TMSN3和PhI(OAc)2来促进反应的进行, 其中叠氮自由基和Cu(Ⅱ)络合物就是由Cu(Ⅰ)与TMSN3和PhI(OAc)2发生氧化还原反应得到的, 之后叠氮自由基加成到烯烃上形成三级碳自由基, 而三级碳自由基在手性Box配体的作用下, 立体选择性地与CuⅡ部分结合, 最后C—O还原消除生成内酯目标产物和CuⅠ催化剂(Scheme 48).
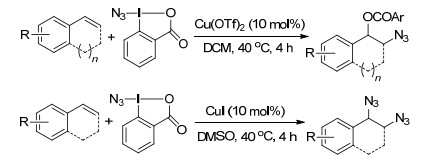
2015年, Loh课题组[61]报道了铜催化烯烃的氧叠氮化反应和二叠氮化反应, Togni试剂为叠氮自由基源, 反应条件温和, 官能团兼容性强, 以较好的产率生成了芳基叠氮化合物(Scheme 49).
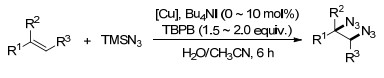
2017年, Bao课题组[62]报道了在乙腈或水溶液中, 无配体的情况下, 以TMSN3为氮源的铜催化的烯烃的二叠氮化反应.反应条件温和, 具有很广的底物范围, 芳基乙烯、未活化的烯烃及双烯类均适用于此反应.其在有机溶剂或水溶液中均可反应的条件, 为实际应用提供了更多的可能(Scheme 50).
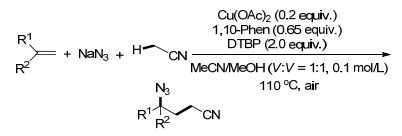
2017年, Zhu课题组[63]报道了首例乙腈和叠氮化钠参与的铜催化的叠氮氰甲基化反应, 通过形成C—N键和C—C键生成γ-叠氮烷基腈.这一目标产物又很容易转化为1, 4-二胺、γ-氨基腈、γ-叠氮酯和γ-内酰胺等在有机合成和药物化学中有用的化合物(Scheme 51).
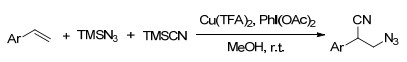
2014年, Wang课题组[64]报道了铜催化芳基烯烃的分子间叠氮氰化反应, 以优良的产率得到了一系列的3-叠氮-2-芳基丙腈类化合物, 为3-氨基-2-芳基丙酸的合成提供了一种潜在的策略(Scheme 52).
2017年, Lin与Liu课题组[65]报道了通过自由基历程的铜催化烯烃的分子间不对称氨氰基化反应和叠氮氰化反应, 这一过程中苄基自由基被手性双噁唑啉配体/Cu(Ⅱ)氰化物的络合物选择性地捕获, 高效地生成各种对映体富集的β-氨基/叠氮化烷基腈.而β-叠氮化烷基腈可以转化成一系列具有价值的光学活性胺基建筑材料和生物活性化合物(Scheme 53).
烯烃的双官能团化反应, 包括氨氧化反应、二氨化反应、氨卤化反应等已经得到了广泛的发展, 相比之下, 共轭烯烃的双官能团化反应还没有引起太多的关注. 2016年, Bao课题组[66]报道了铜催化1, 3-二烯的二酯化反应, 且反应过程可能有烯丙基自由基参与, 后以优良的产率生成了烯丙基二酯衍生物, 这类产物是合成天然产物的重要中间体(Scheme 54).
2016年, Wang课题组[67]报道了铜催化不饱和羧酸的氨基内酯化反应以及类似的分子间烯烃的三组分氨基氧化反应, 反应条件温和, 底物范围广泛, 通过邻苯甲酰羟胺促进烯烃的亲电氨化反应, 生成胺基内酯以及1, 2-氨基醇衍生物(Scheme 55).
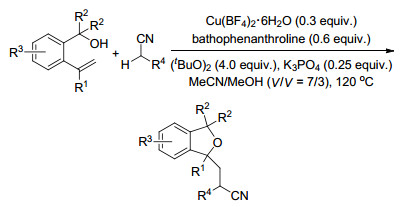
2016年, Zhu课题组[68]报道了烯烃的烷氰基环醚化反应, 铜催化下, 以MeCN/MeOH为溶剂, 烷基氰作为烷基来源, 通过形成C—C键、C—O键生成1, 3-二氢异苯并呋喃化合物, 并且可以通过此方法合成抗抑郁的药物西酞普兰(Scheme 56).
继Warcker反应被发现之后, 过渡金属催化烯烃的双官能团化反应一直是人们研究的热点, 因其显著的高效性, 人们对它的关注度从未降低, 新策略不断被提出, 新颖的底物不断被发现着, 各种催化机理的提出、验证, 都在推动这一领域的发展, 而近几年人们对其研究也从未停断.
2010年, Yoon课题组[69]报道了烯烃分子间的氨羟基化反应, 反应以Fe(acac)3为催化剂, 其中氧杂吖丙啶开环既作为氧源又作为氮源, 在乙腈中与烯烃发生1, 2-加成反应并以较高的产率生成2, 4-取代的噁唑烷化合物.反应具有良好的底物普适性, 可以与各种烷基烯烃、芳基烯烃、共轭烯烃以及共轭烯炔类化合物很好的发生反应得到目标产物, 反应所用的铁催化剂不仅价格便宜且毒性较低, 而且该反应对有机药物的合成具有重要的应用价值(Scheme 57).
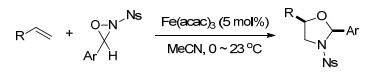
2013, Xu课题组[70]报道了二价铁催化的烯烃与功能化羟胺的分子内氨羟基化反应, 这一高效的分子内反应以优异的选择性(dr>20:1)合成了具有应用价值的氨基醇.反应没有添加其他金属盐或者高价碘来做氧化剂, 只以K4Fe(CN)6作为催化剂, 乙腈作为溶剂, 在70 ℃的条件下生成五元环酰胺酯类化合物.该反应具有良好的底物普适性, 各种烷基烯烃和芳基烯烃都以中等及以上的产率得到了相应的产物.作者认为该反应是先通过铁与配体形成新的络合物, 之后与氨基络合成新的中间体, 可以发生氨羟基化反应生成五元环化合物, 也可以生成氮杂环丙烷(Scheme 58).
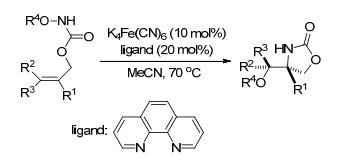
2014年, 该课题组[71]再次报道了铁催化烯烃分子间的氨氧化反应, 以Fe(OTf)2为催化剂, 稳定的功能化的羟胺作为胺化试剂和氧化剂, 这一方法对底物烯烃有很好的普适性, 包括那些对现有的氨氧化反应不适用的烯烃, 也可以得到具有区域和立体化学骨架的氨基醇衍生物(Scheme 59).
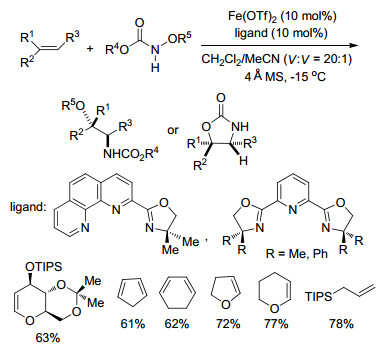
2016年, Xu课题组[72]又报道了铁催化的非对映选择性烯烃的直接二氨化反应, 催化剂和配体与之前相同, 室温反应, 且同样具有很好的底物普适性, 可以得到高达90%的产率和20:1的dr值.反应历经自由基反应历程, 其中路易斯酸的活化作用对第一次叠氮基团的迁移是必不可少的, 而铁催化剂与叠氮基团的第二次迁移相关(Scheme 60).
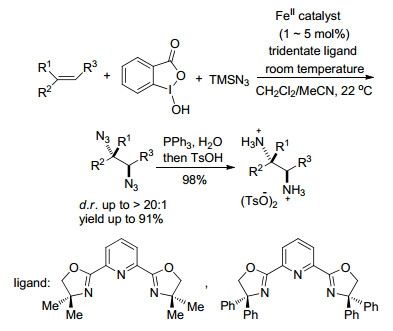
2017年, Zhang与Bao课题组[73]通过一系列反应(自由基加成、氧化和Ritter反应)建立了芳乙烯类的烷基氨化反应和α, β-不饱和酯类的非对映选择性反-烷基氨化反应, 以铁为催化剂, 使简单的烯烃以优良的效率合成了胺类和双取代的β-氨基酸类化合物.而且, 得到的高非对映选择性β-氨基酸化合物可以进一步用于其他的领域.此外, 这一自由基-极性交叉反应是通过铁催化剂和双功能的烷基过氧化物来确保进行的.其中, 烷基二酰基过氧化物可从脂肪酸合成, 作为烷基化试剂和内部氧化剂.且计算研究表明把腈加成到碳正离子上是非对应立体选择性的决定步骤(Scheme 61).
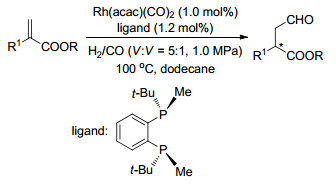
2011年, Buchwald等[74]将具有较大空间位阻的手性双膦配体应用于铑催化的α-烷基丙烯酸酯的不对称氢甲酰化反应中, 获得了较高产率及优良对映选择性的直链醛产物(Scheme 62).
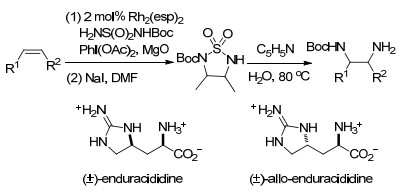
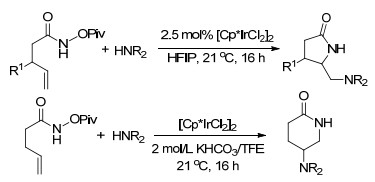
2014年, Bois课题组[75]报道了铑催化烯烃的二氨化反应, 采用一步法串联反应, 包括铑催化形成氮杂环丙烷, 随后NaI促进氮杂环丙烷产物的重排以高产率生成环化磺酰胺.得到的环化磺酰胺化合物在热水和吡啶中可以开环生成不同保护基团的二氨产物.由于环化磺酰胺产物简单温和的开环方式, 使得它具有很好的应用, 如合成一些天然多肽抗生素的氨基酸组分: (±)-endur- acididine、(±)-allo-enduracididine (Scheme 63).
2015年, Rovis课题组[76]报道了铑催化下烯烃的双官能团化反应得到顺式的二取代的芳胺化产物.反应以苯氧基邻苯二甲酰亚胺和三价铑为催化剂, 形成了新的庞大的茂基基团, 也因此改变了反应固有的化学选择性, 其中以甲醇作为溶剂也是至关重要的, 它使邻苯二甲酰亚胺基团经历了原位开环.且机械实验表明, 羰基的碱度通过配位饱和使Rh(Ⅲ)中间体稳定, 从而发生还原消除而不是环丙基化(Scheme 64).
2016年, Gandon课题组[77]报道了铑催化的烯烃与乃春发生的双官能团化反应, 以优良的产率生成了氨氧化产物和二氨化产物.反应先生成氮杂环丙烷, 然后由铑与乃春键合以诱导亲核开环反应(Scheme 65).
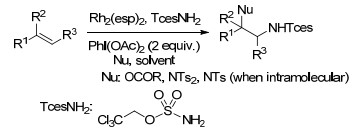
2017年, Lin课题组[78]报道了锰催化烯烃的电化学二氯化反应, 该方法操作简单、可持续且可高效的得到多种邻二氯化合物, 特别是具有氧化活性官能团(如醇、醛、硫化物和胺)的烯烃, 可被转化成具有高化学选择性的邻二氯化合物.这一反应以电为主要能源输入, H2和Mg(OAc)2为得到的唯一副产物(Scheme 66).
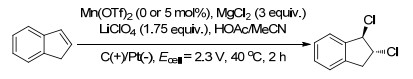
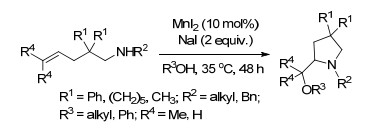
同年, Duan课题组[79]通过MnI2催化非功能化烯烃的分子内碘氨化反应, 随后吖丙啶离子与醇或酚类反应发生的开环反应制备了脯氨醇醚类化合物.在催化量的MnI2和2 equiv. NaI催化下, 乙醇溶剂中, 不同的4-烯-1-氨底物发生分子内氨基烷氧化反应生成产率高达90%的2-(烷氧烷基)吡咯烷产物(Scheme 67).
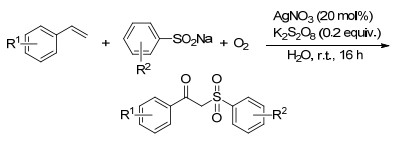
2014年, Yadav课题组[80]报道了温和条件下银催化的烯烃的分子间氧硫化反应, 反应经历自由基历程, 生成β-酮-砜类化合物.反应中, 亚硫酸钠被一价银氧化为亚磺酸基自由基, 之后亚磺酸自由基进攻烯烃形成碳中心的自由基, 其与氧气进一步形成氧中心自由基, 再与碳中心自由基反应并脱去氢自由基得到产物.该反应室温条件下进行, 以水为溶剂, 反应条件温和, 16 h完成就可以得到高产率的产物(Scheme 68).
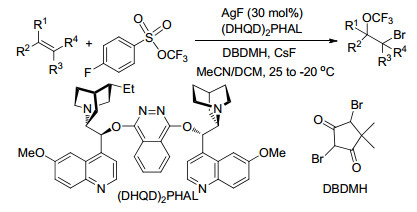
2017年, Tang课题组[81]以三氟甲基芳基磺酸酯(TFMS)为三氟甲氧基化试剂, 报道了银催化烯烃的分子间不对称溴三氟甲基氧化反应, 与其他三氟甲氧基化试剂相比, TFMS不仅合成方便, 热稳定且反应活性好, 能以优良的产率及选择性得到溴-三氟甲氧基化目标产物.另外, 此反应操作简单, 反应条件温和, 底物范围广, 官能团兼容性好, 可以实现克级规模的制备, 还可以用于复杂天然产物或天然产物类似物中双键的衍生化, 在有机合成及药物修饰改造中具有很大的应用价值(Scheme 69).
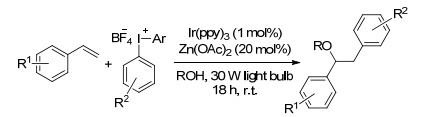
2013年, Greaney课题组[82]报道了一个光催化的三组分串联反应, 以Ir(ppy)3为催化剂, 苯乙烯、芳基化合物及异原子的亲核试剂(如醇、水或腈)反生碳氧/氨化反应, 其中四氟硼酸为碳源, 醇为氧源.该反应不仅反应条件温和, 且底物普适性很好, 无论一级醇还是二级醇都可以得到相对应的产物(Scheme 70).
2016年, Cavallo课题组[83]报道了氮杂环铱复合物作用下未活化烯烃的氢氨化反应, 在这一催化体系下得到了具有极好的对映选择性的吡咯烷化合物(Scheme 71).
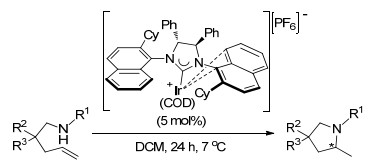
2018年, Rovis课题组[84]报道了温和条件下铱催化烯烃的二氨化反应, 其区域选择性由溶剂和铱络合物决定, 且其反应可以经过两种不同的反应途径, 分别生成五元环和六元环化合物(Scheme 72).
2013年, Liu课题组[85]报道了分子间烯烃的氨基乙酰氧基化反应, 以PdCl2(NCPh)2作为钯催化剂, 邻苯二甲酰亚胺作为氮源, 醋酸碘苯作为氧源, 同时该课题组比较了醋酸碘苯和氧气分别作为氧化剂时反应的不同, 发现醋酸碘苯在改变反应胺钯化步骤的立体化学结构时起决定性的作用.作者推测反应是先发生trans-或cis-胺钯化作用得到Pd(Ⅱ)中间体, 之后醋酸碘苯将其氧化为Pd(Ⅳ)中间体, 最后构型反转还原消除生成产物.且对于末端烯烃或者非末端烯烃反应都可以很好地发生, 该反应具有很好的底物适用范围(Scheme 73).
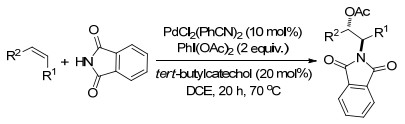
2014年, Studer课题组[86]报道了温和条件下钯催化的苯并呋喃类化合物的1, 2-碳氧化反应, 反应具有高度的非对映选择性, 反应以便宜且易制备的苯并呋喃和芳基硼酸、羧酸为原料, 以2, 2, 6, 6-四甲基哌啶-1-氧化物(TEMPO)为氧化剂, Pd(OAc)2作为催化剂, 底物羧酸作为溶剂, 常温下就可以得到高产率及高非对映选择性、完整的区域选择性的2-烷基-2-芳基-3-酯基-2, 3-二氢苯并呋喃框架的目标产物.这一反应对天然产物及药物的合成具有很大的潜在应用价值(Scheme 74).
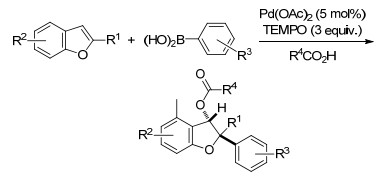
随后, Studer课题组[87]又报道了2-烷基吲哚的1, 2-碳氧化反应, 同样以Pd(OAc)2作为催化剂, TEMPO为温和的氧化剂, 使2-取代的吲哚与商业可用的硼酸发生高度非对映选择性的碳氧化反应, 生成吲哚啉类化合物.该反应在室温条件下4 h就可以完成, 产率也较高, 且酯基可以进一步水解为相应的醇, 这也为此反应的进一步应用提供了条件(Scheme 75).
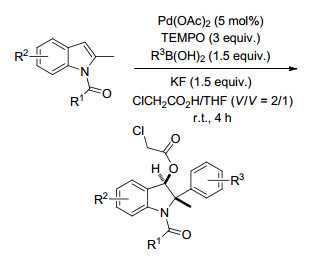
2016年, Liu课题组[88]以氧化稳定的吡啶噁唑啉为手性配体, 钯作为催化剂, NFSI为氮源, 发生的烯烃的分子间氢胺化反应, 其中修饰的手性配体对迁移插入步骤有很好的对映选择性控制, 最终以优良的区域选择性和对映选择性生成各种苄基酰胺类化合物(Scheme 76).
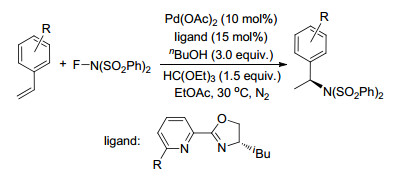
2017年, Cheng课题组[89]报道了钯催化的未活化烯烃与便宜易得的碘代全氟烷烃的芳基全氟烷基化反应, 得到了各种有价值的全氟烷烃化杂环产物.其中芳基底物对亲电子取代基有很好的容忍性, 而且使这一方法成为烯烃双官能团化中极化或自由基引发的芳香环形成的辅助工具.此反应以较高的区域选择性得到了各种全氟烷基化杂环衍生物, 反应条件温和, 且具有很好的底物普适性(Scheme 77).
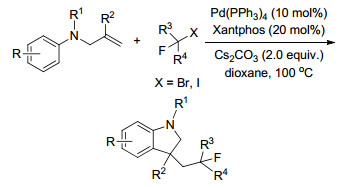
同年, Shao课题组[90]通过使简单易得的烯烃发生氧化芳基乙酰氧基化反应合成了吲哚和吲哚啉衍生物, 以吡咯酰胺为导向基, 肉桂基苯胺通过直接的氧化芳基乙酰氧基化反应得到中等到良好产率的吲哚, 而2-甲基取代的肉桂基苯胺通过连续的氧化芳基乙酰氧基化、水解、氧化等步骤得到了高产率的吲哚啉衍生物(Scheme 78).
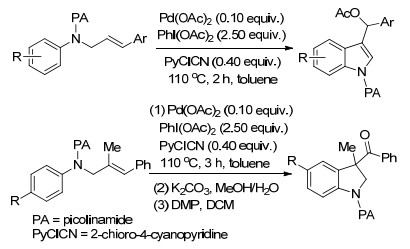
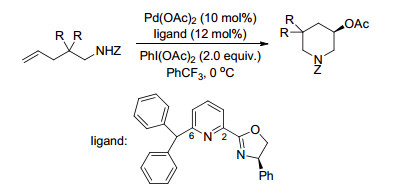
2018年, Liu课题组[91]报道了钯催化未活化烯烃的不对称6-endo氨基乙酰氧基化反应, 在温和的反应条件下, 利用新型的吡啶噁唑啉配体使得反应最终以极好的区域选择性和对映选择性得到了手性的β-乙酰氧基哌啶产物.作者提出, 在吡啶噁唑啉配体的C-6位置引入一个大基团对提高钯催化剂的反应活性以及乙酰氧基哌啶产物的对映选择性都至关重要(Scheme 79).
对于烯烃的双卤化反应, 其产物一般都是反式产物, 即便得到顺式产物, 产率也是很低, 因此关于烯烃顺式双卤化加成反应的研究就很少. 2015年, Denmark课题组[92]首次报道了二苯基二硒催化的脂肪系烃的顺式二氯化反应, 该反应具有广泛的底物普适性, 不饱和酯烯烃、环烷烯烃、反式烯烃、顺式烯烃以及丙烯醇都可以反应, 得到40%~90%的产率, dr值最高达到99:1, 作者也推测了该反应的机理(Scheme 80).
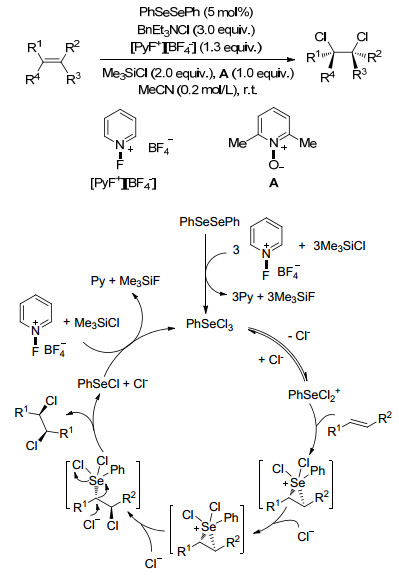
对于烯烃的双官能团化反应, 过渡金属由于其价格昂贵、反应条件要求严格、有反应残留的特点影响了它的应用范围, 而非金属试剂一般毒性较低且便宜易得, 一般符合绿色化学的要求, 因此发展非金属条件下发生的双官能团化也尤为重要.
2015年, Tan和Liu课题组[93]首次研究出用高对映选择性的布朗斯特酸催化烯烃的氢胺化反应, 在温和的反应条件下来获得高产率、高对映选择性的α-三取代碳立体中心的吡咯烷衍生物.这一方法的特性是通过多氢键与布朗斯特酸及双键合作, 使硫脲基团同时作为活化集团和导向集团.而手性合成中间体和有机合成中广泛存在的重要结构的简单构建, 突出了该方法的实用性(Scheme 81).
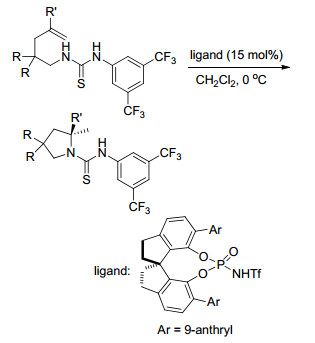
2016年, Liu课题组[94]报道了在有机碱催化下, 通过高效化学选择性完成的未活化烯烃与Togni’s试剂的芳基三氟甲基化及氧三氟甲基化反应.该反应通过改变有机碱催化剂和溶剂来完成化学选择性的转换.并且作者通过机理研究表明, 在DMSO溶剂中发生的芳基三氟甲基化反应经历的是自由基环化历程, 而在DCE溶剂中的氧三氟甲基化反应则经历碳正离子历程(Scheme 82).
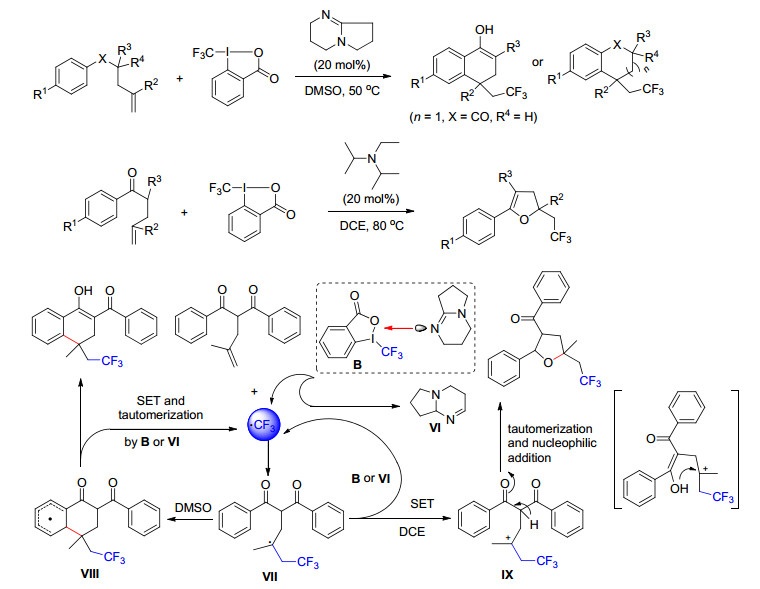
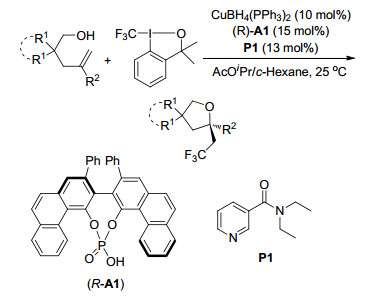
2018年, List课题组[95]报道了一种手性强布朗斯特酸催化的烯烃的分子内不对称氢烷氧化反应.该方法可以快速获得生物活性的1, 1-二取代四氢呋喃, 包括(-)-Boivinianin A, 反应操作简单, 可转化多种烯烃, 且具有很好的收率和对映选择性(Scheme 83).
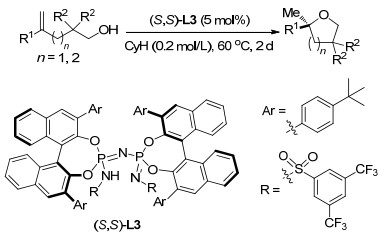
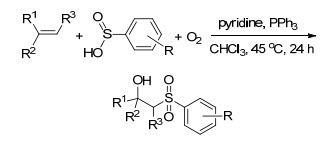
2013年, Lei课题组[96]报道了无金属催化的条件下分子间烯烃的氧磺酰化反应, 该反应没有添加任何的引发剂, 实现了亚磺酸的二氧活化, 这一转变可持续且环保.这一反应有很好的官能团容忍性, 反应条件温和, 以吡啶作为碱, 三苯基膦作为添加剂, 氯仿为溶剂, 40 ℃的条件下反应24 h就能得到高产率的目标产物.作者认为反应过程中, 吡啶首先与芳基亚磺酸作用生成亚磺酸负离子, 之后在氧气的氧化作用下转化为亚磺酸自由基, 而自由基进一步进攻烯烃形成自由基中间体, 而中间体与氧气作用形成过氧基中间体, 最后与吡啶结合的氢形成过氧酸, 并在酸性的环境中脱去水得到最终产物(Scheme 84).
2014年, Chiba课题组[97]通过高价碘试剂(PhI(OCOR)2、PhI(NMs2)2)催化使含脒基的烯烃发生非对映选择性的氨氧化反应与二氨化反应, 而这一反应可以得到各种用于医药、材料、催化应用的多胺化合物(Scheme 85).
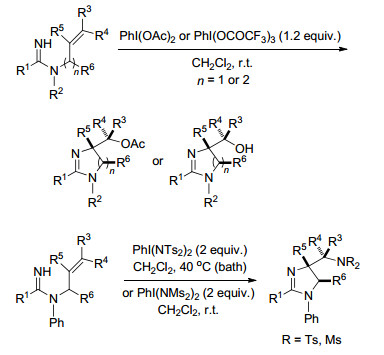
同年, Hong课题组[98]报道了高价碘试剂催化的烯烃的二氨化反应, 室温下生成3-氨基二氢吲哚.该反应利用富电子和缺电子(芳基磺酰胺)氮源的组合, 在胺没有预激活或保护的情况下, 发生烯烃的二氨化反应.反应操作简单, 无金属参与, 具有广泛的官能团兼容性(Scheme 86).

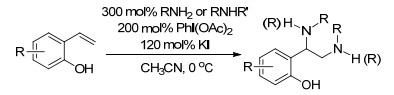
随后, Hong课题组[99]又报道了在高价碘试剂PhI(OAc)2、碘负离子作用下羟基苯乙烯与富电子的二氨化反应, 以双取代的氨与邻或对-羟基苯乙烯反应, 反应具有很好的普适性和官能团兼容性(Scheme 87).
2015年, Xia课题组[100]报道了无金属条件下烯烃的氧叠氮化反应, 以N-羟基邻苯二甲酰亚胺(NHPI)作为氧自由基的前体, TMSN3作为叠氮源, PhI(OAc)2为氧化剂.首先, NHPI在氧化剂作用下产生邻苯二甲酰亚胺氮-氧(PINO)自由基, PINO与烯烃反应产生的自由基又再次被PhI(OAc)2氧化为阳离子, 然后叠氮负离子进攻生成叠氮产物, 且具有良好的官能团兼容性(Scheme 88).
同年, Cheng课题组[101]报道了室温条件下三氟甲磺酸催化的烯烃的氢胺化反应.因2, 2, 2-三氟乙基氨基磺酸盐在有机溶剂及水中都有很好的溶解性, 该反应选择其为氮源, 便于转化与提纯.此反应条件下, 底物官能团兼容性良好, 且以优良的产率得到目标胺化合物(Scheme 89).
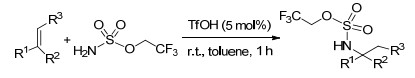
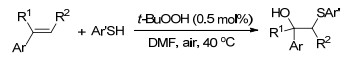
这一年, Zhou课题组[102]报道了芳基烯烃的分子间氧硫化反应, 反应条件温和, 以N, N-二甲基甲酰胺为溶剂, 使烯烃与芳基硫酚在40 ℃的条件下反应, 以中等产率得到氧硫化产物.该反应操作简单, 不需要其它的添加剂或氧化剂, 为β-羟基硫醚化合物的合成提供了一种新的合成方法.得到的β-羟基硫醚化合物可用于药物和有机合成, 也可以转化为其它功能化合物(Scheme 90).
在这一年, Tan和Liu课题组[103]为烯烃的高选择性1, 6-双官能团化反应开发了一种新颖的、简便的非金属自由基串联方法, 通过C—CF3的形成、1, 5-H迁移及远程sp3-C—H键官能团化这一串联过程得到有价值的三氟甲基化α-羟基羰基化合物.另外, 也为未活化烯烃的分子间区域选择性氧三氟甲基化提供了新策略.其中, Togni’s试剂同时作为CF3源和氧化剂.值得注意的是, 这一新的自由基反应不仅在非金属条件下进行, 而且操作简单、条件非常温和(Scheme 91).
2016年, Kuhakarn课题组[104]报道了苯乙烯衍生物的硝化肟化反应, 亚硝酸特丁酯作为反应试剂, 在DMSO溶剂中, 以简便快捷的方式合成了α-硝基肟化合物, 反应条件温和、操作简单、产率优良、无金属试剂且溶剂环保(Scheme 92).
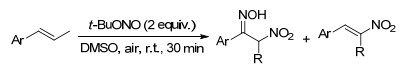
2017年, Dolbier课题组[105]首次报道了光催化的未活化烯烃的氟烷基芳基化反应, 反应条件温和, 经过自由基历程以较好的产率得到了二氟甲基取代的四氢化萘衍生物(Scheme 93).

同年, Li课题组[106]报道了2, 4, 6-三甲基吡啶催化的未活化烯烃的三氟甲基炔基化反应, 以Togni’s试剂为CF3源, 在非金属的条件下与炔基砜类反应生成β-三氟甲基化炔类化合物, 反应底物范围广, 官能团兼容性好, 且反应提出了非链式自由基历程(Scheme 94).
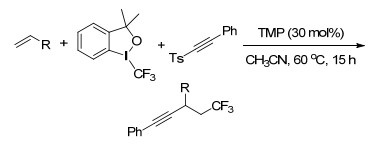
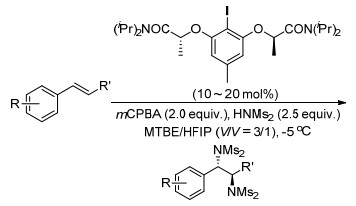
随后, Muiz课题组[107]在分子间反应的控制下, 首次实现了碘(Ⅰ/Ⅲ)催化的苯乙烯的对映选择性二氨化反应.该反应以手性改造的的芳基碘(Ⅰ)为催化剂, 间氯过氧苯甲酸为末端氧化剂, 且环境友好的组合溶剂不仅增加了该工艺的吸引力, 而且减缓了背景反应的速率.得到了91%~98%的ee值.该反应为邻二胺基化合物的合成提供了重要的方法, 为药物研究提供了重要的实体, 且手性芳酰胆碱催化剂的方法将作为相关转化的指南(Scheme 95).
作为一类重要的有机化学反应, 烯烃的双官能团化反应不仅可以经济有效地一步合成多位点反应产物, 而且反应效率高, 为构建化学结构的多样性提供了更多的方法.近十几年来, 烯烃的双官能团化反应迅速发展, 从各种过渡金属催化(如Cu、Fe、Pd、Rh、Ir、Mn、Ag等)到非金属催化的双官能团化反应都被广泛的报道, 且得到了非常广泛的应用.烯烃作为一种非常普遍的前手性基团, 通过双官能团化反应, 可以一步合成含有生物活性或药物活性的产物.烯烃的双官能团化反应为一些有机催化剂或手性配体的发展提供了更好的方法, 而且可以进一步合成高度复杂的化合物.随着研究的进展, 也发展了一系列符合绿色化学要求的非金属催化的双官能团化反应.
随着研究的深入, 烯烃的双官能团化反应仍面临很多问题和挑战.对于不含附属功能基底物的烯烃, 大多数条件下双官能团化反应很难发生.对于金属催化烯烃的双官能团化反应, 催化剂的回收及循环利用问题仍需要更多的研究.随着人们对绿色化学越来越多的关注, 未来烯烃的双官能团化反应也必将努力响应绿色化学的号召.将是该类反应研究的核心内容之一, 并将继续是人们关注的热点.目前, 烯烃的双官能团化反应还缺乏成体系的理论指导, 相关的动力学研究以及对反应历程的理解方面还有待进一步的深入研究.因此, 未来烯烃的双官能团化反应将会有更进一步的发展, 而它在有机领域也将会有更加重要、更加广泛的应用.
(a) Gaich, T.; Baran, P. S. J. Org. Chem. 2010, 75, 4657.
(b) Wender, P. A. Chem. Rev. 1996, 96, 1.
(a) Xu, L.; Mou, X. Q.; Chen, Z. M.; Wang, S. H. Chem. Commun. 2014, 50, 10676.
(b) Beccalli, E. M.; Broggini, G.; Martinelli, M.; Sottocornola, S. Chem. Rev. 2007, 107, 5318.
(c) Jensen, K. H.; Sigman, M. S. Org. Biomol. Chem. 2008, 6, 4083.
(d) Chemler, S. R. Org. Biomol. Chem. 2009, 7, 3009.
(e) Muniz, K. Angew. Chem., Int. Ed. 2009, 48, 9412.
(f) Li, G.; Kotti, S. R. S. S.; Timmons, C. Eur. J. Org. Chem. 2007, 2745.
(a) Zhou, M. B.; Wang, C. Y.; Song, R. J.; Liu, Y.; Wei, W. T.; Li, J. H. Chem. Commun. 2013, 49, 10817.
(b) Mu, X.; Wu, T.; Wang, H. Y. Guo, Y. L.; Liu, G. J. Am. Chem. Soc. 2012, 134, 878.
(c) Wu, T.; Mu, X.; Liu, G. Angew. Chem., Int. Ed. 2011, 50, 12578.
(d) Zhou, S. L.; Guo, L. N.; Wang, S.; Duan, X. H. Chem. Commun. 2014, 50, 3589.
(a) Keith, J. A; Henry, P. M. Angew. Chem., Int. Ed. 2009, 48, 9038.
(b) McDonald, R. I.; Liu, G. S.; Stahl, S. S. Chem. Rev. 2011, 111, 2981.
(a) Zhang, X.; You, S. L. Chem 2017, 3, 919.
(b) Lucet, D.; Le Gall, T.; Mioskowski, C. Angew. Chem., Int. Ed. 1998, 37, 2580.
Yuan, W.; Du, H.; Zhao, B.; Shi, Y. Org. Lett. 2007, 9, 2589. doi: 10.1021/ol071105a
Du, H.; Zhao, B.; Yuan, W.; Shi, Y. Org. Lett. 2008, 10, 4231. doi: 10.1021/ol801605w
Zhao, B.; Peng, X, ; Cui, S.; Shi, Y. J. Am. Chem. Soc. 2010, 132, 11009. doi: 10.1021/ja103838d
Zhao, B.; Peng, X.; Zhu, Y.; Ramirez, T. A.; Comwall, R. G.; Shi, Y. J. Am. Chem. Soc. 2011, 133, 20890. doi: 10.1021/ja207691a
Zhu, Y. G.; Shi, Y. Chem.-Eur. J. 2014, 20, 13901. doi: 10.1002/chem.v20.43
Sequeira, F. C.; Turnpenny, B. W.; Chemler, S. R. Angew. Chem. 2010, 122, 6509. doi: 10.1002/ange.201003499
Wang, Y. F.; Zhu, X.; Chiba, S. J. Am. Chem. Soc. 2012, 134, 3679. doi: 10.1021/ja2120629
Turnpenny, B. W.; Chemler, S. R. Chem. Sci. 2014, 5, 1786. doi: 10.1039/C4SC00237G
Karyakarte, S. D.; Sequeira, F. C.; Zibreg, G. H.; Huang, G. Q.; Matthew, J. P.; Ferreira, M. M. M.; Chemler, S. R. Tetrahedron Lett. 2015, 56, 3686. doi: 10.1016/j.tetlet.2015.01.171
Shen, K.; Wang, Q. Chem. Sci. 2015, 6, 4279. doi: 10.1039/C5SC00897B
Khoder, Z. M.; Wong, C. E.; Chemler, S. R. ACS Catal. 2017, 7, 4775. doi: 10.1021/acscatal.7b01362
Weng, S. S.; Hsieh, K. Y.; Zeng, Z. J. Zhang, J. W. Tetrahedron Lett. 2017, 58, 670. doi: 10.1016/j.tetlet.2017.01.015
Wang, F. L.; Dong, X. Y.; Lin, J. S.; Zeng, Y.; Jiao, G. Y.; Gu, Q. S.; Guo, X. Q.; Ma, C. L.; Liu, X. Y. Chem 2017, 3, 979. doi: 10.1016/j.chempr.2017.10.008
Chen, M. M.; Wang, L. J.; Ren, P. X.; Hou, X. Y.; Zhang, F.; Han, M. Nan.; Li, W. Org. Lett. 2018, 20, 510. doi: 10.1021/acs.orglett.7b03401
Fu, S. M.; Yang, H. H.; Li, G. Q.; Deng, Y. F.; Jiang, H. F.; Zeng, W. Org. Lett. 2015, 17, 1018. doi: 10.1021/acs.orglett.5b00131
Kinnel, R. B.; Gehrken, H. P.; Scheuer, P. J. J. Am. Chem. Soc. 1993, 115, 3376. doi: 10.1021/ja00061a065
Li, S. Q.; Xiong, P.; Zhu, L.; Qian, X. Y.; Xu, H. C. Eur. J. Org. Chem. 2016, 20, 3449.
Shen, K.; Wang, Q. Chem. Sci. 2017, 8, 8265. doi: 10.1039/C7SC03420B
Shen, K.; Wang, Q. J. Am. Chem. Soc. 2017, 139, 13110. doi: 10.1021/jacs.7b06852
Pan, G. H.; Ouyang, X. H.; Hu, M.; Xie, Y. X.; Li, J. H. Adv. Synth. Catal. 2017, 15, 2564. doi: 10.1002/adsc.201700365
张涌灵, 王敏, 曹鹏, 廖建, 化学学报, 2017, 75, 794. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract346081.shtmlZhang, Y, L.; Wang, M.; Cao, P.; Liao, J. Acta Chim. Sinica 2017, 75, 794(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract346081.shtml
Gockel, S. N.; Buchanan, T. L.; Hull, K. L. J. Am. Chem. Soc. 2018, 140, 58. doi: 10.1021/jacs.7b10529
Zeng, W.; Chemler, S. R. J. Am. Chem. Soc. 2007, 129, 12948. doi: 10.1021/ja0762240
Miao, L.; Haque, I.; Manzoni, M. R.; Tham, W. S.; Chemler, S. R. Org. Lett. 2010, 12, 4739. doi: 10.1021/ol102233g
Kaneko, K.; Yoshino, T.; Matsunaga, S.; Kanai, M. Org. Lett. 2013, 15, 2502. doi: 10.1021/ol4009848
Wang, D. H.; Wu, L. Q.; Wang, F.; Wan, X. L.; Chen, P. H.; Lin, Z. Y.; Liu, G. S. J. Am. Chem. Soc. 2017, 139, 6811. doi: 10.1021/jacs.7b02455
Miller, Y.; Miao, L.; Hosseini, A. S.; Chemler, R. S. J. Am. Chem. Soc. 2012, 134, 12149. doi: 10.1021/ja3034075
Zhou, S. L.; Guo, L. N.; Wang, H.; Duan, X. H. Chem.-Eur. J. 2013, 19, 12970. doi: 10.1002/chem.v19.39
Zhou, B.; Hou, W.; Yang, Y.; Feng, H.; Li, Y. Org. Lett. 2014, 167, 1322. doi: 10.1002/chin.201434141
Shi, L.; Wang, Y.; Yang, H.; Fu, H. Org. Biomol. Chem. 2014, 12, 4070. doi: 10.1039/C4OB00576G
Li, X.; Jian, X.; Zhang, P.; Gao, Y.; Wu, J.; Tang, G.; Zhao, Y. Synlett 2014, 25, 2009. doi: 10.1055/s-00000083
Schlosser, M. Angew. Chem., Int. Ed. 2006, 45, 5432. doi: 10.1002/(ISSN)1521-3773
Liang, Z.; Wang, F.; Chen, P.; Liu, G. S. Org. Lett. 2015, 17, 2438. doi: 10.1021/acs.orglett.5b00939
Egami, H.; Shimizu, R.; Kawamura, S.; Sodeoka, M. Angew. Chem., Int. Ed. 2013, 52, 4000. doi: 10.1002/anie.v52.14
Yang, F.; Klumphu, P.; Liang, Y. M.; Lipshutz, B. H. Chem. Commun. 2014, 50, 936. doi: 10.1039/C3CC48131J
Egami, H.; Kawamura, S.; Miyazaki, A.; Sodeoka, M. Angew. Chem., Int. Ed. 2013, 52, 7841. doi: 10.1002/anie.v52.30
Lin, J. S.; Liu, X. G.; Zhu, X. L.; Tan, B.; Liu, X. Y. J. Org. Chem. 2014, 79, 7084. doi: 10.1021/jo5012619
Lin, J. S.; Xiong, Y. P.; Ma, C. L.; Zhao, L. J.; Tan, B.; Liu, X. Y. Chem.-Eur. J. 2014, 20, 1332. doi: 10.1002/chem.v20.5
Lin, J. S.; Dong, X. Y.; Li, T. T.; Jiang, N. C.; Tan, B.; Liu, X. Y. J. Am. Chem. Soc. 2016, 138, 9357. doi: 10.1021/jacs.6b04077
Shen, K.; Wang, Q. Org. Chem. Front. 2016, 3, 222. doi: 10.1039/C5QO00353A
Lin, J. S.; Wang, F. L.; Dong, X. Y.; He, W. W.; Yuan, Y.; Chen, S.; Liu, X. Y. Nat. Commun. 2017, 8, 14841. doi: 10.1038/ncomms14841
Zhu, R.; Buchwald, S. L. J. Am. Chem. Soc. 2012, 134, 12462. doi: 10.1021/ja305840g
Zhu, R.; Buchwald, S. L. Angew. Chem., Int. Ed. 2013, 52, 12655. doi: 10.1002/anie.201307790
Jiang, X. Y.; Qing, F. L. Angew. Chem., Int. Ed. 2013, 52, 14177. doi: 10.1002/anie.201307595
Ye, J. H.; Song, L.; Zhou, W. J.; Ju, T.; Yin, Zh. B.; Yan, S. S.; Zhang, Z.; Li, J.; Yu, D. G. Angew. Chem., Int Ed. 2016, 34, 10022. doi: 10.1002/chin.201651141
Cheng, Y. F.; Dong, X. Y.; Gu, Q. S.; Yu, Z. L.; Liu, X. Y. Angew. Chem. 2017, 30, 9009. doi: 10.1002/ange.201702925
Li, X. T.; Gu, Q. S.; Dong, X. Y.; Meng, X.; Liu, X. Y. Angew. Chem., Int. Ed. 2018, 57, 7668. doi: 10.1002/anie.v57.26
Li, Z. L.; Li, X. H.; Wang, N.; Yang, N. Y.; Liu, X. Y. Angew. Chem., Int. Ed. 2016, 55, 15100. doi: 10.1002/anie.201608198
Liu, Z. C.; Bai, Y. H.; Zhang, J.; Yu, Y. Q.; Tan, Z.; Zhu, G. G. Chem. Commun. 2017, 53, 6440. doi: 10.1039/C7CC02537H
Fu, L.; Zhou, S.; Wan, X. L.; Chen, P. H.; Liu, G. S. J. Am. Chem. Soc. 2018, 140, 10965. doi: 10.1021/jacs.8b07436
(a) Brase, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188.
(b) Drivel, T. G. Org. Biomol. Chem. 2010, 8, 3831.
(c) Fumagalli, G.; Rabet, P. T. G.; Boyd, S.; Greaney, M. F. Angew. Chem., Int. Ed. 2015, 54, 11481.
(a) Rong, J.; Han, J.; Dong, L.; Tan, Y.; Yang, H.; Feng, L.; Wang, Q. W.; Meng, R.; Zhao, J.; Wang, S. Q.; Chen. X. J. Am. Chem. Soc. 2014, 136, 17468.
(b) Gramlich, P. M. E.; Wirges, C. T.; Manetto, A.; Carell, T. Angew. Chem., Int. Ed. 2008, 47, 8350.
Yin, H.; Wang, T.; Jiao, N. Org. Lett. 2014, 16, 2302. doi: 10.1021/ol500793c
Zhu, L.; Yu, H.; Xu, Z.; Jiang, X.; Lin, L.; Wang, R. Org. Lett. 2014, 16, 1562. doi: 10.1021/ol403687k
Zhu, R.; Buchwald, S. L. J. Am. Chem. Soc. 2015, 137, 8069. doi: 10.1021/jacs.5b04821
Lu, M. Z.; Wang, C. Q.; Loh, T. P. Org. Lett. 2015, 17, 6110. doi: 10.1021/acs.orglett.5b03130
Zhou, H.; Jian, W. J.; Qian, B.; Ye, C. Q.; Li, D. L.; Zhou, J.; Bao, H. L. Org. Lett. 2017, 19, 6120. doi: 10.1021/acs.orglett.7b02982
Bunescu, A.; Ha, T. M.; Wang, Q.; Zhu, J. P. Angew. Chem., Int. Ed. 2017, 56, 10555. doi: 10.1002/anie.v56.35
Xu, L.; Mou, X. Q.; Chen, Z. M.; Wang, S. H. Chem. Commun. 2014, 50, 10676. doi: 10.1039/C4CC04640D
Wang, D. H.; Wang, F.; Chen, P. H.; Lin, Z. Y.; Liu, G. S. Angew. Chem., Int. Ed. 2017, 8, 2054. doi: 10.1002/anie.201405937
Qian, Bo.; Xiong, H. G.; Zhu, N. B.; Ye, C. Q.; Jian, W. J.; Bao, H. L. Tetrahedron Lett. 2016, 57, 3400. doi: 10.1016/j.tetlet.2016.06.087
Hemric, B. N.; Shen, K.; Wang, Q. J. Am. Chem. Soc. 2016, 138, 5813. doi: 10.1021/jacs.6b02840
Ha, T. M.; Wang, Q.; Zhu, J. P. Chem. Commun. 2016, 52, 11100. doi: 10.1039/C6CC06356J
Williamson, K. S.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 4570. doi: 10.1021/ja1013536
Liu, G. S.; Zhang, Y. Q.; Yuan, Y. A.; Xu, H. J. Am. Chem. Soc. 2013, 135, 3343. doi: 10.1021/ja311923z
Lu, D. F.; Zhu, C. L.; Jia, Z. X.; Xu, H. J. Am. Chem. Soc. 2014, 136, 13186. doi: 10.1021/ja508057u
Yuan, Y. A.; Lu, D. F.; Chen, Y. R.; Xu, H. Angew. Chem. 2016, 128, 544. doi: 10.1002/ange.201507550
Qian, B.; Chen, S. W.; Wang, T.; Zhang, X. H.; Bao, H. L. J. Am. Chem. Soc. 2017, 139, 13076. doi: 10.1021/jacs.7b06590
Wang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2011, 133, 19080. doi: 10.1021/ja2092689
Olson, D. E.; Su, J. Y.; Roberts, D. A.; Bois, J. D. J. Am. Chem. Soc. 2014, 136, 13506. doi: 10.1021/ja506532h
Piou, T.; Rovis, T. Nature 2015, 527, 86. doi: 10.1038/nature15691
Ciesielski, J.; Dequirez, G.; Retailleau, P.; Gandon, V.; Dauban, P. Chem.-Eur. J. 2016, 22, 9338. doi: 10.1002/chem.201600393
Fu, N. K.; Sauer, G. S.; Lin, S. J. Am. Chem. Soc. 2017, 139, 15548. doi: 10.1021/jacs.7b09388
Sun, H.; Cui, B.; Duan, L. L.; Li, Y. M. Org. Lett. 2017, 19, 1520. doi: 10.1021/acs.orglett.7b00284
Singh, A. K.; Chawla, R.; Yadav, L. D. S. Tetrahedron Lett. 2014, 55, 4742. doi: 10.1016/j.tetlet.2014.06.086
Guo, S.; Cong, F.; Guo, R.; Wang, L.; Tang, P. P. Nat. Chem. 2017, 9, 546. doi: 10.1038/nchem.2711
Fumagalli, G.; Boyd, S.; Greaney, M. F. Org. Lett. 2013, 15, 4398. doi: 10.1021/ol401940c
Sipos, G.; Ou, A.; Skelton, B. W.; Falivene, L.; Cavallo, L.; Dorta, R. Chem.-Eur. J. 2016, 22, 6939. doi: 10.1002/chem.201600378
Conway, J. H.; Rovis, T. J. Am. Chem. Soc. 2018, 140, 135. doi: 10.1021/jacs.7b11455
Martinez, C.; Wu, Y. C.; Weinstein, A. B.; Stahl, S. S.; Liu, G. S.; Muniz, K. J. Org. Chem. 2013, 78, 6309. doi: 10.1021/jo400671q
Hata, K.; He, Z. H.; Daniliuc, C. G.; Itami, K.; Studer, A. Chem. Commun. 2014, 50, 463. doi: 10.1039/C3CC47350C
Ramella, V.; He, Z. H.; Daniliuc, C. G.; Studer, A. Org. Lett. 2015, 17, 664. doi: 10.1021/ol503689r
Yu, F.; Chen, P. H.; Liu, G. S. Chem. Commun. 2016, 52, 11100. doi: 10.1039/C6CC06356J
Zheng, J. H.; Chen, P.; Yuan, Y. F.; Cheng, J. J. J. Org. Chem. 2017, 82, 5790. doi: 10.1021/acs.joc.7b00598
Karnakanti, S.; Zang, Z. L.; Zhao, S.; Shao, P. L.; Hu, P.; He, Y. Chem. Commun. 2017, 53, 11205. doi: 10.1039/C7CC06448A
Qi, X. X.; Chen, C. H.; Hou, Ch. Q.; Fu, L.; Chen, P. H.; Liu, G. S. J. Am. Chem. Soc. 2018, 140, 7415. doi: 10.1021/jacs.8b03767
(a) Cresswell, A. J.; Eey, S. T.-C.; Denmark, S. E. Nat. Chem. 2015, 7, 146.
(b) He, T. X.; Zeng, X. H. Chin. J. Org. Chem. 2017, 37, 798(in Chinese).
(何天雄, 曾祥华, 有机化学, 2017, 37, 798.
Lin, J. S.; Yu, P.; Huang, L.; Zhang, P.; Tan, B.; Liu, X. Y. Angew. Chem., Int. Ed. 2015, 54, 7847. doi: 10.1002/anie.201501762
Yang, N. Y.; Li, Z. L.; Ye, L.; Tan, B.; Liu, X. Y. Chem. Commun. 2016, 52, 9052. doi: 10.1039/C6CC00364H
Tsuji, N.; Kennemur, J. L.; Buyck, T.; Lee, S.; Prevost, S.; Kaib, P. S. J.; Bykov, D.; Fares, C.; List, B. Science 2018, 359, 1501. doi: 10.1126/science.aaq0445
Lu, Q. Q.; Zhang, J.; Wei, F. L.; Qi, Y.; Wang, H. M.; Liu, Z. L.; Lei, A. W. Angew. Chem., Int. Ed. 2013, 52, 7156. doi: 10.1002/anie.201301634
Chen, H.; Kaga, A.; Chiba, S. Org. Lett. 2014, 16, 6136. doi: 10.1021/ol503000c
Hong, K. B; Johnston, J. N. Org. Lett. 2014, 16, 3804. doi: 10.1021/ol501693j
Danneman, M. W.; Hong, K. B.; Johnston, J. N. Org. Lett. 2015, 17, 2558. doi: 10.1021/acs.orglett.5b01177
Xia, X. F.; Gu, Z.; Liu, W. T.; Wang, H. J.; Xia, Y. M.; Gao, H. Y.; Liu, X.; Liang, Y. M. J. Org. Chem. 2015, 80, 290. doi: 10.1021/jo502327r
Fei, J.; Wang, Z.; Cai, Z. R.; Sun, H.; Cheng, X. Adv. Synth. Catal. 2015, 357, 4063. doi: 10.1002/adsc.201500646
Zhou, S. F.; Pan, X. Q.; Zhou, Z. H.; Shoberu, A.; Zou, J. P. J. Org. Chem. 2015, 80, 3682. doi: 10.1021/acs.joc.5b00123
Huang, L.; Zheng, S. C.; Tan, B.; Liu, X. Y. Org. Lett. 2015, 17, 1589. doi: 10.1021/acs.orglett.5b00479
Chumnanvej, N.; Katrun, P.; Pohmakotr, M.; Reutrakul, V.; Soorukram, D.; Kuhakarn, C. Chin. J. Chem. 2016, 34, 830. doi: 10.1002/cjoc.v34.8
Zhang, Z. X.; Martinez, H.; Dolbier. W. R. J. Org. Chem. 2017, 82, 2589. doi: 10.1016/j.jfluchem.2011.05.001
Zhou, S. F.; Song, T.; Chen, H.; Liu, Z. L.; Shen, H. G.; Li, C. Z. Org. Lett. 2017, 19, 698. doi: 10.1021/acs.orglett.6b03870
Muñiz, K.; Barreiro, L.; Romero, R. M.; Martínez, C. J. Am. Chem. Soc. 2017, 139, 4354. doi: 10.1021/jacs.7b01443

图式 2 铜(Ⅰ)催化共轭烯烃的区域选择性二氨化反应
Scheme 2 Cu(Ⅰ)-catalyzed regioselective diamination of conjugated dienes

图式 3 铜催化末端烯烃连续的二氨化作用和脱氢作用
Scheme 3 Cu-catalyzed sequential diamination and dehydrogenation of terminal olefins

图式 14 铜催化下β, γ-不饱和腙的二氨化反应
Scheme 14 Copper-catalyzed diamination of β, γ-unsaturated hydrazones

图式 15 铜催化N-烯丙基脲的分子内环化反应
Scheme 15 Copper-catalyzed intramolecular cyclization of N-alkenylureas

图式 18 铜催化未活化烯烃的氨基叠氮化反应
Scheme 18 Copper-catalyzed aminoazidation of unactivated alkenes

图式 19 铜催化烯烃的分子间氨基烷基化反应
Scheme 19 Copper-catalyzed intermolecular amino-alkylation of alkenes

图式 20 铜催化的苯乙烯的不对称硼胺化反应
Scheme 20 Cu-catalyzed enantioselective aminoboration of styrenes

图式 22 铜催化烯烃的不对称芳氨化反应
Scheme 22 Cu-catalyzed enantioselective carboamination of alkenes

图式 25 铜催化烯烃的分子间不对称芳基氨基化反应
Scheme 25 Asymmetric copper-catalyzed intermolecular aminoarylation of styrenes

图式 26 铜催化烯烃的分子内芳基醚化反应
Scheme 26 Copper-catalyzed intramolecular alkene carboetherification

图式 29 铜催化烯烃的三氟甲基硫氰化反应
Scheme 29 Copper-catalyzed intermolecular trifluoromethylthiocyanation of alkenes

图式 31 铜催化N-芳基丙烯酰胺的芳基三氟甲基化反应
Scheme 31 Copper-catalyzed trifluoromethylation of N-arylacrylamides

图式 32 合成β-三氟甲基胺的三氟甲基化反应
Scheme 32 Trifluoromethylation reactions for the synthesis of β-trifluoromethylamines

图式 33 铜催化未活化烯烃的氨基三氟甲基化反应
Scheme 33 Copper-catalyzed aminotrifluoromethylation of unactivated alkenes

图式 34 铜催化未活化烯烃的氨基三氟甲基化反应
Scheme 34 Copper-catalyzed aminotrifluoromethylation of unactivated alkenes

图式 35 烯烃的不对称自由基氨基三氟甲基化反应
Scheme 35 Asymmetric radical aminotrifluoromethylation of alkenes

图式 37 烯烃的不对称自由基氨基氟烷基化反应和氨基二氟甲基化反应
Scheme 37 Catalytic asymmetric radical aminoperfluoroalkylation and aminodifluoromethylation of alkenes

图式 38 铜催化未活化烯烃的氧三氟甲基化反应
Scheme 38 Copper-catalyzed aminotrifluoromethylation of unactivated alkenes

图式 40 烯丙胺与二氧化碳的选择性氧三氟甲基化反应
Scheme 40 Selective oxytrifluoromethylation of allylamines with CO2

图式 41 烯烃与醇的不对称自由基氧三氟甲基化反应
Scheme 41 Enantioselective radical oxytrifluoromethylation of alkenes with alcohols

图式 42 烯烃肟类化合物的不对称自由基氧三氟甲基化反应
Scheme 42 Asymmetric radical oxytrifluoromethylation of alkenyl oximes

图式 45 铜催化苯乙烯的三氟甲基炔基化反应
Scheme 45 Copper-catalyzed trifluoromethylalkynylation of styrenes

图式 46 铜催化吲哚的氧叠氮化反应和烷氧化叠氮化反应
Scheme 46 Copper-catalyzed oxoazidation and alkoxyazidation of indoles

图式 49 铜催化烯烃的氧叠氮化反应和二叠氮化反应
Scheme 49 Cu-catalyzed oxyazidation and diazidation of styrenes

图式 51 铜催化烯烃的三组分碳叠氮化反应
Scheme 51 Cu-catalyzed three-component carboazidation of alkenes

图式 52 铜催化芳基烯烃的分子间叠氮氰化反应
Scheme 52 Cu-catalyzed intermolecular azidocyanation of aryl alkenes

图式 53 铜催化烯烃的分子间不对称氨氰基化反应和叠氮氰化反应
Scheme 53 Cu-catalyzed intermolecular amino- and azidocyanation of alkenes

图式 56 铜催化烯烃的烷氰基环醚化反应
Scheme 56 Cu-catalyzed cyanoalkylative cycloetherification of clkenes

图式 57 铁催化烯烃的分子间氨羟基化反应
Scheme 57 Iron-catalyzed interrmolecular oxyamination of alkenes

图式 58 铁催化烯烃的分子内氨羟基化反应
Scheme 58 Iron-catalyzed intrarmolecular oxyamination of alkenes

图式 59 铁催化烯烃的分子间氨氧化反应
Scheme 59 Iron-catalyzed interrmolecular aminooxygenation of alkenes

图式 60 铁催化烯烃的非对映选择性二氨化反应
Scheme 60 Iron-catalyzed diastereoselective interrmolecular diamination of alkenes

图式 65 铑催化烯烃与氮宾的双官能团化反应
Scheme 65 Rhodium-catalyzed alkene difunctionalization with nitrenes

图式 66 电催化烯烃与亲核氯源的自由基二氯化反应
Scheme 66 Electrocatalytic radical dichlorination of alkenes with nucleophilic chlorine sources

图式 67 非功能化烯烃的分子内氨基烷氧化反应
Scheme 67 Intramolecular amino alkoxylation of unfunctionalized olefins

图式 68 银催化的烯烃的分子间氧硫化反应
Scheme 68 AgNO3/K2S2O8 catalyzed aerobic oxysulfonylation of alkenes

图式 69 银催化的烯烃的分子间不对称溴三氟甲基氧化反应
Scheme 69 Asymmetric silver-catalysed intermolecular bromotrifluoromethoxylation of alkenes

图式 70 光催化烯烃的碳氧/氨化反应
Scheme 70 Oxyarylation and aminoarylation of styrenes using photoredox catalysis

图式 71 氮杂环铱复合物作用下未活化烯烃的氢氨化反应
Scheme 71 NHC-iridium(Ⅰ) complexes catalyzed intramolecular hydroamination of unactivated aminoalkenes

图式 73 钯催化分子间烯烃的氨基乙酰氧基化反应
Scheme 73 Palladium catalyzed intermolecular aminoacetoxylation of alkenes

图式 74 钯催化苯并呋喃类化合物的1, 2-碳氧化反应
Scheme 74 Palladium-catalyzed oxyarylation of benzo[b]-furans

图式 75 钯催化2-烷基吲哚的1, 2-碳氧化反应
Scheme 75 Palladium-catalyzed diastereoselective oxyarylation of 2-alkylindoles

图式 76 烯烃的分子间不对称氢胺化反应
Scheme 76 Intermolecular enantioselective hydroamination of styenes

图式 77 通过钯催化未活化烯烃的芳基全氟烷基化反应合成杂环化合物
Scheme 77 Pd-Catalyzed arylperfluoroalkylation of unactivated olefins for the synthesis of heterocycles

图式 78 钯催化烯烃的氧化芳基乙酰氧基化反应
Scheme 78 Palladium-catalyzed oxidative arylacetoxylation of alkenes

图式 80 硒催化烯烃的顺式二氯化加成可能反应机理
Scheme 80 Proposed catalytic cycle for the selenium-catalyzed syn-dichlorination of alkenes

图式 81 布朗斯特酸催化烯烃的氢胺化反应
Scheme 81 Br nsted acid catalyzed asymmetric hydroamination of alkenes

图式 82 未活化烯烃的芳基三氟甲基化和氧三氟甲基化反应
Scheme 82 Carbotrifluoromethylation and oxytrifluoromethylation of unactivated alkenes

图式 83 烯烃的分子内不对称氢烷氧化反应
Scheme 83 Catalytic asymmetric intramolecular hydroalkoxylation of unbiased olefins

图式 85 高价碘试剂催化烯烃的非对映选择性的氨氧化反应与二氨化反应
Scheme 85 Diastereoselective amino oxygenation and diamination of alkenes by hypervalent iodine(Ⅲ) reagents

图式 86 高价碘试剂催化的烯烃的二氨化反应
Scheme 86 Hypervalent iodine(Ⅲ) reagents promoted diamination of alkenes

图式 89 三氟甲磺酸催化的分子间烯烃的氢胺化反应
Scheme 89 Intermolecular hydroamination of unfunctionalized alkenes under trifluoromethanesulfonic acid catalysis

图式 92 苯乙烯衍生物与亚硝酸特丁酯的硝化肟化反应
Scheme 92 Nitration-oximization of styrene derivatives with tert-butyl nitrite

图式 93 光催化的未活化烯烃的氟烷基芳基化反应
Scheme 93 Photoredox catalyzed intramolecular fluoro-alkylarylation of unactivated alkenes

图式 94 未活化烯烃的自由基三氟甲基炔基化反应
Scheme 94 Catalytic radical trifluoromethylalkynylation of unactivated alkenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
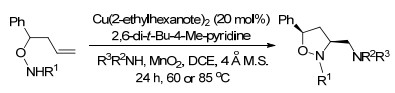



 下载:
下载:







































 下载:
下载: