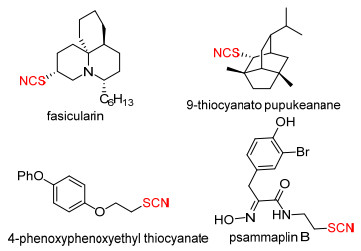
图 1.
一些含有硫氰基团的活性分子
Figure 1.
Selected bioactive molecules containing thiocyanate groups

硫氰酸酯(RSCN)是一类含有硫氰基官能团的重要氮硫类化合物(图 1), 具有多种生物活性, 如抑制酶活性、杀虫活性、抗微生物活性等, 在医药和农药等领域有着重要的应用价值[1~5].此外, 硫氰酸酯类化合物也广泛应用于有机合成领域[6~11].作为重要的有机合成中间体, 硫氰酸酯通过简单转化就可得到硫醇、硫醚、异硫氰酸酯、含硫杂环等多种有机硫化物, 同时它还可作为氰化试剂很好地参与反应.由于硫氰酸酯具有良好的生物活性和广阔的应用前景, 引起了有机合成工作者们的浓厚研究兴趣.
近几十年来, 合成化学家们对硫氰酸酯类化合物的合成与应用展开了较为深入的研究与探索. 2016年, Gulea等[12]对硫氰酸酯的合成及应用做了相关总结.最近, 各种各样的硫氰酸酯类化合物合成新方法、新策略又不断涌现, 有力推动着硫氰酸酯类化合物的制备向高效、绿色合成方向发展.本文着重介绍了近三年来硫氰化物的合成进展, 并对其应用进行了分类讨论, 以期为读者了解硫氰酸酯的研究动态提供帮助.
合成硫氰酸酯主要有直接合成法和间接合成法两种策略, 直接合成法通过底物与硫氰化试剂反应来合成硫氰酸酯, 主要包括—SCN亲核取代反应、+SCN亲电取代反应、•SCN自由基反应; 间接合成法利用有机硫化物的氰化反应来得到硫氰酸酯, 主要包括—CN亲核取代反应、+CN亲电取代反应、•CN自由基反应等.
亲核取代反应是合成硫氰酸酯类化合物的经典方法, 硫氰酸盐(NaSCN、KSCN、NH4SCN)和三甲基甲硅烷基异硫氰酸酯(TMSNCS)是常见的亲核试剂, 容易与带有易离去基团的底物反应得到这类化合物.
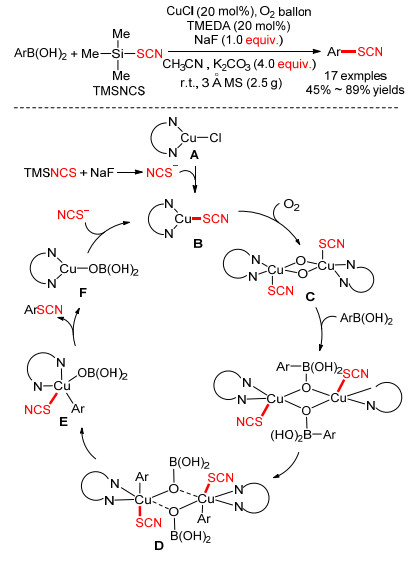
Hu小组[13]于2015年利用Chan-Lam反应成功实现了芳基硼酸与TMSNCS的交叉偶联反应.在室温下, 以氯化亚铜为催化剂, 四甲基乙二胺(TMEDA)为配体, 氧气作氧化剂, TMSNCS可以与芳基硼酸发生氧化偶联反应, 以45%~98%的收率得到各种芳基硫氰酸酯类化合物.通过控制实验, 推测可能的反应机理如下: (1)催化剂A与产生的—SCN阴离子进行配体交换后形成具有反应活性的物种B; (2) B可被氧气氧化得到中间体C; (3) C通过O—B配位键的相互作用与两个芳基硼酸分子结合; (4)芳基转移到铜原子上得到D, 随后分解成两个单体铜(Ⅲ)配合物E; (5) E经历还原消除得到目标产物和一价铜F; (6) F与—SCN阴离子交换以得到B并完成催化循环(Scheme 1).
2015年, Liu课题组[14]以TMSNCS为硫氰基源, Togni试剂为三氟甲基源, 在铜催化下实现了联烯的硫氰化三氟甲基化反应(Eq. 1).该反应条件较为温和, 大部分底物能以良好及以上的收率一步合成含两个重要官能团的产物, 为三氟甲基化硫氰酸酯类化合物的合成提供了新思路.
|
|
(1) |
2017年Wu课题组[15]提出了一条硫氰化钾与(杂)芳烃的硫氰化新路线(Scheme 2).在DMSO溶剂中, 以Cu(OTf)2作催化剂, TMEDA为配体, 三氟化硼为添加剂, 在氧气氛围中加热搅拌反应, 能以62%~95%收率分离得到各种硫氰化产物.该体系底物普适性好, 对于C-4位不含取代基的苯胺可以得到C-4位硫氰化产物.当以4-取代的苯胺为原料时, 会得到2-氨基苯并噻唑类化合物.

2018年, Zhu等[16]以乙腈作为溶剂, 过硫酸钠为氧化剂, 硫氰化钠作硫氰源, 经钯催化实现2-氨基呋喃特定位点sp3-C—H键硫氰化反应(Eq. 2).该方法条件温和, 底物适用范围广, 反应原子经济性较高.
|
|
(2) |
随着绿色环保理念的深入, 化学家们开发了一系列无需过渡金属催化剂、采用清洁能源的绿色合成新路线, 为硫氰酸酯的合成注入了新活力. 2016年, Rad[17]在N-(对甲苯磺酰基)咪唑(TsIm)环境中实现了醇的硫氰化反应, 合成了一系列烷基硫氰酸酯(Eq. 3).在70 ℃无水N, N-二甲基甲酰胺(DMF)中, 以三乙胺作碱, 加入少量TsIm, 经6~12 h后能以最高95%的收率完成反应.值得一提的是, 该方法具有良好的选择性, 即使有仲醇存在, 硫氰化钾也能与伯醇特定结合.
|
|
(3) |
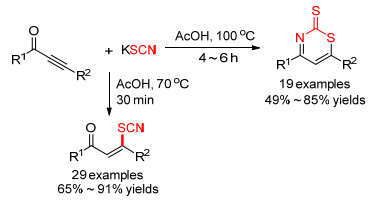
2017年, Reddy等[18]在70 ℃的醋酸溶剂中, 实现了硫氰化钾与炔酮的选择性硫氰化反应, 以良好到优异的收率分离得到硫氰化产物.升高温度并延长反应时间, 硫氰化烯酮可发生分子内环化得到噻嗪-2-硫酮衍生物(Scheme 3).该方法的底物适应性较好, 且收率不错.
另外, Patel课题组[19]使用异硫氰化物充当硫氰化亲核试剂, 实现了一种甲基咪唑促进的酮类化合物α-位硫氰化反应(Eq. 4).之后2017年, 该课题组[20]研究发现仅需催化量的甲基咪唑就可实现环氧乙烷的硫氰化酯化反应(Scheme 4), 反应条件温和, 原子利用率100%, 收率在75%~92%, 底物普适性也很好.
|
|
(4) |

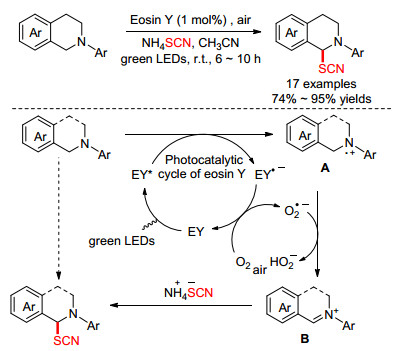
Yadav课题组[21]以Eosin Y作光催化剂, 在绿光的照射下促进叔胺α位的硫氰化反应.该方法仅需要1 mol%的Eosin Y, 在空气氛围中, 室温下搅拌反应6~10 h, 就可以74%~95%的收率获得一系列硫氰化四氢异喹啉产物.他们提出了一种可能的机理: (1) EY吸收光能进入激发态(EY*); (2)原料和EY*之间进行单电子转移得到A和RB•—; (3)氧气氧化RB•—得到基态EY, 并产生O2•—; (4) O2•—从A的α位攫取质子, 形成亚胺离子B; (5) B受到—SCN的进攻, 得到最终产物(Scheme 5).
2018年, Bolm课题组[22]合成一种新型的N-SCN亚砜亚胺试剂, 并很好地将其应用于烯烃的双官能团化反应中(Eq. 5).在二氯甲烷(DCM)溶剂中, N-Br亚砜亚胺与NH4SCN在常温下反应16 h便可制得这种新型硫氰化试剂.以Ir为催化剂, 在蓝光照射下, 该试剂能与烯烃以38%~81%的收率反应得到一系列新颖的硫氰化亚胺化产物.
|
|
(5) |
同年, Liang等[23]以非均相固体盐Amberlyst-15(H) [A-15(H)]为质子催化剂, 采用恒电流电解的方法来合成有机硫氰化物(Eq. 6).值得一提的是, 这种电化学氧化硫氰化路径无需添加外部导电盐, 并且固体盐可以重复使用多达五次, 效率没有显著降低.
|
|
(6) |
富电子芳环与硫氰化亲电试剂发生亲电取代反应是在芳香化合物中引入硫氰根官能团比较实用的方法, 现已开发出多种新型亲电硫氰化体系.较为经典的是氯代琥珀酰亚胺(NCS)和硫氰酸盐体系, NCS与硫氰酸盐反应生成的中间体硫氰代琥珀酰亚胺(NTS)是关键的亲电硫氰源. 2016年, Besset小组[24]报道了一种NCS促进下的苯胺N原子上引入硫氰根的新方法(Eq. 7).以四氢呋喃(THF)为溶剂和硫氰酸银作硫氰源, 在温和的条件下以71%~87%收率获得目标产物.
|
|
(7) |
Wang等[25]于2016年提出了一锅两步法合成3-氰硫基咪唑并[1, 2-a]吡啶(Scheme 6).他们使用商业化的溴代苯乙酮与2-氨基吡啶为原料在乙醇中反应得到中间产物2-苯基咪唑并[1, 2-a]吡啶, 随后经硫氰化亲电取代反应, 以64%~90%的收率合成得到一系列硫氰化咪唑并[1, 2-a]吡啶类化合物.该方法简单快速, 对环境友好且易于实施, 为这类硫氰酸酯的简便合成提供新思路.

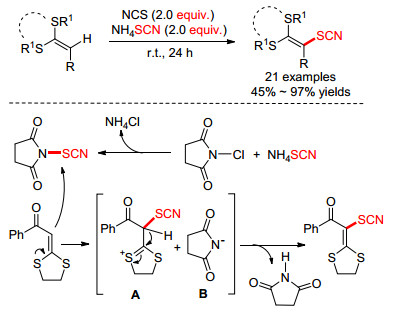
2017年, Wang小组[26]报道了氯代琥珀酰亚胺(NCS)促进下的烯酮二硫缩醛的直接硫氰化反应.该小组对反应机理进行了推测: (1) NCS和NH4SCN反应生成硫氰代琥珀酰亚胺(NTS); (2)烯酮二硫缩醛与NTS结合, 得到中间体A和B; (3) A和B作用, 经去质子化得到目标产物(Scheme 7).
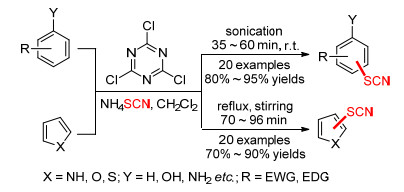
众所周知, 1, 1, 1, 3, 3, 3-六氟-2-丙醇(HFIP)在有机中作为一种绿色溶剂而倍受欢迎, 并且它具有一些独特的性质, 如低亲核性、高离子化能力等. Wang课题组[27]认为HFIP可以通过氢键结合激活NCS, 从而促进了N—Cl键的裂解而形成ClSCN.该课题组发现, 在超声辅助下, 在HFIP溶剂中, NCS和硫氰酸铵能与富电子芳烃(苯酚、苯胺或吲哚)发生硫氰化反应(Scheme 8).反应只需5~30 min, 即可以最高99%的收率得到目标产物.

此外, 其他亲电硫氰化体系也有报道. 2016年, Rajanna等[28]在室温下的二氯甲烷溶剂中, 以三聚氰氯(TCTA)作氯化剂, 实现了(杂)芳环的硫氰化取代反应(Scheme 9).对比发现反应在超声辅助下, 可平稳且快速地完成, 并能以良好到优异的收率得到产物.
2018年, Chen小组[29]就β-酮羰基化合物的不对称硫氰酸酯化提出一条新路径.他们以N-硫氰基邻苯二甲酰亚胺为硫氰化亲电试剂, 在金鸡纳生物碱的催化下完成底物不对称α-硫氰酸酯化反应(Eq. 8).可反应的底物种类较多, 且大部分产物的收率都在90%以上.同时方法具有高对映体选择性, 实现合成具有季碳中心的各种手性α-氰硫基β-酮酯, 但反应需在-78 ℃下进行.
|
|
(8) |
最近, Chen课题组[30]以甜味添加剂糖精为原料, 分两步合成一种新型的硫氰化亲电试剂.首先将糖精和次氯酸叔丁酯在甲醇中处理5 min, 得到N-氯代糖, 随后其与AgSCN在CH2Cl2溶剂中反应30 min可得到N-硫氰酸基糖精.他们研究发现这种新硫氰化试剂能很好用于吲哚类、羟吲哚类、酚类、β-酮羰基化合物, 芳香胺类和芳香酮等化合物的硫氰化反应, 并且表现出较为优异的反应性能(Scheme 10).
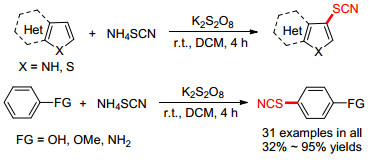
自由基硫氰化反应是合成有机硫氰化物的一种重要途径, 传统的反应中通常需要加入过量的氧化剂, 如过硫酸盐、醋酸锰、硝酸铈铵等促进反应发生. 2015年Guo等[31]开发了一种无需金属催化的烯烃酰胺与硫氰酸钾的串联硫氰化/分子内环化反应.以容易获得烯烃酰胺为原料, 在温和的条件下通过一步反应构建C—O和C—S键, 调控酰胺底物, 可以分别得到苯并六元杂环和苯并五元杂环.该课题组对反应机理进行了阐述: (1) K2S2O8氧化硫氰根阴离子产生硫氰根自由基A; (2) A加成到原料的碳碳双键上并产生B; (3) B进行自由基环化得到C; (4)氧化剂将自由基C进一步氧化成相应的碳正离子, 其经去质子化得到产物(Scheme 11).
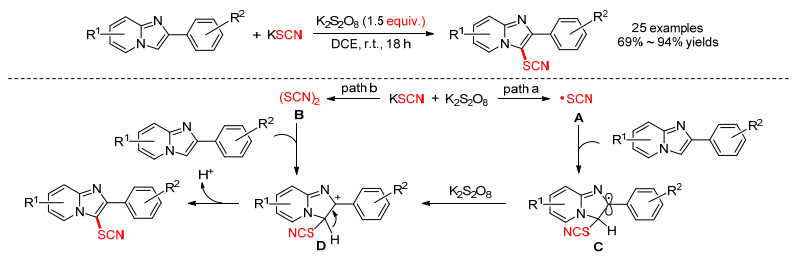
2015年, Wang课题组[32]报道了一种咪唑并吡啶硫氰化的方法.在室温下, 1, 2-二氯乙烷(DCE)溶剂中, 以过硫酸钾为氧化剂, 硫氰化钾作硫氰源, 实现C-3位硫氰化咪唑并吡啶的有效合成.此外, 该方法也能很好地应用于其他杂环和芳香族(咪唑并[2, 1-b]噻唑、吲哚和N, N-二甲基苯胺)的硫氰化反应.对此过程他们提出了两条可能的途径:路径a: (1)首先KSCN被氧化产生•SCN自由基A; (2) A与咪唑并吡啶反应得到自由基C; (3) C被K2S2O8进一步氧化成碳正离子D; (4) D去质子化得到硫氰化产物.路径b: (1) K2S2O8与KSCN产生一种亲电子的二硫氰化物(SCN)2; (2) B与咪唑并吡啶进行亲电加成产生D, 随后失去质子生成产物(Scheme 12).
Yu小组[33]于2016年发现在室温下的DCE溶剂中, 以过硫化钾为氧化剂, 烯胺酮与硫氰化钾顺利进行硫氰化/环化反应(Eq. 9).他们认为烯胺中的二甲基氨基可作活化基团, 可促进硫氰化过程, 并在环化过程中作为离去基团离去.
|
|
(9) |
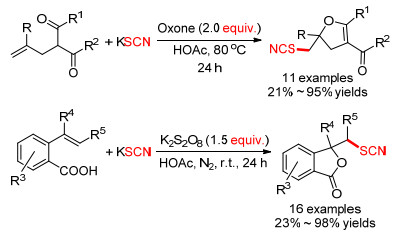
2016年, Guo等[34]报道了一种在无过渡金属催化的烯烃硫氰化/环化反应(Scheme 13).这种串联分子间硫氰化和分子内环化的过程, 为合成含有硫氰基团的二氢呋喃和内酯化合物提供了一种易于操作, 原子经济好, 收率高的新方法.但是他们发现当R为不同取代的苯环时, 空间位阻效应对反应收率有较大影响.
2017年, Xu等[35]开发了一种铜催化8-氨基喹啉衍生物的硫/硒氰化反应(Eq. 10).优化条件时他们发现以1, 2-二氯乙烷(DCE)为溶剂, KSCN/KSeCN作为硫/硒氰化试剂, 过硫酸钾作氧化剂的条件下, 反应收率表现较为一般.但加入少量相转移催化剂四丁基碘化铵(TBAI)可有效提高收率.
|
|
(10) |
2017年, Bhat课题组[36]开发了一种过硫酸钾氧化下直接用于苯酚、苯胺和吲哚、噻吩等杂环的硫氰化路径(Scheme 14).他们利用DCM为溶剂, 过硫化钾作氧化剂, 在温和条件下实现了上述芳香化合物的高效硫氰化反应.
含硫氰基团的杂环化合物具有一定药理活性, 这类化合物通常是杂环直接硫氰化得到, 最近科学家们开发出了一种串联硫氰化/环化的方案来制备含硫氰基团的杂环化合物. Li等[37]于2017年发展了一种以铁为催化剂和过硫酸钾作氧化剂, 在氩气保护下合成硫氰化噁唑啉的新方法(Eq. 11).他们利用β, γ-不饱和肟的硫化/环化过程, 实现特定位点以最高84%的收率引入硫氰基团.
|
|
(11) |
2018年, Chen小组[38]报道了一种硫氰化银为原料的N-芳基丙烯酰胺的硫氰基自由基加成环化反应, 最高能以91%的收率合成3-硫氰甲基化-2-吲哚酮类化合物(Eq. 12).该方法条件温和, 底物适用性范围广, 但使用硫氰化银作为硫氰化试剂, 合成成本较高.
|
|
(12) |
Wang小组[39]开发了一种合成3-硫氰基香豆素的新方法(Eq. 13).在二甲基亚砜(DMSO)溶剂中, 以硝酸铈铵[Ce(NH4)2(NO3)6]作氧化剂, 实现硫氰酸银与3-苯基丙炔酸苯酯的自由基环化反应.采用硫氰酸银作为硫氰化试剂, 合成成本较高, 而且产品的收率也表现较为一般.
|
|
(13) |
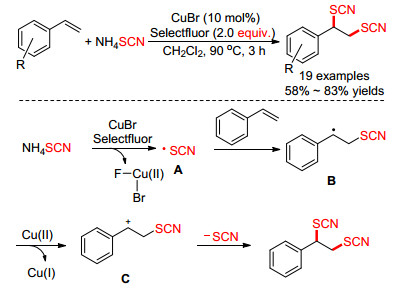
2016年, Lv小组[40]实现了铜催化下苯乙烯的双硫氰化反应.在二氯甲烷溶剂中, 以Selectfluor为氧化剂, 加热回流, 反应能以中等到良好的收率得到目标产物.由于二硫氰酸酯类化合物显示出良好的抑制真菌活性, 合成这类化合物的方法也备受关注, 该方法为二硫氰酸酯类化合物的合成提供了新途径.他们对反应机理进行了推测: (1)在CuBr存在下, 由Selectfluor氧化硫氰根阴离子产生硫氰酸根自由基A; (2) A加成到苯乙烯上产生以自由基B; (3)其进一步氧化成碳阳离子C; (4) SCN—与C进行亲核反应形成的产物(Scheme 15).
2018年, Zhang课题组[41]开发了一种烯烃硫氰化膦酰化反应(Eq. 14).以铜作催化剂和醋酸锰为氧化剂, 实现二苯基酰膦和TMSNCS与烯烃的自由基加成反应.机理实验表明烯烃首先与膦酰基自由基进行加成, 之后在铜催化下完成硫氰化.该方法条件温和, 底物普适性好, 链状烯烃和环状烯烃都能很好地完成反应, 得到相应产物.
|
|
(14) |
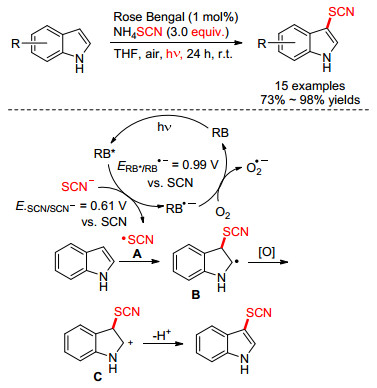
近几年来, 化学家们又发展了一系列高效节能的光催化硫氰化反应, 相对于传统的自由基硫氰化反应, 这类反应可以直接以空气为氧化剂, 更为绿色环保. Li课题组[42]于2014年提出了一种可见光促进吲哚β位引入硫氰基策略.使用廉价易得的Rose Bengal作光催化剂, 以四氢呋喃为溶剂, 经白光照射24 h后完成吲哚的硫氰化反应.该小组对反应机理进行了推测: (1)经可见光照射, Rose Bengal转变成激发的RB*; (2) —SCN和RB*之间进行单电子转移得到•SCN自由基A并产生RB•—自由基阴离子; (3)氧气氧化RB•—得到基态Rose Bengal; (4) A和吲哚进行亲电加成, 得到中间体B; (5) B被氧化后产生阳离子C; (6) C失去质子最终得到产物(Scheme 16).
2015年, Hajra等[43]报道了蓝光照射Eosin Y催化咪唑杂环的硫氰化反应(Eq. 15).在空气和室温下的乙腈溶剂中, 通过蓝光催化实现NH4SCN与原料的反应.底物拓展研究表明, 该方法可以高产率合成具有广泛官能团的3-硫氰基咪唑并[1, 2-a]吡啶类化合物, 底物上无论有强供电子基还是吸电子基, 都能很好地适应该体系顺利完成.
|
|
(15) |
2016年He小组[44]采用TiO2/MoS2纳米材料为复合光催化剂, 催化吲哚的硫氰化反应(Eq. 16).大部分底物收率表现良好, 当有底物有吸电子基团存在时, 则收率较差.催化剂的可重复使用性是本方法的一大优点.
|
|
(16) |
Singh等[45]于2018年以Eosin Y为光催化剂, 以异腈和硫氰酸铵为原料, 在乙腈溶剂中, 经绿光照射8~16 h后合成了一系列6-硫氰菲啶类化合物(Eq. 17).不同异腈底物均能较好地适用该反应条件, 产物收率最高可达94%, 为6-硫氰菲啶的有效合成提供了新思路.
|
|
(17) |
随着直接硫氰化路线的发展, 对于不易直接硫氰化的化合物, 化学家们也在尝试间接方法来合成硫氰化物.最常见的是通过氰化试剂与含硫底物反应得到硫氰酸酯.
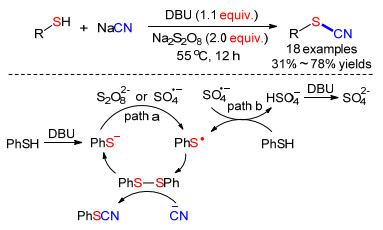
2017年, Xia等[46]利用过硫酸钠和1, 5-二氮杂二环[5.4.0]十一-5-烯(DBU)促进硫酚、硫醇与氰化钠直接发生氰化反应.实验结果显示, 各种硫酚和硫醇都可以较好地参与反应, 能以31%~78%收率分离得到相应的硫氰酸酯类化合物.他们提出了两种可能机理.路径a: (1)苯硫酚被DBU脱去质子得到PhS—; (2) PhS—被Na2S2O8氧化得到PhS•; (3)两分子PhS•偶联得到PhSSPh; (4)氰根对PhSSPh进行亲核取代反应得到产物.路径b: (1) Na2S2O8分解产生的SO4•—攫取PhSH的氢自由基生成HSO4—和自由基PhS•; (2)两分子PhS•偶联得到PhSSPh, HSO4—被DBU中和; (3) CN—与PhSSPh反应得到产物(Scheme 17).
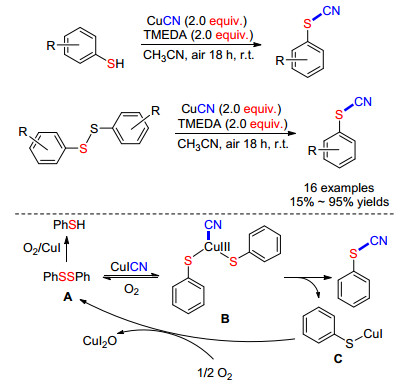
2014年, Suffert课题组[47]实现了苯硫酚或二芳基二硫化物的氰化反应.他们使用氰化亚铜为氰源和氧气作氧化剂, 在室温条件下进行反应, 产物收率在15%~95%.该课题组对机理进行了推测: (1)开始时苯硫酚几乎瞬时完全转化为二硫化物A; (2)然后氰化亚铜经氧化后插入到S—S键以形成铜(Ⅲ)中间体B; (3)中间体B经历还原消除得到苯基硫氰酸酯以及硫醇铜(Ⅰ) C; (4)硫醇铜(Ⅰ) C在氧气作用下生成A和铜(Ⅰ)完成循环(Scheme 18).
2018年, Guo等[48]通过直接可见光催化的S—H键氰化过程, 实现了新型C—S键断裂和重建, 开发了一种由无机硫氰酸盐和硫醇合成硫氰酸酯的新策略(Eq. 18).该反应体系无需金属、碱、配体、无过氧化物等参与, 官能团容忍性好, 目标产物收率在24%~96%, 反应放大至克级别, 产品收率也令人满意.
|
|
(18) |
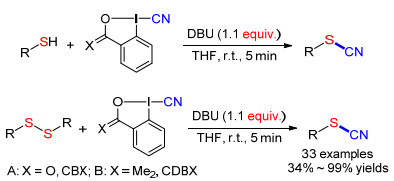
2014年Waser等[49]使用低毒的1-氰基-1, 2-苯碘酰- 3-(1H)酮(CBX)或1-氰基-3, 3-二甲基-3-(1H)-1, 2-苯碘酰(CDBX)为氰源, 实现了硫醇或二硫化物的氰化反应(Scheme 19).在THF溶剂中, 以CBX或CDBX为亲电试剂, DBU作碱, 反应几分钟即可获得各种芳香族、脂肪族类的硫氰酸酯化合物.
2015年, Shi小组[50]报道了新型无过渡金属催化的硫醚的去甲基化氰化反应.使用较低毒性的芳基(氰基)-碘鎓三氟甲磺酸盐为氰源, 在氮气保护下, 实现了硫醚与芳基(氰基)碘鎓三氟甲磺酸盐的交叉偶联.该方法对芳香硫醚有很好的反应性, 而二烷基硫醚反应活性较差.该课题组对反应机理进行了推测: (1)芳基(氰基)碘鎓三氟甲磺酸盐会氧化硫醚会产生中间体A和芳基碘化物; (2)三氟甲磺酸酯(—OTf)与A发生亲核取代反应, 形成产物并释放三氟甲磺酸烷基酯(Scheme 20).

Cheng课题组[51]于2014年发现以碘化亚铜为催化剂, 偶氮二异丁腈(AIBN)和二硫化物能发生氰化反应(Eq. 19).在乙腈溶剂中, 以碳酸氢钾作碱, 经加热回流后反应能以良好的收率进行.值得注意的是, (ArS)2中的两个ArS单元在反应中都能使用.他们认为该反应经历了自由基历程.
|
|
(19) |
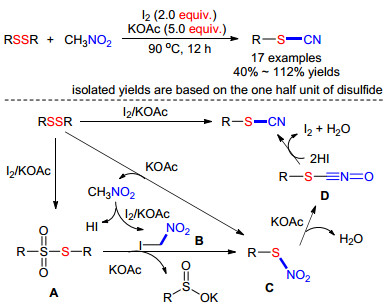
2015年, Tang小组[52]发现当碘和醋酸钾共同存在时, 硝基甲烷和二硫化物可在90 ℃下发生氰化反应.使用硝基甲烷作为氰基源代替高毒的氰化试剂, 拓宽了氰化试剂的来源.他们对反应机理进行了推测: (1)在碘和KOAc存在下二硫化物被氧化成A; 同时, 碘会与MeNO2反应, 得到碘代硝基甲烷B和HI; (2) A与B反应, 生成中间体C和副产物亚磺酸钾(中间体C还有可能直接由MeNO2与二硫化物的反应形成); (3)中间体C脱水得到D; (4)通过HI还原D得到相应的有机硫氰化物(Scheme 21).
硫氰酸酯类化合物广泛应用于有机合成领域.作为重要的有机合成中间体, 硫氰酸酯通过简单转化就可得到硫醇、硫醚、二硫醚、含硫杂环、硫代磷/硫酸酯、异硫氰酸酯等多种有机硫化物, 同时它还可作为氰化试剂很好地参与反应.
2009年Peters课题组[53]在合成硫代酯类抗癌醚脂质(S-ProAEL)的方案中, 有涉及将硫氰化物还原成硫醇的步骤(Eq. 20).在乙醚中, 加入LiAlH4还原硫氰化物, 能以96%的收率得到硫醇.
|
|
(20) |
2015年Brown等[54]报道了一种有趣的方法来修复肽或蛋白质的三维构象(Scheme 22), 具体是将硫醇与1, 2-二氯四嗪结合, 这使得两个半胱氨酸结合在一起, 形成一种桥形结构.随后经紫外光照射, 这种结构被选择性地破坏, 得到相应的二硫氰酸酯肽, 其在游离的半胱氨酸作用下可以生成硫醇.值得注意的是, 在经过这一系列转变后肽的结构未发生改变.
2011年, Zhou课题组[55]开发了使用硫氰化钾和卤代烷烃合成硫醚的反应(Eq. 21).以水为溶剂, 碳酸铯为碱, 1, 10-菲咯啉作配体, 铜为催化剂和四丁基氟化铵作相转移催化剂, 历经48 h后得到产物.此外, 当加入两种不同卤代芳烃能以优异的收率合成不对称硫醚.他们认为首先卤代芳烃和硫氰化钾反应产生关键中间体苯基硫氰酸酯, 再经铜催化与卤代芳烃发生交叉偶联反应得到硫醚产物.
|
|
(21) |
Hajipour小组[56]于2014年研发了一种新型钯催化剂[DBNT][PdCl4], 在DMSO溶剂中, 以卤代芳烃和硫氰化钾为原料, 氢氧化钾作碱, 以良好到优异的收率得到硫醚产物(Eq. 22).反应机理与Zhou课题组[55]类似.
|
|
(22) |
2015年Werz小组[57]开发了一条硫氰酸酯同时作为氰化试剂和硫化试剂的高原子经济利用率的新路径(Eq. 23).以乙腈为溶剂, 氟化铯作碱, Xantphos为配体, 钯为催化剂和氧气作氧化剂, 一步构建C—SAr和C—CN键, 以42%~81%的收率得到产物.
|
|
(23) |
Wang课题组[58]于2014年发现硫脲、烷基卤化物和硫氰酸酯在一定条件下, 可合成苄基烷基二硫化物(Eq. 24).在水中, 以磷酸钾作碱, 加入碘化钾和四丁基卤化铵(TBAH)后, 经微波处理15 min以最高90%的收率得到产物.
|
|
(24) |
2009年, Gotor等[59]发现在甲醇溶液中加入硼氢化钠, 不仅实现羰基部分还原, 而且形成新五元杂环, 得到2-亚氨基-1, 3-氧硫杂环戊烷及其衍生物(Eq. 25).他们认为存在氢化物和甲醇盐时, 羟基硫氰酸盐中间体的OH基能去质子化, 而这过程恰恰是环化步骤基础.
|
|
(25) |
苯并异噻唑-3(2H)-酮衍生物可用于制药领域, 因其具有抑制真菌和细菌的作用, 通常采用2, 2'-二硫代水杨酸与胺反应[60].由于其在药物市场中的强烈需求, 开发出原料易得且收率较优的方法显得十分必要. 2012年, Xi课题组[61]以水为溶剂, 1, 4-二氮杂双环[2.2.2]辛烷(DABCO)作碱, 邻菲罗啉作配体和铜为催化剂的条件下, 实现邻溴苯甲酰胺衍生物与硫氰酸钾反应(Eq. 26).通过一锅两步法, 先形成S—C键, 再环化构建S—N键, 此法可用于合成苯并异噻唑-3(2H)-酮衍生物.
|
|
(26) |
四氮唑能用于制药、高能材料等领域, 关于合成它的方法大部分基于Finnegan等的工作[62], 腈在叠氮化钠和氯化铵存在下形成四氮唑.虽然在此基础上已有化学家提出改进方案[63~65], 但这些方法条件较为苛刻且反应时间较长. 2014年, Myznikov小组[66]开发了一条将硫氰酸酯转化为四唑衍生物的新路径(Eq. 27).在异丙醇溶剂中, 加入氯化锌和叠氮化钠, 搅拌1.5 h即可得到四氮唑产物.该方法条件温和, 反应时间短, 底物适用性广, 为四氮唑类化合物的合成提供新策略.
|
|
(27) |
硫代磷酸酯越来越广泛地用于生物领域, 主要包括害虫防治酶稳定的磷酸类似物, 以及用于化学疗法的不可水解核糖核苷酸模拟物[67, 68]. 2002年, Mioskowski等[69]在硫代磷酸酯的合成中获得新途径(Eq. 28).他们以二取代膦氧化物和硫氰酸酯为原料, 在温和无水条件下, 经碱催化能以中等到优异的收率得到硫代磷酸酯.
|
|
(28) |
2013年, Janeba课题组[70]报道了在室温下的N, N-二甲基甲酰胺(DMF)溶剂中, 实现(杂)芳族硫氰酸酯制备硫代硫酸酯反应(Eq. 29).经亚硫酸钠处理两小时后以最高74%收率得到重要的化工中间体Bunte盐.
|
|
(29) |
Gonda等[71]发现某些硫氰酸盐可通过[3, 3]-σ-重排选择性地得到3-(S)-异硫氰酸根合-3-脱氧-3-C-乙烯基葡萄糖(Scheme 22).值得一提的是, 即使底物中存在不同的立体异构体, 经过重排后均得到同一种产物.此外作者通过密度泛函理论(DFT)计算认为, 产物的立体选择性是硫氰根与1, 2-O-异亚丙基在空间上相互作用的结果.

腈类化合物存在于天然产品, 是药品中关键部分, 能作为染料和除草剂等[72].过渡金属催化卤代芳烃与氰化物反应广泛用于制备芳基腈化合物.但通常氰化反应使用剧毒的无机氰化物, 不利于绿色生产发展. 2006年, Liebeskind等[73]发现在噻吩-2-羧酸铜(Ⅰ) (CuTC)和钯共同催化作用下, 硫氰酸苄酯可作为很好的氰化试剂与芳基硼酸进行交叉偶联反应(Eq. 30).
|
|
(30) |
Chung课题组[74]于2007年以对甲苯基硫氰酸酯为氰化试剂, 与炔烃氰化反应(Eq. 31).以苯为溶剂和钯作催化剂, 经微波处理, 仅需1 h即能以62%~83%收率得到双官能团化烯烃产物.
|
|
(31) |
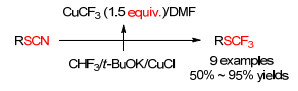
2014年, Rozen等[75]利用硫氰酸酯为原料, 开发了一种合成三氟甲基硫化物的新方法(Scheme 23).以三氟甲烷为起始原料, 在强碱叔丁醇钾作用下与氯化亚铜反应得到三氟甲基铜, 接着其与硫氰酸酯反应得到产物.该方法为大气污染物氟利昂类化合物的有利转化提供了很好的借鉴思路.
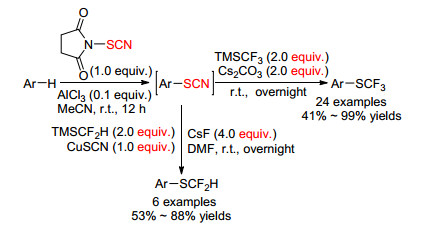
2015年, Goossen小组[76]发现由N-硫氰酰琥珀酰亚胺(NTS)与芳烃得到的硫氰酸酯, 可进一步转化得到三氟甲基硫醚或二氟甲基硫醚(Scheme 24).首先富电子芳烃在AlCl3催化下实现硫氰化反应, 中间产物硫氰酸酯在碳酸铯作用下与TMSCF3反应得到三氟甲基硫醚; 此外硫氰酸酯在铜催化下与TMSCF2H反应生成二氟甲基硫醚.他们发现这两个过程均在富电子且空阻最小位置上进行.
近几年来, 各种各样的硫氰酸酯类化合物合成新方法、新策略层出不穷.直接硫氰化反应是合成硫氰酸酯类化合物的重要方法.此外, 各种有机硫化物的氰化反应大大拓宽了硫氰酸酯类化合物的合成途径.随着硫氰酸酯合成方法研究的不断推进, 这类化合物的应用研究也发展迅速.然而, 在应用的过程中也面临着底物原子利用率低、经济性差等问题.以硫氰酸酯作为双官能团合成子不仅可以提高原子利用率, 而且可以丰富氮硫化合物的分子库, 为这类化合物的应用与研究指明了新方向.
Elhalem, E.; Bailey, B. N.; Docampo, R.; Ujvary, I.; Szajnman, S. H.; Rodriguez, J. B. J. Med. Chem. 2002, 45, 3984. doi: 10.1021/jm0201518
Kokorekin, V. A.; Terent'ev, A. O.; Ramenskaya, G. V.; Grammatikova, N. E.; Rodionava, G. M.; llovaiskii, A. I. Pharm. Chem. J. 2013, 47, 422. doi: 10.1007/s11094-013-0973-7
Dutta, S.; Abe, H.; Aoyagi, S.; Kibayashi, C.; Gates, K. S. J. Am. Chem. Soc. 2005, 127, 15004. doi: 10.1021/ja053735i
Capon, R. J.; Skene, C.; Liu, E. H.-T.; Lacey, E.; Gill, J. H.; Heiland, K.; Friedel, T. J. Org. Chem. 2001, 66, 7765. doi: 10.1021/jo0106750
Yasmana, Y.; Edrada, R. A.; Wray, V.; Proksch, P. J. Nat. Prod. 2003, 66, 1512. doi: 10.1021/np030237j
Billard, T.; Large, S.; Langlois, B. R. Tetrahedron Lett. 1997, 38, 65. doi: 10.1016/S0040-4039(96)02216-2
Johnson, T. B.; Douglass, I. B. J. Am. Chem. Soc. 1939, 61, 2548. doi: 10.1021/ja01878a085
Sabacky, M. J.; Johnson, S. M.; Martin, J. C.; Paul, I. C. J. Am. Chem. Soc. 1969, 91, 7542. doi: 10.1021/ja01054a073
Aureggi, V.; Sedelmeier, G. Angew. Chem. Int. Ed. 2007, 46, 8440. doi: 10.1002/(ISSN)1521-3773
Ciszek, J. W.; Stewart, M. P.; Tour, J. M. J. Am. Chem. Soc. 2004, 126, 13172. doi: 10.1021/ja0472477
Still, I. W. J.; Toste, F. D. J. Org. Chem. 1996, 61, 7677. doi: 10.1021/jo961029h
Castanheiro, T.; Suffert, J.; Donnard, M.; Gulea, M. Chem. Soc. Rev. 2016, 45, 494. doi: 10.1039/C5CS00532A
Sun, N.; Che, L.-S.; Mo, W.-M.; Hu, B.-X.; Shen, Z.-L.; Hu, X.-Q. Org. Biomol. Chem. 2015, 13, 691. doi: 10.1039/C4OB02208D
Zhu, N.; Wang, F.; Chen, P.-H.; Ye, J.-X.; Liu, G.-S. Org. Lett. 2015, 17, 3580. doi: 10.1021/acs.orglett.5b01677
Jiang, H.-F.; Yu, W.-T.; Tang, X.-D.; Li, J.-X.; Wu, W.-Q. J. Org. Chem. 2017, 82, 9312. doi: 10.1021/acs.joc.7b01122
Chen, Y.-T.; Wang, S.-F.; Jiang, Q.-W.; Cheng, C.-G.; Xiao, X.-H.; Zhu, G.-G. J. Org. Chem. 2018, 83, 716. doi: 10.1021/acs.joc.7b02700
Rad, M. N. S. J. Chem. Res. 2016, 40, 583. doi: 10.3184/174751916X14736925997854
Dwivedi, V.; Rajesh, M.; Kumar, R.; Kantc, R.; Reddy, M. S. Chem. Commun. 2017, 53, 11060. doi: 10.1039/C7CC06081E
Palsuledesai, C. C.; Murru, S.; Sahoo, S. K.; Patel, B. K. Org. Lett. 2009, 11, 3382. doi: 10.1021/ol901561j
Modi, A.; Ali, W.; Patel, B. K. Org. Lett. 2017, 19, 432. doi: 10.1021/acs.orglett.6b03430
Yadav, A. K.; Yadav, L. D. S. Tetrahedron Lett. 2015, 56, 6696. doi: 10.1016/j.tetlet.2015.10.048
Zhang, D.; Wang, H.; Bolm, C. Chem. Commun. 2018, 54, 5772. doi: 10.1039/C8CC03178A
Liang, S.; Zeng, C.-C.; Tian, H.-Y.; Sun, B.-G.; Luo, X.-G.; Ren, F.-Z. Adv. Synth. Catal. 2018, 360, 1444. doi: 10.1002/adsc.v360.7
Xiong, H.-Y.; Pannecoucke, X.; Besset, T. Org. Chem. Front. 2016, 3, 620. doi: 10.1039/C6QO00064A
Zhang, H.-L.; Wei, Q.; Wei, S.-Q.; Qu, J.-P.; Wang, B.-M. Eur. J. Org. Chem. 2016, 20, 3373.
Chen, Q.; Lei, Y.-J.; Wang, Y.-F.; Wang, C.; Wang, Y.-N.; Xu, Z.-Q.; Wang, H.; Wang, R. Org. Chem. Front. 2017, 4, 369. doi: 10.1039/C6QO00676K
Wang, Z.-H.; Wang, L.; Chen, Q.; He, M.-Y. Synth. Commun. 2018, 40, 76.
Venkanna, P.; Rajanna, K. C.; Kumar, M. S.; Venkateswarlu, M.; Ali, M. M. Synlett 2016, 27, 237.
Qiu, J.-S.; Wu, D.; Karmaker, P. G.; Yin, H.-Q.; Chen, F.-X. Org. Lett. 2018, 20, 1600. doi: 10.1021/acs.orglett.8b00342
Wu, D.; Qiu, J.-S.; Karmaker, P. G.; Yin, H.-Q.; Chen, F.-X. J. Org. Chem. 2018, 83, 1576. doi: 10.1021/acs.joc.7b02850
Yang, H.; Duan, X.-H.; Zhao, J.-F.; Guo, L.-N. Org. Lett. 2015, 17, 1998. doi: 10.1021/acs.orglett.5b00754
Yang, D.-S.; Yan, K.-L.; Wei, W.; Li, G.-Q.; Lu, S.-L.; Zhao, C.-X.; Tian, L.-J.; Wang, H. J. Org. Chem. 2015, 80, 11073. doi: 10.1021/acs.joc.5b01637
Zhang, X.-Z.; Ge, D.-L.; Chen, S.-Y.; Yu, X.-Q. RSC Adv. 2016, 6, 66320. doi: 10.1039/C6RA13303G
Guo, L.-N.; Gu, Y.-R.; Yang, H.; Hu, J. Org. Biomol. Chem. 2016, 14, 3098. doi: 10.1039/C6OB00221H
Chen, J.-C.; Wang, T.-Y.; Wang, T.; Lin, A.-J.; Yao, H.-Q.; Xu, J.-Y. Org. Chem. Front. 2017, 4, 130. doi: 10.1039/C6QO00590J
Mete, T. B.; Khopade, T. M.; Bhat, R. G. Tetrahedron Lett. 2017, 58, 415. doi: 10.1016/j.tetlet.2016.12.043
Ji, F.; Fan, Y.; Yang, R.; Yang, Y.-Y.; Yu, D.-W.; Wang, M.-Y.; Li, Z.-Y. Asian J. Org. Chem. 2017, 6, 682. doi: 10.1002/ajoc.v6.6
Karmaker, P. G.; Qiu, J.; Wu, D.; Yin, H.; Chen, F. X. Synlett 2018, 29, 954. doi: 10.1055/s-0036-1591927
Zeng, Y.-F.; Tan, D.-H.; Chen, Y.-Y.; Lv, W.-X.; Liu, X.-G.; Li, Q.-J.; Wang, H.-G. Org. Chem. Front. 2015, 2, 1511. doi: 10.1039/C5QO00271K
Lv, Y.-H.; Pu, W.-Y.; Cui, H.; He, J.-L.; Zhang, Q.-M. Synth. Commun. 2016, 46, 1223. doi: 10.1080/00397911.2016.1194997
Tao, Z.-K.; Li, C.-K.; Zhang, P.-Z.; Shoberu, A.; Zou, J.-P.; Zhang, W. J. Org. Chem. 2018, 83, 2418. doi: 10.1021/acs.joc.7b02929
Fan, W.-G.; Yang, Q.; Xu, F.-S.; Li, P.-X. J. Org. Chem. 2014, 79, 10588. doi: 10.1021/jo5015799
Mitra, S.; Ghosh, M.; Mishra, S.; Hajra, A. J. Org. Chem. 2015, 80, 8275. doi: 10.1021/acs.joc.5b01369
Wang, L.; Wang, C.-C.; Liu, W.-J.; Chen, Q.; He, M.-Y. Tetrahedron Lett. 2016, 57, 1771. doi: 10.1016/j.tetlet.2016.03.028
Singh, M.; Yadav, A. K.; Yadav, L. D. S.; Singh, R. K. P. Synlett 2018, 29, 176. doi: 10.1055/s-0036-1590921
李伟, 杨超, 高国林, 夏吾炯, 有机化学, 2017, 37, 480. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345747.shtmlLi, W.; Yang, C.; Gao, G.-L.; Xia, W.-J. Chin. J. Org. Chem. 2017, 37, 480 (in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345747.shtml
Castanheiro, T.; Gulea, M.; Donnard, M.; Suffert, J. Eur. J. Org. Chem. 2014, 7814.
Guo, W.; Tan, W.; Zhao, M.; Zheng, L.; Tao, K.; Chen, D.; Fan, X. J. Org. Chem. 2018, 83, 6580. doi: 10.1021/acs.joc.8b00887
Frei, R.; Courant, T.; Wodrich, M. D.; Waser, J. Chem.-Eur. J. 2014, 20, 1. doi: 10.1002/chem.201390210
Zhu, D.; Chang, D.-H.; Shi, L. Chem. Commun. 2015, 51, 7180. doi: 10.1039/C5CC00875A
Teng, F.; Yu, J.-T.; Yang, H.-T.; Jiang, Y.; Cheng, J. Chem. Commun. 2014, 50, 12139. doi: 10.1039/C4CC04578E
Wang, Z.-H.; Ji, X.-M.; Hu, M.-L.; Tang, R.-Y. Tetrahedron Lett. 2015, 56, 5067. doi: 10.1016/j.tetlet.2015.07.054
Linderoth, L.; Fristrup, P.; Hansen, M.; Melander, F.; Madsen, R.; Andresen, T. L.; Peters, G. H. J. Am. Chem. Soc. 2009, 131, 12193. doi: 10.1021/ja901412j
Brown, S. P.; Smith, A. B. Ⅲ J. Am. Chem. Soc. 2015, 137, 4034. doi: 10.1021/ja512880g
Ke, F.; Qu, Y.-Y.; Jiang, Z.-Q.; Li, Z.-K.; Wu, D.; Zhou, X.-G. Org. Lett. 2011, 13, 454. doi: 10.1021/ol102784c
Hajipour, A. R.; Pourkaveh, R.; Karimi, H. Appl. Organomet. Chem. 2014, 28, 879. doi: 10.1002/aoc.v28.12
Pawliczek, M.; Garve, L. K. B.; Werz, D. B. Org. Lett. 2015, 17, 1716. doi: 10.1021/acs.orglett.5b00494
Lu, X.-G.; Wang, H.-M.; Gao, R.-L.; Sun, D.-M.; Bi, X.-J. RSC Adv. 2014, 4, 28794. doi: 10.1039/C4RA03592E
Bisogno, F. R.; Cuetos, A.; Lavandera, I.; Gotor, V. Green Chem. 2009, 11, 452. doi: 10.1039/b900137a
Sano, T.; Takagi, T.; Gama, Y.; Shibuya, I.; Shimizu, M. Synthesis 2004, 1585.
Wang, F.; Chen, C.; Deng, G.; Xi, C.-J. J. Org. Chem. 2012, 77, 4148. doi: 10.1021/jo300250x
Finnegan, W. G.; Henry, R. A.; Lofquist, R. J. Am. Chem. Soc. 1958, 80, 3908. doi: 10.1021/ja01548a028
Roh, J.; Artamonova, T. V.; Vavrova, K.; Koldobskii, G. I.; Hrabalek, A. Synthesis 2009, 2175.
Roh, J.; Vavrova, K.; Hrabalek, A. Eur. J. Org. Chem. 2012, 31, 6101.
Yoneyama, H.; Usami, Y.; Komeda, S.; Harusawa, S. Synthesis 2013, 45, 1051. doi: 10.1055/s-00000084
Vorona, S.; Artamonova, T.; Zevatskii, Y.; Myznikov, L. Synthesis 2014, 46, 781.
Marshall, W. S.; Caruthers, M. H. Science 1993, 259, 1564. doi: 10.1126/science.7681216
Cieslak, J.; Jankowska, J.; Stawinski, J.; Kraszewski, A. J. Org. Chem. 2000, 65, 7049. doi: 10.1021/jo000729q
Renard, P.-Y.; Schwebel, H.; Vayron, P.; Josien, L.; Valleix, A.; Mioskowski, C. Chem.-Eur. J. 2002, 8, 2910. doi: 10.1002/1521-3765(20020703)8:13<2910::AID-CHEM2910>3.0.CO;2-R
Jansa, P.; Cechova, L.; Dracínsky, M.; Janeba, Z. RSC Adv. 2013, 3, 2650. doi: 10.1039/c2ra21975a
Gonda, J.; Martinková, M.; Raschmanová, J.; Balentová, E. Tetrahedron: Asymmetry 2006, 17, 1875. doi: 10.1016/j.tetasy.2006.06.032
Sundermeier, M.; Zapf, A.; Beller, M.; Sans, S. Tetrahedron Lett. 2001, 42, 6707. doi: 10.1016/S0040-4039(01)01390-9
Zhang, Z.-H.; Liebeskind, L. S. Org. Lett. 2006, 8, 4331. doi: 10.1021/ol061741t
Lee, Y. T.; Choi, S. Y.; Chung, Y. K. Tetrahedron Lett. 2007, 48, 5673. doi: 10.1016/j.tetlet.2007.06.041
Potash, S.; Rozen, S. J. Fluorine Chem. 2014, 168, 173. doi: 10.1016/j.jfluchem.2014.09.026
Jouvin, K.; Matheis, C.; Goossen, L. J. Chem.-Eur. J. 2015, 21, 14324. doi: 10.1002/chem.201502914

图式 2 铜催化(杂)芳环化合物的硫氰化反应
Scheme 2 Copper-catalyzed thiocyanation of aromatics/het- eroaromatics

图式 3 炔酮的硫氰化反应和分子内环化反应
Scheme 3 Thiocyanation of ynones and concomitant intramolecular cyclizations

图式 4 有机催化环氧乙烷的硫氰化酰化反应
Scheme 4 Organocatalytic thiocyanation and acylation of oxiranes

图式 6 一锅法制备咪唑并吡啶硫氰酸酯
Scheme 6 Thiocyanation of imidazopyridines in a sequential one-pot process

图式 8 超声辅助下苯酚、苯胺和吲哚的硫氰化反应
Scheme 8 Ultrasonic assisted thiocyanation of phenols, anilines and hydrazines

图式 9 三聚氰氯/NH4SCN促进的芳烃硫氰化反应
Scheme 9 Cyanuric chloride/NH4SCN triggered thiocyanation of aromatic compounds

图式 14 苯酚、苯胺和杂环区域选择性的硫氰化
Scheme 14 Regioselective thiocyanation of phenols, anilines and heterocycles

图式 18 CuCN与苯硫酚或二芳基二硫化物直接氰化反应
Scheme 18 CuCN-mediated cyanation of thiophenols or diaryl disulfides

图式 19 CBX或CDBX与苯硫酚或二芳基二硫化物的氰化反应
Scheme 19 Cyanation of thiophenols or diaryl disulfides with CBX or CDBX

图式 20 硫醚与芳基(氰基)碘鎓三氟甲磺酸盐交叉偶联反应
Scheme 20 Cross-coupling of thioethers with aryl(cyano)io- donium triflates

图式 21 以硝基甲烷为氰化试剂合成硫氰酸酯
Scheme 21 Nitromethane as a cyanating reagent for the synthesis of thiocyanates

图式 23 硫氰酸烯丙酯[3, 3]-σ-重排合成α-异硫氰酸酯
Scheme 23 Synthesis of α-quaternary isothiocyanate by [3, 3]- σ-rearrangement of allyl thiocyanate

图式 24 铜催化硫氰根转化为三氟甲基化硫化物
Scheme 24 Copper-mediated transformation of thiocyanate into trifluoromethylated sulfides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



































 下载:
下载:












 下载:
下载: