











图式 1.
钯催化导向的芳烃硝化反应机理
Scheme 1.
Proposed mechanism for the palladium-catalyzed chelation-assisted C—H nitration of arenes

芳烃硝基化合物作为有机合成中重要的中间体, 在医药、农药、精细化工、染料、炸药等领域有着广泛的应用[1].近年来, 人们越来越重视利用硝基化合物作为有机合成中的活性中间体.硝基的活化作用在许多有机反应中被运用, 并且容易转化成各种官能团, 拓宽了硝基化合物在合成复杂分子中的重要性[2].芳香族化合物的硝化是最广泛研究的反应之一, 芳烃亲电取代硝化是合成硝基芳烃的经典合成方法[1a, 3].传统的亲电硝化对于芳香族化合物通常伴随着几种持续性的缺点, 例如(1)区域选择性较差, 由于取代位置不同, 容易导致产生异构硝化产物, 特别对一些单取代芳烃的硝基化, 如苯胺、苯酚等; (2)化学选择性差, 因为硝化过程难以控制, 普遍存在过度硝化现象, 而且硝化剂也是良好的氧化剂, 硝化化合物通常伴随着氧化产物; (3)官能团兼容性较差, 主要是因为亲电硝化反应一般在酸性与氧化性较强的体系中进行, 从而影响到底物与官能团的兼容性, 而且容易造成环境污染问题; (4)受取代基定位规则制约, 由于定位规则限制, 传统亲电硝化方法不能区域专一性地合成邻硝基芳酮、邻硝基芳醛等硝基化合物.此外, 水洗过程会产生大量含无机酸的废水, 给环境带来污染问题.基于对环境因素和生产成本方面的考虑, 寻找一类能够用于芳香族化合物硝化反应并提高产物选择性的绿色环保催化体系就显得尤为重要.因此, 温和与区域选择性硝化方法的发展具有重要的实际意义[4].
过去几十年, 过渡金属催化导向基团辅助的C—H键活化反应取得了令人鼓舞的成就, 已成为区域选择性形成碳-碳和碳-杂原子键的重要合成方式[5].近些年来, 过渡金属催化的导向基团辅助邻位C—H键硝化反应研究取得了重要进展.根据不同的过渡金属催化剂, 本文进行分类综述导向基团辅助的惰性C—H键硝化反应的合成研究进展及其反应机理, 并就该研究领域的局限性和未来的发展前景进行了总结和展望.
2010年, 刘运奎和徐振元课题组[6]合作首次报道用喹喔啉作为导向基团的C—H键硝化反应(Eq. 1).该反应采用醋酸钯作为催化剂, 在亚硝酸银和过硫酸钾的共同作用下, 高效地实现了C—H键硝基化反应, 以较高的产率选择性地得到一元硝基化的目标产物.该催化体系具有广泛的底物范围及良好的官能团兼容性.最后, 作者通过自由基捕获的方法进一步证明了该反应是由硝基自由基参与的反应机理.
|
|
(1) |
2013年, 刘运奎课题组[7]进一步详细研究了喹喔啉、吡啶、肟醚、喹啉和吡啶导向的C—H键硝化反应(Eq. 2).其中, 在以肟醚作为导向基团时, 需要在三氟乙酸钯的催化下进行, 实现了区域专一性的邻位C—H键定向硝化反应[8].在酸性条件下, 肟醚导向的硝化产物通过水解移除导向基团, 可以区域专一性地合成一系列的邻硝基芳基酮化合物.该方法不受取代基定位规则限制, 具有区域专一性, 并且具有高效、底物普适性好、避免了使用浓酸造成的环境污染等优点.
|
|
(2) |
2014年, 刘运奎课题组[9]将反应底物扩大到2-苯氧基吡啶导向的硝化反应(Eq. 3).该反应以二氯乙烷(DCE)为溶剂, 110 ℃下反应48 h成功实现了吡啶导向的邻位C—H键硝化反应.此外, 2-苯氧基嘧啶作为导向基团也可以实现邻位C—H键硝化反应.以无水甲苯为溶剂, 在100 ℃下与MeOTf反应2 h, 然后在Na/MeOH体系中回流15 min, 经过这两步可以很容易地脱除导向基团.因此, 通过引入导向基团吡啶、C—H键硝化和去除导向基团三步, 可以区域选择性地合成邻硝基苯酚.
|
|
(3) |
2014年, 刘运奎课题组[10]将反应底物进一步扩大到偶氮基作为导向邻位芳香C—H键硝化反应(Eq. 4, Condition A).该反应可以实现由苯胺类化合物区域专一性地制备邻苯二胺类化合物的新方法.随后, Sun课题组[11]报道了二氧化氮作为硝基源, 醋酸钯催化偶氮苯邻位C—H键硝化反应(Eq. 4, Condition B).该反应具有宽广的底物范围, 优秀的产率, 良好的官能团兼容性等优点.为了显示该反应的实用价值, 作者通过锌粉的还原作用进一步将目标产物转化为一系列三唑类化合物.通过Zn/HCO2H体系, 偶氮导向的邻位芳烃C—H键硝化产物能够同时还原偶氮基和硝基, 可以高效地合成邻苯二胺类化合物. 2014年, Ranu课题组[12]以亚硝酸叔丁酯为硝化来源、丰富的空气为氧化剂也成功实现了偶氮苯邻位C—H键硝化反应(Eq. 4, Condition C).
|
|
(4) |
2015年, 焦宁课题组[13]开发了分子氧为氧化剂自由基参与的芳基C—H键官能团化反应(Eq. 5).他们以亚硝酸叔丁基酯为硝基的来源、溴代四丁基铵(TBAB)为添加剂, 实现了Pd(OAc)2催化吡啶、嘧啶、肟基及偶氮导向基团诱导的芳烃C—H键硝基化反应.该反应经历了PdⅡ/PdⅣ的反应历程.
|
|
(5) |
同年, 刘运奎课题组[14]报道了钯催化8-甲基喹啉为导向基团的sp3-C—H键硝化反应(Eq. 6).该反应以亚硝酸叔丁酯为硝化试剂、氧气为氧化剂的条件下, 成功实现了8-甲基喹啉sp3-C—H键硝化反应.该反应具有较高的选择性、较好的底物范围及产率高等优点.
|
|
(6) |
2015年, 吴养洁课题组[15]开发了一种有效的钯催化2-芳基苯并噁唑邻位C—H键硝化的方法(Eq. 7).这种硝化反应具有很高的区域选择性, 而且苯环可以耐受如卤素、甲基及甲氧基等许多官能团.在这个催化体系下, 苯环上无论反应底物含有吸电子取代基还是吸电子基, 均能反应, 以中等至良好的产率生成邻硝化产物.此外, 2-芳基苯并噻唑类底物也能很好地进行反应.
|
|
(7) |
2013年, 李亚明等[16]以苯胺、硝酸铋和乙酸酐为原料, 开发了一种有效合成N-苯基乙酰胺硝化的合成方法(Eq. 8).该反应在温和条件下以中等偏上的产率合成邻硝基乙酰苯胺, 实验操作比较简单, 但区域选择性较差, 往往伴随对位硝化的副产物. 2015年, Xu等[17]开发了一种有效而实用的Pd(OAc)2催化二级和三级芳香胺邻对位二硝基化的方法:以三氟乙醇(TFE)和三氟乙酸(TFA)为混合溶剂, Bi(NO3)3•5H2O为硝化试剂.该产物可用于偶氮染料5-氨基-N-甲基苯并咪唑酮的合成.乙酸钯促进了硝酰正离子的生成, 该反应机理是通过路易斯酸催化芳烃亲电取代进行的.
|
|
(8) |
2016年, Kapur等[18]报道了钯催化2-氨基嘧啶作为导向基团的C—H键邻位硝化反应(Eq. 9).此方法以AgNO2为硝化试剂、K2S2O8为氧化剂80 ℃的DCE中反应, 可以顺利实现芳香C—H键邻位硝化反应.作者通过三乙基硅烷还原、N2H4/AcOH处理两步, 可以成功地移除导向基团, 高产率合成了邻硝基苯胺类化合物.
|
|
(9) |
2017年, 姜玉波等[19]报道了一种有效的钯催化1, 2, 3-三唑导向的区域选择性芳烃邻位硝化反应(Eq. 10).该反应以Pd(OAc)2为催化剂, 廉价的NaNO2为硝化源, 以良好至优秀的产率合成一系列含1, 2, 3-三唑的硝基化产物.该催化体系显示出较高的区域选择性和化学选择性, 具有良好的官能团耐受性, 为1, 2, 3-三唑硝基化衍生物的合成提供一种有效的方法.
|
|
(10) |
2017年, 张梦扬等[20]报道了Pd(PPh3)4催化α-磺酰基甲基苯乙烯的硝化反应(Eq. 11).通过邻硝化过程, 该反应可以合成一系列磺酰基邻硝基苯乙烯.目标产物的结构通过X射线晶体学证实.
|
|
(11) |
目前报道的Pd催化导向基团辅助的C—H硝化反应主要有以下两种反应机制[6~20], 如Scheme 1所示.第一种是Pd(Ⅱ)/Pd(Ⅳ)催化的反应历程:首先, 二价钯催化剂和反应底物配位, 通过协同质子脱氢形成单核环钯芳基钯物种A, 然后通过自由基的氧化形成的Pd(Ⅳ)物种B; 最后, 物种B通过还原消除形成硝化产物, 同时二价钯催化剂再生.另外一个反应机制是通过Pd(Ⅱ)/Pd(Ⅲ)催化循环:首先, 二价钯催化剂和反应底物配位, 通过协同质子脱氢形成双核环钯物种C, 然后通过自由基的氧化形成的Pd(Ⅲ)-Pd(Ⅲ)或Pd(Ⅱ)/Pd(Ⅳ)物种; 最后, 通过配体交换和还原消除形成硝化产物, 同时二价钯催化剂再生.
2011年, 毕锡和和刘群等[21]报道了首例廉价的铜催化吡啶导向的硝化反应(Eq. 12).富电子的底物能很好地适用该反应体系, 而缺电子的底物反应效果较差, 溶剂1, 2, 3-三氯丙烷对反应起到了至关重要的作用.相对于钯催化体系, 该反应需要的催化剂用量较高, 底物普适性相对较差.该反应可能经历了四元环状过渡状态协同的反应过程.
|
|
(12) |
2013年, 王梅祥课题组[22]报道了以亚硝酸钾为硝化试剂Cu(OTf)2催化吡啶导向的C—H键硝化反应(Eq. 13).虽然该反应在室温条件反应, 但需要化学计量的催化剂, 同时伴随着大量的C—H键羟基化副产物.此外, 底物的普适性以及官能团的兼容性也相对较差.
|
|
(13) |
2014年, Gooen课题组[23]开发了CuNO3(PPh3)2催化喹啉酰胺双齿导向的芳基C—H键邻位硝化反应(Eq. 14, Condition A).该反应以AgNO2作为硝基的来源, NMO作为氧化剂, 在50 ℃条件下成功实现了芳环邻位C—H键硝化反应.该一反应不仅适用于芳基甲酰喹啉酰胺底物, 而且对于五元杂环、六元杂环底物也能适用.随后, Tan课题组[24]在以Cu(OAc)2•H2O为催化剂、K2HPO4为碱的条件下, 反应48 h以中等的产率得到邻位单硝基化产物(Eq. 14, Condition B).相较于Goo en[23]报道的反应体系需要当量的铜盐, 该反应体系采用NaNO2作为硝化试剂, 相对较为廉价.在碱性或酸性条件下, 目标产物通过水解可以高效合成邻硝基苯甲酸类化合物.
|
|
(14) |
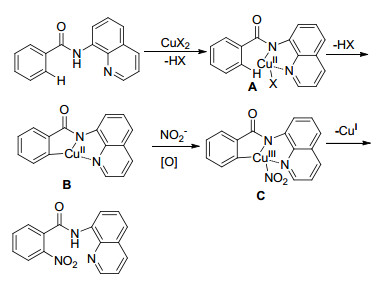
虽然该类反应机理还不明确, 目前看来合理的反应机理如Scheme 2所述[23, 24]:首先, Cu(Ⅱ)与反应底物进行配体交换产生铜络合物A, 接下来经历C—H活化产生芳基Cu(Ⅱ)物质B; 随后, 在硝化试剂存在的条件下, 物种B被空气中的氧气氧化或者Cu(OAc)2促进B氧化生成硝酸Cu(Ⅲ)络合物C; 最后, 物种C还原消除产生目标硝化化合物和一价铜.
2014年, Rodrguez等[25]报道了硝酸为硝化试剂, 硝酸铜催化N-酰基苯胺C—H键硝化反应(Eq. 15).采用氧气作为氧化剂, 在乙腈中100 ℃反应1 h就顺利得到硝化产物.但是该反应受底物取代基位置的影响较大, 区域选择性较差:对位取代的苯胺底物生成邻位硝基化产物, 邻位取代的苯胺底物生成对位硝基化产物, 而间位取代的苯胺底物则成邻位和对位硝化产物的混合物.
|
|
(15) |
2015年, Sadhu等[26]使用N-芳基-1-芳基-1H-四唑-5-胺作为底物, 发现一种有效的铜催化的芳烃选择性硝化反应(Eq. 16).在室温反应条件下, 他们使用硝酸铁(Ⅲ)作为硝化源, 与CuCl2•2H2O一起搅拌, 在1, 2-二氯乙烷中反应11 h, 成功地实现了邻位C—H键硝化反应.在碱性条件下, 硝化产物可以发生水解以较高的产率得到邻硝基苯胺衍生物.
|
|
(16) |
2015年, 燕红等[27]使用亚硝酸叔丁酯作为硝化试剂, 10%的Cu(NO3)2•3H2O高效催化N-芳基酰胺邻位硝化反应(Eq. 17).该方法可以很好地耐受各种官能团, 一系列的苯胺和丙烯酰胺底物都可以顺利反应, 但是伴随对位硝化的副产物.丙烯酰胺类底物在室温下反应可以选择性得到(E)式丙烯酰胺硝化产物.该合成方法具有原料价格低廉、反应条件温和及高效等优点.机理研究表明:叔丁基亚硝酸酯首先发生均裂, 在氧气条件下产生的硝基自由基参与两个硝化过程.
|
|
(17) |
2016年, 章鹏飞、张新迎及Ribas三个课题组分别报道了喹啉酰胺喹啉环上5位C—H键硝化反应(Eq. 18).章鹏飞课题组[28]发展了铜催化喹啉酰胺喹啉环上C—H键硝化反应(Eq. 18, Condition A).在室温条件下, 该反应以NaNO2为硝化试剂, 二乙酸碘苯为氧化剂及新戊酸为添加剂, 以良好的产率快速和有效地构建了一系列硝化喹啉衍生物.当喹啉环上5位被取代时, 则生成喹啉环上7位C—H键硝化产物.该方法克服了以前的硝化策略中报道的使用昂贵的金属催化剂或使用有毒和不稳定的硝化剂等缺点.张新迎等[29]使用经济无毒的Fe(NO3)3•9H2O作为催化剂和硝化试剂实现了喹啉酰胺喹啉环上5位C—H键硝化反应(Eq. 18, Condition B).当添加CuCl2•2H2O作为催化剂时, 主要进行二元硝基化得到5, 7-二硝基-8-氨基喹啉酰胺.其中, Fe(NO3)3•9H2O同时起到螯合促进剂和硝化试剂的双重作用. Ribas课题组[30]发展了钴催化喹啉酰胺喹啉环上C—H键硝化反应(Eq. 18, Condition C).该反应采用叔丁基亚硝酸酯为硝化试剂、氧气为氧化剂室温反应18 h, 以中等产率得到喹啉环5位和7位一元硝化混合物.该实验反应温度比较温和, 但是区域选择性和底物官能团的兼容性较差.
|
|
(18) |
2017年, Koley等[31]报道了一种高效的铜催化8-氨基喹啉酰胺5-位C—H键硝化反应(Eq. 18, Condition D).该反应以亚硝酸叔丁酯为硝化剂、氧气为氧化剂, 5 mol%的氯化铜为催化剂, 在室温下实现了N-芳烃、杂芳烃及脂肪酰基底物的硝化反应. 2017年, 侯红卫课题组[32]在研究喹啉酰胺作为配体构建过渡金属配位聚合物时, 发现了一个令人意外的实验结果:硫酸锌催化喹啉5, 7-位双硝化反应(Eq. 18, Condition E).该反应经过不断尝试, 最终实现了硫酸锌(Ⅱ)在酸性条件下催化喹啉环硝化的合成.这是首例报道的二价锌盐催化芳香C—H键活化反应, 成为一个简便环保合成硝基喹啉衍生物的重要策略.
该类反应是通过单电子转移的机理进行的(Scheme 3)[28~32].首先, 中心金属和底物配位、氧化生成硝化三价金属络合物A; 随后, 物种A在酸性介质中通过发生单电子转移过程产生阳离子喹啉基和二价金属络合物B; 最后, 原位产生的硝基自由基对喹啉发起进攻通过自由基偶联的形式生成物种C, 通过协同质子转移、脱金属步骤提供了硝化产物, 同时二价金属盐再生.

2017年, Chandrasekharam等[33]利用苯甲酰胺作为导向基团在铜催化C—H键邻位硝化反应(Eq. 19).在温度为83 ℃的实验条件下, 该反应以TMSN3为添加剂、1, 2-二氯乙烷为溶剂实现了区域专一性的邻位硝化反应.该方法不受取代基定位规限制, 具有区域专一性, 并且具有高效、底物普适性好等优点.
|
|
(19) |
2017年, Chandrasekharam等[34]开发了一种通用且高效的铜催化2-芳基苯并噁唑和苯并噻唑导向的邻位芳基硝化反应(Eq. 20).在空气氛围中, 他们使用廉价丰富的氯化铜作为催化剂、Fe(NO3)3•9H2O作为硝化试剂、Ag2O作为氧化剂, 在DCE中回流24 h, 以较高的区域选择性实现了2-芳基苯并噻唑的硝化反应.该反应不仅操作简单, 而且官能团的耐受性较好.反应机理如Scheme 4所示, 首先, 氯化铜与2-芳基的氮配位得到中间体A, 通过阴离子交换形成含硝酸根离子的铜(Ⅱ)配合物B; 然后, 中间体B经历单电子转移途径与芳环配位形成自由基阳离子中间体C, 物种C随后被存在于附近的硝基捕获转化为阳离子物种D; 最后, 中间体D去质子化生成目标硝化产品.或者通过四元环状过渡状态E断裂C—H和N—O键形成硝化产物.
|
|
(20) |

2017年, Sarma等[35]报道了一种简单实用的铜催化咔唑氮导向的芳烃邻位C—H键硝化反应(Eq. 21).该反应以氯化铜为催化剂、硝酸铁为硝化试剂, 以优秀的位置选择性和产率实现了9-(吡啶-2-基)-9H-咔唑的硝化反应.此外, 他们还完成了该类底物的C—H键三氟甲基化和氰基化反应.
|
|
(21) |
2018年, 姜超等[36]报道了一种高效廉价的铜介导吲哚C—H键碘化和硝化多米诺反应(Eq. 22).在温和的有氧条件下, 该反应以吲哚为起始原料一步法合成3-碘-2-硝基吲哚, 具有高区域选择性和广泛的底物范围.经过季铵化、醇解两步以中等偏上的产率脱除导向基团, 生成3-碘-2-硝基吲哚.该反应通过碘化亚铜和叔丁基亚硝酸酯反应生成Cu(Ⅲ)物种, 对吲哚的3位进行亲电加成-还原消除形成3-碘吲哚中间体, 随后经过C—H活化、硝基自由基氧化加成和还原消除等步骤生成目标产物.
|
|
(22) |
2018年, 孙丽萍等[37]报道了一种新型高效的铜催化芳基脲邻位C—H键硝化反应(Eq. 23).该反应以CuCl2•2H2O为催化剂, 以脲为导向基团、硝酸铁为硝基源及p-TSA为催化剂添加剂, 以中等至优良的产率合成了硝化芳基脲.该方法不仅具有高度的区域选择性, 而且显示了良好的官能团容忍性.芳基脲邻位C—H键硝化反应通过合成各种硝化芳基尿衍生物, 这是尿素化学的有效延伸和补充.
|
|
(23) |
2016年, 张翱课题组[38]首次报道了通过Ru3(CO)12催化芳烃间位C—H键硝化反应(Eq. 24).他们以Cu(NO3)2•3H2O为硝化试剂, Oxone和AgTFA作为氧化剂, 顺利实现了吡啶导向的芳烃C—H键间位硝化反应.应用这一方法, 得到的硝化产物可以通过一步反应方便地转变为间位氨基化、烷氨基化、磺酰胺化产物, 也能转化为吲哚和苯并噻唑等杂环.同时, 这一新方法可以简捷、高效地实现治疗基底细胞癌药物Vismodegib及CDK/CK1双靶抑制剂(R)-DRF053的合成.
|
|
(24) |
反应机理如Scheme 5所示, 首先催化剂与底物通过芳烃C—H活化步骤形成活性Ru络合物A; 随后, 原位生成的硝基自由基对Ru-CAr的σ键对位碳发起亲核进攻生成物种B, 通过阴离子交换产生的Cu(CF3COO)NO3可能促进物种B的去质子化, 生成更稳定的复合物C; 最后, 复合物C与反应底物进行配体交换生成硝化产品, 同时复合物A再生.

随后, 该课题组[39]又报道了肟导向芳烃C—H键间位硝化反应(Eq. 25).在非酸性催化体系下, 采用AgNO3作为硝化试剂、O2和PhI(TFA)2作为共氧化剂.在酸性条件下, 硝化产物水解生成3-硝基苯乙酮.在ZrCl4/NaBH4的还原条件下, 可以转化为胺类化合物.此外, 该方法还可以应用于药物分子Diazepam和Fluvoxamine的后期硝化, 这是常规的硝化方法所不能实现的.
|
|
(25) |
最近, 该课题组[40]实现了嘌呤导向6-芳烃C—H键间位硝化反应(Eq. 26).通过筛选11种不同的配体, 作者发现大位阻的膦配体是实现该催化反应的关键因素.不同位置取代的6-芳烃嘌呤及核苷底物均能专一性的得到间位硝化产物.作者将该方法应用于p-38激酶抑制剂的合成, 这进一步说明该方法的实用性.
|
|
(26) |
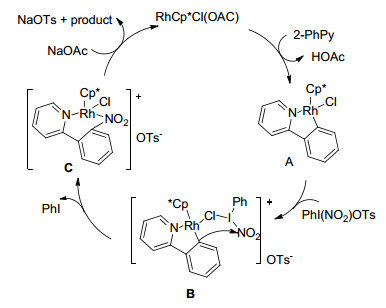
2013年, 李兴伟课题组[41]报道了以硝酸钠为硝基化试剂, PhI(OTs)OH为氧化剂铑催化吡啶导向芳烃C—H键邻位硝化反应(Eq. 27).实验结果表明:无论给电子基还是吸电子基的底物都能以较高的产率合成硝化产物.芳环上邻位, 间位及对位不同位置取代的底物均能顺利反应.此外, 嘧啶、吡唑等其他含氮杂环也能适用该催化体系.作者提出如下反应机理:首先, 预催化剂进行配体交换生成的活性催化剂RhCp*Cl(OAC)与底物2-苯基吡啶发生配位环金属化过程生成芳基铑(Ⅲ)中间体A; 然后, 物种A与原位生成的三价碘硝化试剂进行作用, 经过中间体B发生亲电硝化生成中间体C和一分子PhI; 最后, 物种C与乙酸根进行配位交换转化为硝化产物, 同时催化剂再生.
|
|
(27) |
2017年, Das等[42]开发了2-氨基吡啶作为导向基团的C—H键硝化反应(Eq. 28).该反应以AgNO2为硝化试剂, 氧气为氧化剂, THF为溶剂, 在Co(OAc)2•4H2O催化下于80 ℃反应6 h, 成功实现了导向基团的邻位C—H键硝化反应.这种不常见的催化体系具有广泛的底物范围, 优异的官能团耐受性和较高的区域选择性, 可以应用于生物活性化合物的快速合成, 为具有挑战性的邻硝基苯胺类衍生物的合成提供了一个有吸引力的方法.为了突出该方法的有效性, 作者通过嘧啶和吡啶环的裂解去除导向基团, 可以得到邻硝基苯胺和苯-1, 2-二胺类化合物.经过机理研究实验和计量, 作者认为该反应是过质子耦合电子转移的途径进行的.
|
|
(28) |
2018年, Kapur等[43]开发了一种简单有效的钴催化3-取代吲哚2位C(sp2)—H键硝化反应(Eq. 29).该反应使用经济、无毒的Co(NO3)2·6H2O作为催化剂和亚硝酸叔丁酯(TBN)作为硝基来源, 具有良好的官能团耐受性.利用可脱除的t-Boc作为导向基团, 提高了该方法的合成效用.在温和的反应条件下, 各种3-取代吲哚底物均能以中等至良好的产率选择性地生成2-硝基-3-取代吲哚衍生物, 有望成为一种合成2-氨基吲哚的独特方法.
|
|
(29) |
2017年, 郭凯等[44]以亚硝酸叔丁酯为硝基源、廉价的镍盐作为催化剂, 有效地实现了2-芳基噁唑啉酰胺底物远程C—H键硝化反应(Eq. 30).该反应底物范围广泛, 区域选择性较高, 并能够实现克级合成.目标产物可以进行多样性转化, 酰胺键碱性水解可以得到2-(4, 5-二氢噁唑-2-基)-4-硝基苯胺, 酸性条件下进一步彻底水解可以转化为2-氨基-5-硝基苯甲酸.值得注意的是, 通过简单地提高亚硝酸叔丁酯的当量, 可以较高的产率得到双硝化产品.
|
|
(30) |
2016年, Chandrasekharam等[45]采用廉价易得且无毒的硝酸铁为硝化试剂, 实现了苯胺类和芳香族磺酰胺芳烃邻位硝化反应(Eq. 31).该反应产生各种具有中等到优异的产率得到的单硝化产物, 但是伴随着少量对位硝化副产物.该反应具有较宽的底物范围和官能团兼容性, 为芳香硝化合物的合成提供了一种温和实用方法. 2018年, 高杨等[46]采用N-羟基邻苯二甲酰亚胺(NHPI)为添加剂、二氯乙烷和六氟异丙醇为溶剂在温和的条件下高选择性实现该类反应(Eq. 32). Fe(NO3)3•9H2O起到反应促进剂和硝化试剂的双重作用.该类反应是通过单电子转移的机理进行的:首先, 硝酸铁与反应底物的乙酰胺基进行导向配位形成络合物; 然后, 通过热分解产生的二氧化氮自由基对苯环进行加成; 最后, 通过去芳构化的过程生成硝化产物.
|
|
(31) |
|
|
(32) |
过渡金属催化导向基团辅助的芳烃C—H键硝化反应能够将硝基区域专一性地引入芳烃的特定位置, 可以较好地控制在单硝化阶段以及在中性的反应条件下进行, 且其化学选择性、底物和官能团的兼容性均较好.近年来, 过渡金属催化导向基团辅助的芳烃C—H键硝化反应研究, 已经取得了重要的进展.但是, 芳烃C—H键硝化反应仍存在较多的问题, 如想在芳烃的绿色化选择性硝化方面取得突破需要从以下几个方面入手: (1)该类反应多数为氮导向及双齿导向的反应, 底物比较受限, 更为广阔的导向基团急需研究; (2)目前较多采用价格较高的金属硝化物为硝化试剂和昂贵的过渡金属催化剂, 寻找到原料来源广、成本低、无污染的新型绿色硝化试剂和开发可回收、高选择性及反应活性的廉价催化剂是未来研究的方向.
(a) Ono, N. The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001.
(b) Feuer, H.; Nielson, A. T. Nitro Compounds: Recent Advances in Synthesis and Chemistry, VCH, New York, 1990.
(c) Olah, G. A.; Malhotra, R.; Narang, S. C. Nitration: Methods and Mechanisms, VCH, Weinheim, 1989.
White, E. Org. Synth. 1973, 47, 44.
For review on synthesis of nitro compounds, see: Yan, G.; Yang, M. Org. Biomol. Chem. 2013, 11, 2554.
Prakash, G. K.; Mathew, T. Angew. Chem., Int. Ed. 2010, 49, 1726. doi: 10.1002/anie.v49:10
For reviews on ortho CAr-H activation, see: (a) Yeung, C. S.; Dong, V. M. Chem. Rev. 2011, 111, 1215.
(b) Sun, C. L.; Li, B. J.; Shi, Z. J. Chem. Rev. 2011, 111, 1293.
(c) Liu, C.; Zhang, H.; Shi, W.; Lei, A. W. Chem. Rev. 2011, 111, 1780.
(d) Davies, H. L.; Bois, J. D.; Yu, J. Q. Chem. Soc. Rev. 2011, 40, 1855.
(e) Li, B. J.; Shi, Z. J. Chem. Soc. Rev. 2012, 41, 5588.
(f) Neufeldt, S. R.; Sanford, M. S. Acc. Chem. Res. 2012, 45, 936.
(g) Arockiam, P. B.; Bruneau, C.; Dixneuf, P. H. Chem. Rev. 2012, 112, 5879.
(h) Colby, D. A.; Tsai, A. S.; Bergman, R. G.; Ellman, J. A. Acc. Chem. Res. 2012, 45, 814.
(i) Engle, K. M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Acc. Chem. Res. 2012, 45, 788.
(j) Neufeldt, S. R.; Sanford, M. S. Acc. Chem. Res. 2012, 45, 936.
(k) Li, B.; Dixneuf, P. H. Chem. Soc. Rev. 2013, 42, 5744.
(l) Ackermann, L. Acc. Chem. Res. 2014, 47, 281.
(m) Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Org. Chem. Front. 2015, 2, 1107.
(n) Moselage, M.; Li, J.; Ackermann, L. ACS Catal. 2016, 6, 498.
(o) Wang, Y.; Chen, G.; Cui, X. Chin. J. Org. Chem. 2012, 32, 2018 (in Chinese).
(王勇, 程国林, 崔秀灵, 有机化学, 2012, 32, 2018.)
(p)Zhang, W.; Zhang, J.; Liu, Y. Chin. J. Org. Chem. 2014, 34, 36 (in Chinese).
(张巍, 张家慧, 刘运奎, 有机化学, 2014, 34, 36.)
(r) Zhang, B.; Guan, H.; Liu, B.; Shi, B. Chin. J. Org. Chem. 2014, 34, 1487 (in Chinese).
(张博, 管晗曦, 刘斌, 史炳锋, 有机化学, 2014, 34, 1487.)
(s) Tang, H. M.; Huo, X. H.; Meng, Q. H.; Zhang, W. B. Acta Chim. Sinica 2016, 74, 219 (in Chinese).
(汤淏溟, 霍小红, 孟庆华, 张万斌, 化学学报, 2016, 74, 219.)
Liu, Y.-K.; Lou, S.-J.; Xu, D.-Q.; Xu, Z.-Y. Chem.-Eur. J. 2010, 16, 13590. doi: 10.1002/chem.v16.46
Zhang, W.; Lou, S.; Liu, Y.; Xu, Z. J. Org. Chem. 2013, 78, 5932. doi: 10.1021/jo400594j
Zhang, W.; Wu, D.; Zhang, J.; Liu, Y. Eur. J. Org. Chem. 2014, 26, 5827.
Zhang, W.; Zhang, J.; Ren, S.; Liu, Y. J. Org. Chem. 2014, 79, 11508. doi: 10.1021/jo502145v
Zhang J.; Zhang, W.; Ren, S.; Cui, J.; Liu, Y. Chin. J. Org. Chem. 2015, 35, 647. doi: 10.6023/cjoc201410042
Dong, J.; Jin, B.; Sun, P. Org. Lett. 2014, 16, 4540. doi: 10.1021/ol502090n
Majhi, B.; Kundu, D.; Ahammed, S.; Ranu, B. C. Chem.-Eur. J. 2014, 20, 9862. doi: 10.1002/chem.201403325
Liang, Y.-F.; Li, X.; Wang, X. C.; Yan, Y.; Feng, P.; Jiao, N. ACS Catal. 2015, 5, 1956. doi: 10.1021/cs502126n
Zhang, W.; Ren, S.; Zhang, J.; Liu, Y. J. Org. Chem. 2015, 80, 5973. doi: 10.1021/acs.joc.5b00735
Qiao, H.-J.; Yang, F.; Wang, S.-W.; Leng, Y.-T.; Wu, Y.-J. Tetrahedron 2015, 71, 9258. doi: 10.1016/j.tet.2015.10.035
Lu, Y.; Li, Y.; Zhang, R.; Jin, K.; Duan, C. Tetrahedron 2013, 69, 9422. doi: 10.1016/j.tet.2013.08.076
Feng, Y. S.; Mao, L.; Bu, X. S.; Dai, J. J.; Xu, H. J. Tetrahedron 2015, 71, 3827. doi: 10.1016/j.tet.2015.04.013
Pawar, G. G.; Brahmanandan, A.; Kapur, M. Org. Lett. 2016, 18, 448. doi: 10.1021/acs.orglett.5b03493
Zhao, F.; Chen, Z.; Huang, S. J.; Jiang, Y. B. Synthesis 2016, 48, 2105. doi: 10.1055/s-00000084
Chang, M.; Chen, H.; Wang, H. J. Org. Chem. 2017, 82, 10601. doi: 10.1021/acs.joc.7b02311
Zhang, L.; Liu, Z.; Li, H.; Fang, G.; Barry, B.-D.; Belay, T. A.; Bi, X.; Liu, Q. Org. Lett. 2011, 13, 6536. doi: 10.1021/ol2028288
Zhang, H.; Zhao, L.; Wang, D. Org. Lett. 2013, 15, 3836. doi: 10.1021/ol401453f
Katayev, D.; Pfister, K. F.; Wendling, T.; Gool en, L. J. Chem.-Eur. J. 2014, 20, 9902. doi: 10.1002/chem.201403363
Liu, J.; Zhuang, S.; Gui, Q.; Chen, X.; Yang, Z.; Tan, Z. Adv. Synth. Catal. 2015, 357, 732. doi: 10.1002/adsc.v357.4
Hernando, E.; Rafael, R.; Castillo, R. R; Rodrguez, N. Chem.-Eur. J. 2014, 20, 13854. doi: 10.1002/chem.v20.43
Sadhu, P.; Alla, S. K.; Punniyamurthy, T. J. Org. Chem. 2015, 80, 8245. doi: 10.1021/acs.joc.5b01021
Ji, Y.-F.; Yan, H.; Jiang, Q.-B. Eur. J. Org. Chem. 2015, 2051.
He, Y.; Zhao, N.; Qiu, L.; Zhang, X.; Fan, X. Org. Lett. 2016, 18, 6054. doi: 10.1021/acs.orglett.6b02998
Zhu, X.; Qiao, L.; Ye, P.; Ying, B.; Xu, J.; Shen, C.; Zhang, P. RSC Adv. 2016, 6, 89979. doi: 10.1039/C6RA19583K
Planas, O.; Company, A.; Ribas, X. Adv. Synth. Catal. 2016, 358, 1679. doi: 10.1002/adsc.v358.10
Khan, B.; Khan, A. A.; Bora, D.; Verma, D.; Koley, D. ChemistrySelect 2017, 2, 260. doi: 10.1002/slct.201601917
Wang, Y.; Yu, F.; Han, X.; Li, M.; Tong, Y.; Ding, J.; Hou, H. Inorg. Chem. 2017, 56, 5953. doi: 10.1021/acs.inorgchem.7b00653
Vinayak, B.; Chandrasekharam, M. Org. Lett. 2017, 19, 3528. doi: 10.1021/acs.orglett.7b01489
Vinayak, B.; Ashok, A.; Chandrasekharam, M. Eur. J. Org. Chem. 2017, 7127.
Reddy, G. M.; Rao, N. S.; Sarma, M. H. Asian J. Org. Chem. 2017, 6, 59. doi: 10.1002/ajoc.v6.1
Tu, D.; Luo, J.; Jiang, C. Chem. Commun. 2018, 54, 2514. doi: 10.1039/C8CC00267C
Wang, C. M.; Tang, K. X.; Gao, T. H.; Chen, L.; Sun, L. P. J. Org. Chem. 2018, 83, 8315. doi: 10.1021/acs.joc.8b01016
Fan, Z.; Ni, J.; Zhang, A. J. Am. Chem. Soc. 2016, 138, 8470. doi: 10.1021/jacs.6b03402
Fan, Z.; Li, J.; Lu, H.; Wang, D.-Y.; Wang, C.; Uchiyama, M.; Zhang, A. Org. Lett. 2017, 19, 3199. doi: 10.1021/acs.orglett.7b01297
Fan, Z.; Lu H.; Zhang, A. J. Org. Chem. 2018, 83, 3245. doi: 10.1021/acs.joc.8b00149
Xie, F.; Qi, Z. S.; Li, X. W. Angew. Chem., Int. Ed. 2013, 52, 11862. doi: 10.1002/anie.v52.45
Rao, D. N.; Rasheed, S. K.; Raina, G.; Ahmed, Q. N.; Jaladanki, C. K.; Bharatam, P. V.; Das, P. J. Org. Chem. 2017, 82, 7234. doi: 10.1021/acs.joc.7b00808
Saxena, P.; Kapur, M. Chem. Asian J. 2018, 13, 861. doi: 10.1002/asia.201800036
Wan, L.; Qiao, K.; Yuan X.; Zheng, M.-W.; Fan, B.-B.; Di, Z. C.; Zhang, D.; Fang, Z.; Gu, K. Adv. Synth. Catal. 2017, 359, 2596 doi: 10.1002/adsc.v359.15
Botla, V.; Ramana, D. V.; Chiranjeevi, B.; Chandrasekharam, M. ChemistrySelect 2016, 1, 3974. doi: 10.1002/slct.201600906
Gao, Y.; Mao, Y.; Zhang, B.; Zhan, Y.; Huo, Y. Org. Biomol. Chem. 2018, 16, 3881. doi: 10.1039/C8OB00841H

图式 1 钯催化导向的芳烃硝化反应机理
Scheme 1 Proposed mechanism for the palladium-catalyzed chelation-assisted C—H nitration of arenes

图式 2 铜催化导向的苯甲酸邻位硝化反应机理
Scheme 2 Proposed mechanism for copper-catalyzed chelation-assisted ortho-nitration of benzoic acid derivatives

图式 3 8-氨基喹啉酰胺导向5-位C—H键硝化反应机理
Scheme 3 Proposed mechanism for the nitration of C-5–H of 8-aminoquinoline amides

图式 4 CuCl2催化导向的芳烃邻位C—H键硝化反应机理
Scheme 4 Plausible mechanism for the copper-catalyzed chelation assisted ortho-nitration of arenes

图式 5 Ru3(CO)12催化芳烃间位C—H键硝化反应机理
Scheme 5 Plausible mechanism for Ru3(CO)12-catalyzed meta C—H nitration of arenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





















 下载:
下载:

 下载:
下载: