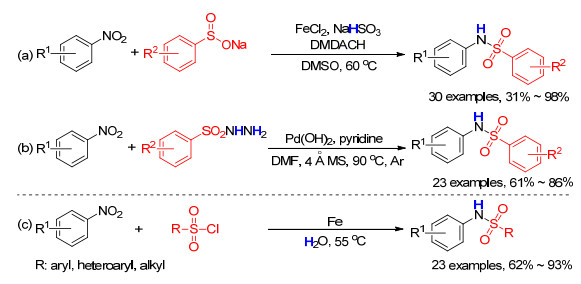
Scheme 1.
Synthesis of sulphonamides from nitroarenes
Direct Synthesis Sulfonamides from Nitroarenes and Sulfonyl Chlorides in Water
Huilan Yue , Pengli Bao , Leilei Wang , Xiaoxia Lü , Daoshan Yang , Hua Wang , Wei Wei

As one of the most important structural moieties, sulfonamides are frequently found in various natural bioactive compounds as well as synthetic drugs.[1] Consequently, extensive efforts have been dedicated toward constructing sulfonamides over the past decades.[2] In most synthetic methods for such motifs, anilines were used as the nitrogen sources, [3] which are odorous and potentially toxic. Moreover, anilines are often prepared from the corresponding nitroarenes through hydrogenation reduction. Compared to anilines, nitroarenes are much cheaper, more abundant and more readily accessible. Thus, a direct synthesis of sulfonamides from nitroarenes would be a better fitted and an eco-friendly industrial application. In 2015, Luo et al.[4] reported an elegant work for iron salts/trans-N, N'-dime- thyl-1, 2-diaminocyclohexane (DMDACH) synergetic catalyzed construction of N-arylsulfonamide from sodium arylsulfinates with nitroarenes by using of NaHSO3 as the reductant and hydrogen source (Scheme 1a). In 2016, Deng and co-workers developed a Pd(OH)2-cata- lyzed method for the synthesis of N-arylsulfonamide from arylsulfonyl hydrazides and nitroarenes via the hydrogen transfer strategy (Scheme 1b).[5] Both well developed reactions, although effective, but suffer from the limited commercial availability of sulfonating reagents that are too expensive for larger-scale synthesis. Arylsulfonyl hydrazides and sodium arylsulfinates are usually prepared from the corresponding sulfonyl chlorides in the presence of hazardous sulfinate salts and strong bases[6] or dangerous hydrazine hydrate[7] under relatively harsh reaction conditions. Tedious multistep operations might limit their wide applications in the field of organic synthesis and pharmaceutical chemistry. Therefore, the development of a simple, direct, economic and especially, environmentally friendly method for synthesis of sulfonamides from nitroarenes and readily available sulfonyl chlorides is still highly desirable.
On the other hand, water is not only environmentally friendly reaction media, [8] but it also a “green” hydrogen source for reduction reaction. Over the past decades, considerable efforts have been made in the development of sustainable and economical chemical transformation with water as the hydrogen source.[9] Up to date, there is still no report for the aqueous synthesis of sulfonamides with water as the terminal hydrogen source. Following our interest in environmentally friendly synthesis, [10] herein we report a sustainable, step-economical and practical method for the construction of sulfonamides from sulfonyl chlorides and nitroarenes with water as the reaction media as well as the hydrogen source (Scheme 1c).
At the start of our investigations, we attempted the sulfonamidation of nitrobenzene (1a%) and tosyl chloride (2a) (1.5 equiv.) with zinc dust (3 equiv.) as the reductant in water (0.5 mL) under ultrasound irradiation (USI). An 18% yield of the desired sulfonamidation product 3a was formed (Table 1, Entry 1). In addition, a trace amount of p-toluenesulfonic acid was observed. This can possibly be attributed to the side reactions of hydrolysis of 2a. Considering a large amount of 1a remained completely unconsumed, the reaction outcome of zinc loading was next performed. On increasing the reductant loading to 3.5 and 4 equiv., the 3a was obtained with the same yields (Table 1, Entries 2 and 3). Therefore, the loading of zinc duct was optimized at 3.5 equiv. using iron powder instead of zinc dust led to an increase in the yield of 3a (Table 1, Entry 4). Adjusting the amount of 2a to 2 equiv. resulted in the desired product 3a with 55% yield (Table 1, Entry 5). Further increasing the loading of 2a did not provide improved the yield of the desired product 3a (Table 1, Entry 6). The results revealed that ultrasound irradiation was not appropriate for this present reaction. Choosing the best ultrasound irradiation conditions (Table 1, Entry 5), this transformation was further optimized under traditional heating conditions. To our delight, traditional heating used under the same reaction parameters led to 68% yield (Table 1, Entry 7). Further investigations on the effect of reaction temperature suggested that 55 ℃ was ideal temperature for this reaction (Table 1, Entries 8~10). Thus, the optimal reaction conditions are nitrobenzene (1a), 2 equiv. of tosyl chloride (2a), 3.5 equiv. of Fe powder and water.
 下载:
导出CSV
下载:
导出CSV
 |
|||||
| Entry | Reductant (equiv.) | 2a/equiv. | Conditions | Conv./% | Yieldb/% |
| 1 | Zn dust (3) | 1.5 | USI, 1.5 h | 25 | 18 |
| 2 | Zn dust (3.5) | 1.5 | USI, 1.5 h | 33 | 29 |
| 3 | Zn dust (4) | 1.5 | USI, 1.5 h | 34 | 29 |
| 4 | Fe powder (3.5) | 1.5 | USI, 1.5 h | 48 | 40 |
| 5 | Fe powder (3.5) | 2 | USI, 1.5 h | 63 | 55 |
| 6 | Fe powder (3.5) | 2.5 | USI, 1.5 h | 61 | 54 |
| 7 | Fe powder (3.5) | 2 | 25 ℃, 40 h | 75 | 68 |
| 8 | Fe powder (3.5) | 2 | 45℃, 40 h | 84 | 76 |
| 9 | Fe powder (3.5) | 2 | 55℃, 40 h | 100 | 93 |
| 10 | Fe powder (3.5) | 2 | 65℃, 40 h | 100 | 84 |
| a Reaction conditions: 1a (0.2 mmol) and 2a (0.6 mmol), reductant (3~4 equiv.), solvent (1.5 mL), temperature (25~65 ℃), reaction time (1.5~40 h). b Isolated yields based on 1a. | |||||
Next, the scope and limitations of the present protocol were studied under the optimal reaction conditions (Table 2). Arylsulfonyl chlorides bearing with either electron-donating or electron-withdrawing substituents, such as methyl, methoxy, halide (F, Cl and Br), trifluoromethyl, cyan and acetyl groups underwent the sulfonamidation reactions efficiently to afford the desired products in good to excellent yields (3a~3i). Raised steric hindrances of the substituents of arylsulfonyl chlorides resulted in a low yield of sulfonamide product (3b vs. 3k). Naphthalenesulfonyl and thiophene-2-sulfonyl chloride also displayed good reactivity to afford the corresponding products in good yields (3l and 3m). This method was also extended to aliphatic sulfonyl chlorides, such as methanesulfonyl chloride and butanesulfonyl chloride. These results revealed that aliphatic sulfonyl chlorides were also well tolerated under the standard reaction conditions to deliver the desired products in good yields (3n and 3o). However, no product was afforded when trifluoromathanesulfonyl chloride was utilized as the substrates. Next, the substrate scope of the nitroarenes in this sulfonamidation reaction has also been investigated by the employment of TsCl as a test substrate. Good to excellent yields were generally obtained for nitrobenzenes bearing not only electron-donating but also electron-withdrawing substituents (3p~3v). β-Nitronaphthalene could also be transferred into the desired product in good yield (3w). No reaction occurred when nitroethane was used as the substrate.
To demonstrate the practicality and reliability of the present synthetic method, the gram-scale experiment was performed under the standard reaction conditions, furnishing the desired products 3a in 79% isolated yield (Eq. 1). However, decreasing the loading of TsCl and iron powder leads to a significant decrease in the yield of the desired product 3a.
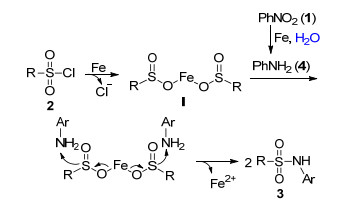
To understand the reaction mechanism of the present sulfonamidation reaction, some control experiments were carried out, as shown in Scheme 2. When the reaction of nitrobenzene (1a) with tosyl chloride (2a) was stopped after 12 h, it was found that in addition to the sulfonamidation product 3a, unexpectedly, both aniline (4a) and N-phenylhy- droxylamine (5a) were observed. When 4a and 5a were subjected to the optimal reaction conditions, the desired product 3a was obtained in 90% and 87% NMR yields, respectively. These experimental results suggested that this present reaction might involve 4a and 5a as the reactive intermediate. Treatment of iron(Ⅱ) 4-methyl- benzene-sulfinate 6a[11] with 1a or 4a under the optimal reaction conditions gave the desired product 3a in 85% NMR yield, which indicates that 6a might also be the reaction intermediate.
The sulfonamidation reaction may follow a similar reaction pathway to that of the sulfonation of quinoline N-oxides with sulfonyl chlorides reported by He et al., [12] with the difference being aniline instead of quinoline N-oxides (Scheme 3). First, the nitrobenzene 1 was deep reduced with iron powder to generate aniline 4. Meanwhile, the sulfonyl chloride 2 was reduced by iron powder into an iron bis-sulphinate compound I. Subsequently, the intermediate I reacted with aniline 4 to produce the desired product sulfonamide 3 with release of the Fe2+.
|
|
(1) |
In summary, we have developed a new and general route for the synthesis of various functionalized sulfonamides from nitroarenes and sulfonyl chlorides in the presence of iron powder as the sole reductant in water (green solvent) under open-air conditions. In the reaction, both starting materials were reduced by inexpensive and nontoxic iron at the same time. From an economic and industrial perspective, this chemical process has the potential for important impact given the single-step sulfonamidation reaction of readily accessible reagents, the mild reaction conditions, and the excellent functional group tolerance of the method. Thus, this strategy would certainly be valuable from both academic research and industry.
Sulfonyl chlorides (0.6 mmol), nitroarenes (0.3 mmol) and Fe powder (1.15 mmol) were added in H2O (1.5 mL). The mixture was allowed to react in a 10 ml round-bottom flask at 55 ℃ under air for 40 h. After cooling to room temperature, the mixture was extracted with dichloromethane (5 mL×3). The combined organic layer was dried with anhydrous MgSO4. The solvent was removed. Products were separated by short flash chromatography on a silica gel column.
4-Methyl-N-phenylbenzenesulfonamide (3a): White solid (68.9 mg, 93% yield), m.p. 104~105 ℃ (lit.[4] 104~105 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.70 (d, J=8.4 Hz, 2H), 7.28 (brs, 1H), 7.23 (d, J=8.8 Hz, 4H), 7.14 (d, J=7.7 Hz, 3H), 2.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 143.9, 136.7, 136.0, 129.7, 129.3, 127.3, 125.3, 121.5, 21.6.
N-Phenylbenzenesulfonamide (3b): White solid (59.4 mg, 85%), m.p. 105~106 ℃ (lit.[4] 106~107 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.80 (d, J=7.6 Hz, 2H), 7.58 (t, J=7.2 Hz, 1H), 7.50 (t, J=7.6 Hz, 2H), 7.30 (d, J=8.0 Hz, 2H), 7.20 (t, J=7.2 Hz, 1H), 7.10 (d, J=7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 139.2, 136.4, 133.2, 129.6, 129.2, 127.4, 125.5, 122.2.
4-Methoxy-N-phenylbenzenesulfonamide (3c): White solid (51.3 mg, 65% yield), m.p. 108~109 ℃ (lit.[4] 109~110 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.72 (td, J=8.1, 7.5, 3.6 Hz, 2H), 7.22 (q, J=6.7, 5.4 Hz, 2H), 7.16~6.96 (m, 4H), 6.93~6.78 (m, 2H), 3.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.2, 136.7, 130.6, 129.5, 129.4, 125.3, 121.6, 114.3, 55.7.
4-Fluoro-N-phenylbenzenesulfonamide (3d): White solid (55.7 mg, 74% yield), m.p. 109~110 ℃ (lit.[5] 109~110 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.78 (dd, J=8.9, 5.2 Hz, 2H), 7.28~7.24 (m, 2H), 7.15~7.05 (m, 5H), 6.61 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ: 166.6, 164.1, 136.2, 135.0, 135.1, 130.2, 130.0, 129.5, 125.8, 121.9, 116.6, 116.3.
4-Chloro-N-phenylbenzenesulfonamide (3e): White solid (60.9 mg, 76% yield), m.p. 102~103 ℃ (lit.[5] 104~105 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.68 (d, J=8.0 Hz, 2H), 7.46 (d, J=8.4 Hz, 2H), 7.32 (dd, J=14.7, 8.4 Hz, 3H), 7.20 (q, J=7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 139.7, 137.4, 136.2, 129.6, 129.4, 128.8, 125.6, 121.8.
4-Bromo-N-phenylbenzenesulfonamide (3f): White solid (75.6 mg, 81% yield), m.p. 113~115 ℃ (lit.[5] 116~117 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.58~7.52 (m, 4H), 7.24~7.21 (m, 2H), 7.22~7.15 (m, 1H), 7.09~7.04 (m, 2H), 6.61 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ: 138.1, 136.1, 132.4, 129.4, 128.7, 128.2, 125.9, 122.4.
N-Phenyl-S-4-trifluoromethylphenyl sulfonamide (3g): White solid (56.9 mg, 63%), m.p. 120~121 ℃ (lit.[4] 120~122 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.81 (d, J=8.4 Hz, 2H), 7.68 (d, J=8.0 Hz, 2H), 7.31 (t, J=7.6 Hz, 2H), 7.22 (t, J=7.6 Hz, 1H), 7.12 (d, J=7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 142.3, 136.1, 134.3 (JC−F=33.2 Hz), 129.2, 127.3, 126.5 (JC−F=3.6 Hz), 126.3, 123.5 (JC−F=271.7 Hz), 122.1. 19F NMR (376 MHz, CDCl3) δ: -63.4.
4-Cyano-N-phenylbenzenesulfonamide (3h): White solid (55.0 mg, 71% yield), m.p. 112~113 ℃ (lit.[7] 110~111 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.83 (d, J=8.4 Hz, 2H), 7.68 (d, J=8.4 Hz, 2H), 7.32 (d, J=7.2 Hz, 2H), 7.23 (t, J=7.8 Hz, 1H), 7.08 (d, J=7.6 Hz, 2H), 6.72 (brs, 1H). 13C NMR (100 MHz, CDCl3) δ: 143.2, 135.6, 132.6, 129.7, 127.7, 126.7, 122.3, 117.3, 116.8.
4-Acetyl-N-phenylbenzenesulfonamide (3i): White solid (51.2 mg, 62%), m.p. 98~99 ℃ (lit.[8] 99~102 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.98 (d, J=8.4 Hz, 2H), 7.88 (d, J=8.3 Hz, 2H), 7.45 (brs, 1H), 7.24 (t, J=7.7 Hz, 2H), 7.14~7.09 (m, 3H), 2.61 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 197.1, 142.8, 140.2, 135.9, 129.5, 128.9, 127.6, 125.9, 121.9, 26.9.
3-Methyl-N-phenylbenzenesulfonamide (3j): White solid (60.8 mg, 82% yield), m.p. 103~104 ℃ (lit.[4] 101~102 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.62~7.53 (m, 2H), 7.36 (d, J=6.4 Hz, 2H), 7.21 (t, J=7.6 Hz, 2H), 7.18 (brs, 1H), 7.12 (d, J=7.6 Hz, 3H), 2.32 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 139.5, 139.1, 136.6, 133.9, 129.6, 129.1, 127.7, 125.4, 124.5, 121.7, 21.4.
N-Phenyl-S-2-methylphenyl sulfonamide (3k): White solid (46.7 mg, 63%), m.p. 124~126 ℃ (lit.[4] 126~127 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.82 (d, J=8.4 Hz, 1H), 7.44~7.33 (m, 2H), 7.26~7.21 (m, 2H), 7.23 (t, J=7.2 Hz, 2H), 7.20 (d, J=8.4 Hz, 3H), 2.70 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 137.5, 137.4, 136.8, 133.2, 132.3, 130.5, 129.8, 126.4, 124.9, 120.1, 20.7.
N-Phenylnaphthalene-2-sulfonamide (3l): White solid (65.4 mg, 77% yield), m.p. 131~133 ℃ (lit.[7] 131~133 ℃). 1H NMR (400 MHz, CDCl3) δ: 8.41 (brs, 1H), 7.96~7.74 (m, 4H), 7.61 (dt, J=20.6, 7.2 Hz, 2H), 7.26~7.01 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 136.4, 136.2, 135.1, 132.1, 129.6, 129.6, 129.4, 129.1, 128.2, 127.6, 125.7, 122.3, 121.8.
N-Phenylthiophene-2-sulfonamide (3m): White solid (63.8 mg, 89% yield), m.p. 82~83 ℃ (lit.[7] 81~83 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.55 (d, J=4.4 Hz, 2H), 7.31~7.24 (m, 3H), 7.22~7.15 (m, 3H), 6.73 (d, J=4.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 139.3, 136.4, 133.2, 132.8, 129.8, 127.4, 125.5, 122.2.
N-Phenylmethanesulfonamide (3n): White solid (34.4 mg, 67% yield), m.p. 98~100 ℃ (lit.[3g] 99~100 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.41 (t, J=7.6 Hz, 2H), 7.21~7.19 (m, 3H), 6.94 (brs, 1H), 3.04 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 136.8, 129.7, 125.6, 120.9, 39.6.
N-Phenylbutane-1-sulfonamide (3o): White solid (56.9 mg, 89% yield), m.p. 108~110 ℃ (lit.[3g] 108~110 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.36 (d, J=8.6 Hz, 2H), 7.23~7.14 (m, 3H), 6.38 (brs, 1H), 3.07 (t, J=8.0 Hz, 2H), 1.85~1.75 (m, 2H), 1.41 (m, 2H), 0.91 (t, J=7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 136.9, 129.8, 125.3, 120.5, 51.4, 25.6, 21.6, 13.6.
N-(p-Tolyl)-S-phenyl sulfonamide (3p): White solid (67.4 mg, 91%), m.p. 116~117 ℃ (lit.[3g] 116~117 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.81 (d, J=7.6 Hz, 2H), 7.61 (t, J=7.2 Hz, 1H), 7.53 (t, J=7.6 Hz, 2H), 7.07 (d, J=8.2 Hz, 2H), 6.87 (d, J=8.0 Hz, 2H), 6.51 (brs, 1H), 2.23 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 139.5, 135.2, 133.3, 133.2, 130.4, 129.3, 127.7, 122.4, 21.2.
N-(4-Cyanophenyl)benzenesulfonamide (3q): Yellow solid (55.7 mg, 72%), m.p.: 147~149 ℃ (lit.[3g] 147~149 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.86 (d, J=7.8 Hz, 2H), 7.60 (t, J=7.3 Hz, 1 H), 7.54~7.48 (m, 4H), 7.19 (d, J=8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 141.0, 138.7, 133.9, 133.8, 129.6, 127.3, 119.6, 118.5, 108.1.
N-(4-Methoxyphenyl)-S-phenylsulfonamide (3r): White solid (51.3 mg, 65%), m.p. 87~88 ℃ (lit.[3h] 88~89 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.68 (d, J=7.6 Hz, 2H), 7.53 (t, J=7.4 Hz, 1H), 7.40 (t, J=7.2 Hz, 2H), 7.02 (d, J=8.4 Hz, 2H), 6.72 (d, J=8.4 Hz, 2H), 3.71 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 158.6, 139.6, 133.3, 129.5, 128.3, 127.5, 125.3, 114.5, 55.6.
N-(4-Fluorophenyl)-S-phenyl sulfonamide (3s): White solid (58.7 mg, 78%), m.p. 111~112 ℃ (lit.[3h] 110~111 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.81 (d, J=7.6 Hz, 2H), 7.47 (t, J=7.2 Hz, 1H), 7.43 (t, J=7.6 Hz, 2H), 7.12 (m, 2H), 6.90 (t, J=8.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 160.5 (JC−F=243.8 Hz), 138.2, 133.5, 132.7, 129.2, 127.1, 124.9 (JC−F=8.5 Hz), 116.3 (JC−F=22.4 Hz); 19F NMR (376 MHz, CDCl3) δ: −116.5.
N-(4-Chlorophenyl)benzenesulfonamide (3t): White solid (66.5 mg, 83% yield), m.p. 104~105 ℃ (lit.[4] 104~105 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.75 (d, J=8.6 Hz, 2H), 7.41 (d, J=8.4 Hz, 2H), 7.28 (d, J=14.8 Hz, 3H), 7.14 (q, J=7.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 139.7, 137.4, 136.2, 129.5, 129.5, 128.8, 125.8, 121.9.
N-(4-Bromophenyl)benzenesulfonamide (3u): White solid (75.6 mg, 81% yield), m.p. 114~116 ℃ (lit.[4] 116~117 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.74 (d, J=7.6 Hz, 2H), 7.55 (t, J=7.4 Hz, 1H), 7.44 (t, J=7.6 Hz, 2H), 7.34 (d, J=7.8 Hz, 2H), 6.97 (d, J=7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 137.9, 136.2, 132.6, 129.7, 128.8, 128.2, 126.2, 121.0.
N-(3-Chlorophenyl)-S-phenylsulfonamide (3v): White solid (60.9 mg, 76%), m.p. 116~117 ℃ (lit.[5] 115~117 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.88 (d, J=7.6 Hz, 2H), 7.62 (t, J=7.2 Hz, 1H), 7.44 (t, J=7.2 Hz, 2H), 7.18−7.15 (m, 2H), 7.07 (d, J=8.0 Hz, 1H), 6.93 (d, J=7.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 138.6, 137.7, 135.2, 133.7, 130.2, 129.7, 127.4, 125.6, 121.2, 19.8.
N-(2-Naphthyl)-S-phenyl sulfonamide (3w): White solid (62.8 mg, 74%), m.p. 97~99 ℃ (lit.[4] 98~99 ℃). 1H NMR (400 MHz, CDCl3) δ: 7.83 (d, J=7.8 Hz, 2H), 7.78~7.67 (m, 3H), 7.55 (s, 1H), 7.50 (d, J=7.6 Hz, 1H), 7.47~7.36 (m, 4H), 7.24 (d, J=8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 139.1, 134.0, 133.7, 133.2, 131.3, 129.5, 129.2, 127.8, 127.7, 127.4, 126.8, 12
Supporting Information 1H NMR and 13C NMR spectra of compounds 3a~3w. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(a) Supuran Claudiu, T.; Scozzafava, A.; Casini, A. Med. Res. Rev. 2002, 23, 146.
(b) Andrea, S.; Takashi, O.; Antonio, M.; Claudiu, T. S. Curr. Med. Chem. 2003, 10, 925.
(c) Smith, D. A.; Jones, R. M. Curr. Opin. Drug Discovery Dev. 2008, 11, 72.
(d) Mujumdar, P.; Poulsen, S.-A. J. Nat. Prod. 2015, 78, 1470.
(a) Zhu, H.; Shen, Y.; Deng, Q.; Tu, T. Chem. Commun. 2015, 51, 16573. (b) Mokhtari, B.; Nematollahi, D.; Salehzadeh, H. Green Chem. 2018, 20, 1499.
(a) Deng, X.; Mani, N. S. Green Chem. 2006, 8, 835.
(b) Bahrami, K.; Khodaei, M. M.; Soheilizad, M. J. Org. Chem. 2009, 74, 9287.
(c) Powell, D. A.; Fan, H. J. Org. Chem. 2010, 75, 2726.
(d) Mokhtar, M.; Saleh, T. S.; Ahmed, N. S.; Al-Thabaiti, S. A.; Al-Shareef, R. A. Ultrason. Sonochem. 2011, 18, 172.
(e) Zhao, J.; Xu, J.; Chen, J.; Wang, X.; He, M. RSC Adv. 2014, 4, 64698.
(f) Pan, X.; Gao, J.; Liu, J.; Lai, J.; Jiang, H.; Yuan, G. Green Chem. 2015, 17, 1400.
(g) Yang, K.; Ke, M.; Lin, Y.; Song, Q. Green Chem. 2015, 17, 1395.
(h) Wei, W.; Liu, C.; Yang, D.; Wen, J.; You, J.; Wang, H. Adv. Synth. Catal. 2015, 357, 987.
(i) Jiang, Y.-y.; Wang, Q.-Q.; Liang, S.; Hu, L.-M.; Little, R. D.; Zeng, C.-C. J. Org. Chem. 2016, 81, 4713.
(j) Zhu, M.; Wei, W.; Yang, D.; Cui, H.; Wang, L.; Meng, G.; Wang, H. Org. Biomol. Chem. 2017, 15, 4789.
(k) Sheykhan, M.; Khani, S.; Abbasnia, M.; Shaabanzadeh, S.; Joafshan, M. Green Chem. 2017, 19, 5940.
(l) Yu, H.; Zhang, Y. Chin. J. Chem. 2016, 34, 359.
(m) Zhao Y.; Ren X.; Liu H.; Tang Z. Chin. J. Org. Chem. 2014, 34, 1218.
(n) Wei M.; Gao X.; Zhang H.; Li X. Chin. J. Org. Chem. 2015, 35, 439.
Zhang, W.; Xie, J.; Rao, B.; Luo, M. J. Org. Chem. 2015, 80, 3504. doi: 10.1021/acs.joc.5b00130
Zhao, F.; Li, B.; Huang, H.; Deng, G.-J. RSC Adv. 2016, 6, 13010. doi: 10.1039/C5RA26588F
Du, B.; Qian, P.; Wang, Y.; Mei, H.; Han, J.; Pan, Y. Org. Lett. 2016, 18, 4144. doi: 10.1021/acs.orglett.6b02289
Sheykhan, M.; Khani, S.; Abbasnia, M.; Shaabanzadeh, S.; Joafshan, M. Green Chem. 2017, 19, 5940. doi: 10.1039/C7GC03141F
(a) Tang, L.; Yang, Y.; Wen, L.; Yang, X.; Wang, Z. Green Chem. 2016, 18, 1224.
(b) Wei, W.; Wen, J.; Yang, D.; Liu, X.; Guo, M.; Dong, R.; Wang, H. J. Org. Chem. 2014, 79, 4225.
(c) Lu, G.-p.; Cai, C.; Chen, F.; Ye, R.-l.; Zhou, B.-J. ACS Sustainable Chem. Eng. 2016, 4, 1804.
(d) Wei, W.; Wen, J.; Yang, D.; Du, J.; You, J.; Wang, H. Green Chem. 2014, 16, 2988.
(e) Wen, J.; Wei, W.; Xue, S.; Yang, D.; Lou, Y.; Gao, C.; Wang, H. J. Org. Chem. 2015, 80, 4966.
(f) Uday Kumar, R.; Reddy, K. H. V.; Anil Kumar, B. S. P.; Satish, G.; Reddy, V. P.; Nageswar, Y. V. D. Tetrahedron Lett. 2016, 57, 637.
(g) Li, Y.; Huang, Y.; Gui, Y.; Sun, J.; Li, J.; Zha, Z.; Wang, Z. Org. Lett. 2017, 19, 6416.
(h) Chen, D.; Feng, Q.; Yang, Y.; Cai, X.-M.; Wang, F.; Huang, S. Chem. Sci. 2017, 8, 1601.
(i) Wei, W.; Cui, H.; Yang, D.; Yue, H.; He, C.; Zhang, Y.; Wang, H. Green Chem. 2017, 19, 5608.
(j) Kong, D.-L.; Lu, G.-P.; Wu, M.-S.; Shi, Z.-F.; Lin, Q. ACS Sustainable Chem. Eng. 2017, 5, 3465.
(k) Wei, W.; Bagzo, P.; Yue, H.; Liu, S.; Wang, L.; Li Y.; Yang, D. Org. Lett. 2018, 20, 7125.
(a) Wu, C.; Yang, P.; Fu, Z.; Peng, Y.; Wang, X.; Zhang, Z.; Liu, F.; Li, W.; Li, Z.; He, W. J. Org. Chem. 2016, 81, 10664.
(b) Liu, W.; Wang, H.; Li, C.-J. Org. Lett. 2016, 18, 2184.
(c) Deng, T.; Wang, C.-Z. Catal. Sci. Technol. 2016, 6, 7029.
(d) Cui, H.; Wei, W.; Yang, D.; Zhang, J. Xu, Z.; Wen, J.; Wang, H. RSC Adv. 2015, 5, 84657.
(e) Li, W.; Yin, G.; Huang, L.; Xiao, Y.; Fu, Z.; Xin, X.; Liu, F.; Li, Z.; He, W. Green Chem. 2016, 18, 4879.
(f) Wu, C.; Xin, X.; Fu, Z.-M.; Xie, L.-Y.; Liu, K.-J.; Wang, Z.; Li, W.; Yuan, Z.-H.; He, W.-M. Green Chem. 2017, 19, 1983.
(g) Hou, S.; Yang, H.; Cheng, B.; Zhai, H.; Li, Y. Chem. Commun. 2017, 53, 6926.
(h) Xie, L.-Y.; Duan, Y.; Lu, L.-H.; Li, Y.-J.; Peng, S.; Wu, C.; Liu, K.-J.; Wang, Z.; He, W.-M. ACS Sustainable Chem. Eng. 2017, 5, 10407.
(i) Wu, C.; Lu, L.-H.; Peng, A.-Z.; Jia, G.-K.; Peng, C.; Cao, Z.; Tang, Z.; He, W.-M.; Xu, X. Green Chem. 2018, 20, 3683.
(j) Luo, F.; Long, Y.; Li, Z.; Zhou, X. Acta Chim. Sinica 2016, 74, 805.
(k) Wu, J.; Liu, Y.; Ma, X.; Liu, P.; Gu, C.; Dai, B. Chin. J. Chem. 2017, 35, 1391.
(l) Zhou, Z.; Duan, J.; Mu, X.; Xiao, S. Chin. J. Org. Chem. 2018, 38, 585(in Chinese). (周曌, 段建凤, 穆小静, 肖尚友, 有机化学, 2018, 38, 585. http://www.cnki.com.cn/Article/CJFDTotal-YJHU201803005.htm
(a) Wei, W.; Liu, C.; Yang, D.; Wen, J.; You, J.; Suo, Y.; Wang, H. Chem. Commun. 2013, 49, 10239.
(b) Wei, W.; Liu, X.; Yang, D.; Dong, R.; Cui, Y.; Yuan F.; Wang, H. Tetrahedron Lett. 2015, 56, 1808.
(c) Cui, H.; Liu, X.; Wei, W.; Yang, D.; He, C.; Zhang, T.; Wang, H. J. Org. Chem. 2016, 81, 2252.
(d) Liu, X.; Cui, H.; Yang, D.; Dai, S.; Zhang, T.; Sun, J.; Wei, W.; Wang, H. RSC Adv. 2016, 6, 51830.
(e) Liu, C.; Zhu, M.; Wei, W.; Yang, D.; Cui, H.; Liu, X.; Wang, H. Org. Chem. Front. 2015, 2, 1356.
(f) Yi, D.; Fu, Q.; Chen, S. Y.; Gao, M.; Yang, L.; Zhang, Z. J.; Liang, W.; Zhang, Q.; Ji, J. X.; Wei, W. Tetrahedron Lett. 2017, 58, 2058.
(g) Fu, Q.; Yi, D.; Zhang, Z. J.; Liang, W.; Chen, S. Y.; Yang, L.; Zhang, Q.; Ji, J. X.; Wei, W. Org. Chem. Front. 2017, 4, 1385.
(h) Liang, W.; Zhang, Q.; Yi, D.; Fu, Q.; Chen, S. Y.; Yang, L.; Du, F. T.; Ji, J. X.; Wei, W. Chin. J. Chem. 2017, 35, 1378.
(i) Zhang, Z. J.; Yi, D.; Fu, Q.; Liang, W.; Chen, S. Y.; Yang, L.; Du, F. T.; Ji, J. X.; Wei, W. Tetrahedron Lett. 2017, 58, 2417.
(j) Wang, L.; Yue, H.; Yang, D.; Cui, H.; Zhu, M.; Wang, J.; Wei, W.; Wang, H. J. Org. Chem. 2017, 82, 6857.
(k) Wei, W.; Cui, H.; Yang, D.; Liu, X.; He, C.; Dai, S.; Wang, H. Org. Chem. Front. 2017, 4, 26.
(l) Wei, W.; Cui, H.; Yue, H.; Yang, D. Green Chem. 2018, 20, 3197.
(m) Cui, H.; Wei, W.; Yang, D.; Zhang, Y.; Zhao, H.; Wang, L.; Wang, H. Green Chem. 2017, 19, 3520.
Chumachenko, N.; Sampson, P. Tetrahedron 2006, 62, 4540. Xie, L.-Y.; Li, Y.-J.; Qu, J.; Duan, Y.; Hu, J.; Liu, K.-J.; Cao, Z.; He, W.-M. Green Chem. 2017, 19, 5642.
Table 1. Screening the optimal reaction conditionsa
|
|
|||||
| Entry | Reductant (equiv.) | 2a/equiv. | Conditions | Conv./% | Yieldb/% |
| 1 | Zn dust (3) | 1.5 | USI, 1.5 h | 25 | 18 |
| 2 | Zn dust (3.5) | 1.5 | USI, 1.5 h | 33 | 29 |
| 3 | Zn dust (4) | 1.5 | USI, 1.5 h | 34 | 29 |
| 4 | Fe powder (3.5) | 1.5 | USI, 1.5 h | 48 | 40 |
| 5 | Fe powder (3.5) | 2 | USI, 1.5 h | 63 | 55 |
| 6 | Fe powder (3.5) | 2.5 | USI, 1.5 h | 61 | 54 |
| 7 | Fe powder (3.5) | 2 | 25 ℃, 40 h | 75 | 68 |
| 8 | Fe powder (3.5) | 2 | 45℃, 40 h | 84 | 76 |
| 9 | Fe powder (3.5) | 2 | 55℃, 40 h | 100 | 93 |
| 10 | Fe powder (3.5) | 2 | 65℃, 40 h | 100 | 84 |
| a Reaction conditions: 1a (0.2 mmol) and 2a (0.6 mmol), reductant (3~4 equiv.), solvent (1.5 mL), temperature (25~65 ℃), reaction time (1.5~40 h). b Isolated yields based on 1a. | |||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们






 下载:
下载: