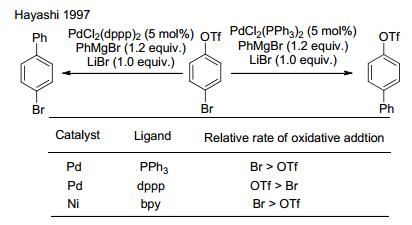
图式 1.
Ar—X/OTf选择性氧化加成
Scheme 1.
Selective oxidative addition of aryl halides and triflates

亲核试剂与亲电试剂的偶联反应被广泛应用于有机分子的合成中, 亲核试剂一般采用有机金属试剂, 如RMgX, RZnX, RB(OH)3等.这些亲核试剂大多是由卤代烃制备而来, 并且多数对空气、水敏感, 与亲电试剂相比, 能直接购买得到的亲核试剂也相对较少.因此, 改进金属试剂的制备方法或避免使用有机金属试剂将是有机合成中非常重要的研究领域.而通过亲电试剂的交叉偶联反应可以有效避免金属亲核试剂的使用[1].早期的亲电试剂偶联反应有Wurtz反应[2]、Ullman反应[3]等, 主要用于自身偶联反应.然而, 亲电试剂的交叉偶联的发展仍远远落后于亲核-亲电试剂的偶联反应, 这主要源于亲电试剂偶联缺乏选择性, 生成交叉偶联产物的同时也有相当一部分自身偶联的副产物.这与亲电-亲核偶联反应中的亲电试剂被金属氧化加成再与亲核试剂金属交换过程不同, 亲电试剂交叉偶联反应在两种亲电试剂活性相近的情况下, 都会与金属催化剂发生氧化加成, 从而导致大量自身偶联反应的副产物生成.因此, 要选择性地取得交叉偶联的产物将面临非常大的挑战[4].本文将主要介绍近些年通过亲电试剂交叉偶联形成C(sp2)—C(sp2)及C(sp2)—C(sp3)键的研究进展.
早期的亲电试剂偶联反应采用单一的金属试剂[1b, 5], 当两种芳基亲电试剂活性相当时, 偶联反应没有选择性.通过采用活性不同的亲电试剂[5], 并且改变亲电试剂的化学计量比[1b], 由此可增加交叉偶联产物并抑制较活泼亲电试剂的自身偶联.
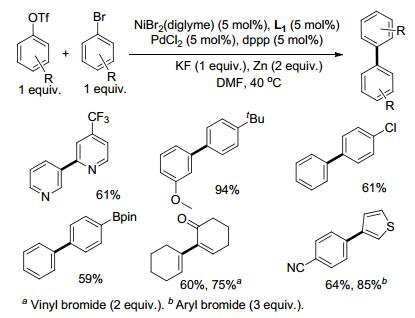
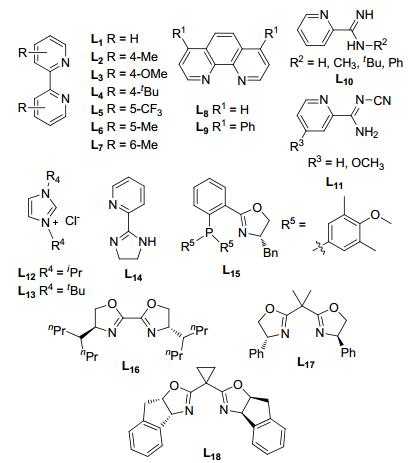
芳基上离去基团的不同, 在金属催化的反应中会有活性的差异.如芳基卤化物与芳基三氟甲磺酸酯在不同的金属催化剂条件下显现出不同的活性.钯催化下, 通过调整膦配体可选择性地对芳基卤化物或三氟甲磺酸酯氧化加成[6], 镍催化剂则更容易对芳基溴化物氧化加成(Scheme 1)[1b]. 2015年, Weix课题组[6d]采用Pd/Ni双金属催化体系, 通过Ni活化溴代芳烃1, Pd活化三氟甲磺酸酯2, 可以高选择性地得到交叉偶联产物(Scheme 2). Ni/Pd分别选择性地对Ar1—Br/Ar2—OTf氧化加成形成Ar1—NiII(bpy)—Br (3) (bpy=L1, 图 1)与Ar2—PdII—OTf (4).钯中间体4比较稳定, 并不与自身进行偶联.镍中间体3形成后将快速与4金属交换生成Ar2— PdII—Ar1 (5), 随后进行还原消除即得到交叉偶联产物Ar1—Ar2. TfO—NiII—Br (6)通过Zn粉还原重新进入催化循环(Scheme 3).由于钯中间体4相比镍中间体3更稳定, 体系中前者的含量相比后者更多, 因此3将快速地与相对高浓度的4金属交换, 3由于浓度较低, 与自身碰撞反应的机会也大大降低, 从而抑制了卤代芳烃的自身偶联.反应过程中芳环上连接的氯、硼酸酯、氰基等基团不受影响, 杂环化合物也能以该方法进行偶联.
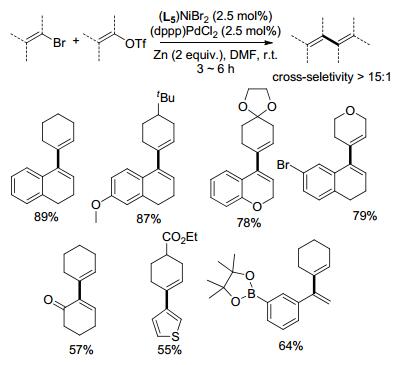
2018年, Weix课题组[7]成功将Ni/Pd双金属催化体系应用于溴代烯烃与烯基三氟甲基磺酸酯的偶联, 制备多取代的1, 3-二烯烃(Scheme 4).适当调整镍催化剂的配体, 可以改变烯烃自身偶联与交叉偶联的比例, 提高交叉偶联产物的收率.链状烯烃与环烯烃都能进行偶联, 卤代杂环化合物, 如卤代的噻吩、苯并噻吩、2H-色烯等也都能与烯基三氟甲基磺酸酯反应, 反应过程中底物连有的官能团如Boc保护的胺基、烯酮、硼酸酯、氰基以及芳基溴等不受影响.通过4-溴-1, 2-二氢萘与环己烯三氟甲基磺酸酯的偶联反应, 用I2或酸淬灭分别得到4-碘-1, 2-二氢萘与1, 2-二氢萘, 且二者的浓度均远高于体系中催化量的镍的浓度, 推测偶联反应经由Vinyl-ZnII中间体生成[8], 而不仅是Viny-NiII中间体. Vinyl-NiII中间体与Zn作用形成Vinyl-ZnII中间体之后再与钯中间体金属交换进行C—C偶联, Zn在双金属催化体系中具体的作用机制有待进一步研究.
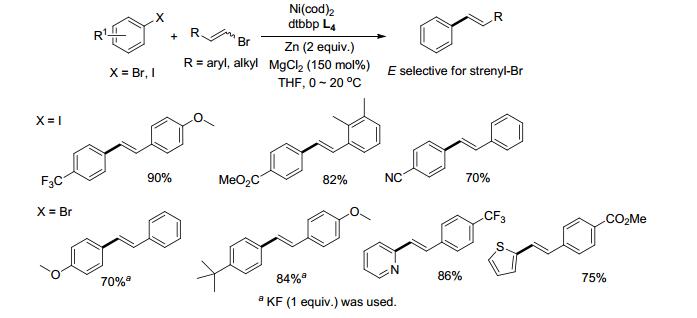
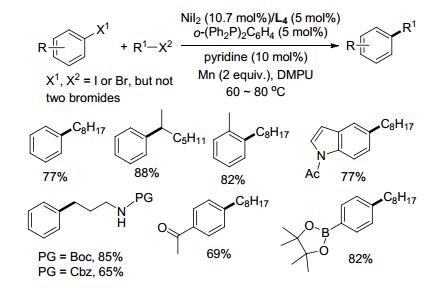
苯乙烯基结构单元在天然产物、药物分子、聚合物中广泛存在[9], 它的合成可以通过卤代芳烃与卤代烯烃还原偶联实现[10]. 2016年, Gong课题组[11]报道了在Ni(cod)2/L4催化下, 卤代芳烃与β-溴苯乙烯偶联, 反应有很好的立体选择性, (Z/E)-卤代烯烃的混合物都主要生成E型产物(Scheme 5).溴代芳烃与碘代芳烃参与偶联时表现出不同的官能团耐受性.电中性、吸电子基取代的碘代芳基与电中性或含供电子基的卤代烯烃均可以较高的收率得到偶联产物.卤代烯烃的芳环上若连有吸电子基收率相对较低, 有供电子基取代的碘代芳烃则难以进行反应.当采用Ar—Br, 添加KF, 供电子取代的溴代芳烃以及溴代杂环化合物可与β-溴苯乙烯很好地进行偶联反应.烷基取代的溴乙烯也能与溴代芳烃偶联, 反应需添加LiCl作添加剂.
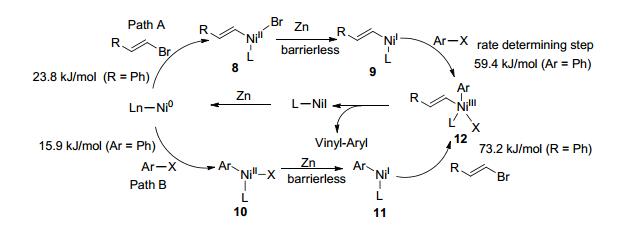
Gong课题组对该芳烃-烯烃的偶联反应提出了一个可能的机理(Scheme 6, Path A/B), 首先Ni0对卤代芳烃或卤代烯烃进行氧化加成得Ar—NiII—X (10)或Vinyl—NiII—X (8), 随即被锌粉还原成Ar—NiI (11)或Vinyl—NiI (9), 再分别对卤代烯烃或卤代芳烃氧化加成生成中间体Ar—NiIII—Vinyl (12), 最后还原消除即得到偶联产物.根据密度泛函理论(DFT)计算以path A进行反应所需能垒相对path B较低, 但由取代基效应、离去基团的不同(Br/I)、添加剂等对反应的影响方面推测反应更倾向于以path B进行.烯烃自身偶联的能垒更高(82.0 kJ/mol), 因此交叉偶联相比较而言更容易发生.对中间体Ar—NiII—X (10)的研究实验表明, 在等物质的量的碘代芳烃与溴代烯烃存在下, 中间体10更容易与后者进行偶联反应, 芳烃的自身偶联产物较难以生成.
由于Pd/Ni双金属催化剂对氧化加成的底物选择性不同, 钯倾向于氧化加成Ar—X[12], 而镍催化剂更倾向于与烷基R—X键反应[13].因此, Weix课题组[14]最初采用Pd/Ni双金属催化剂催化卤代芳烃与卤代烷烃的偶联, 然而实验发现, 采用双金属催化体系与单独采用镍得到的结果相同.镍催化下, 当同时采用4, 4'-二叔丁基联吡啶(L4)与膦配体1, 2-双二苯基膦苯时, 产率明显提高.单采用L4主要生成自身偶联产物, 交叉偶联产物较少; 而单采用配体1, 2-双二苯基膦苯时, 芳烃自身偶联降低的同时交叉偶联的收率也会降低.另外, 这种双配体的镍催化体系选择性虽好, 却有大量的卤代烷烃β-H消除的副产物, 而通过添加吡啶可以抑制β-H消除.反应过程中硼酸酯及有质子氢的酰胺(BocNH, CbzNH)不受影响, 伯/仲卤代烃都能发生反应, 其中卤素X1/X2可以为Br/I (Scheme 7).
卤代烷烃参与的偶联反应以自由基机理进行[15], 首先NiII在还原剂Mn、Zn等作用下还原生成Ni0, 随后在C(sp2)—X与C(sp3)—X之间, 选择性地对C(sp2)—X进行氧化加成, 生成中间体C(sp2)—NiII—X (13).氧化加成这一步为决速步骤, 不活泼的卤代芳烃如氯苯在这一步将难以氧化加成, 因此氯苯偶联时收率较低.通过分离纯的C(sp2)—NiII—X (13)发现, 在无还原剂情况下, 13能与碘代烷烃反应形成交叉偶联产物, 却并不与卤代芳烃偶联(Scheme 8).通过(R)-β-溴代丁酸乙酯、环丙基甲基溴与碘苯的偶联反应分别生成外消旋与重排产物(Eqs. 1, 2), 由此推断镍催化下卤代烷烃以自由基机理进行反应, 且烷基自由基的形成与后续反应分别通过不同的镍中间体进行.通过15形成烷基自由基, 再与13作用生成14, 最后还原消除得到交叉偶联产物与15, 后者继续进入催化循环.

|
|
卤代芳烃与卤代烷烃的偶联反应若同时采用溴代物, 即以溴代芳烃与溴代烷烃为底物, 由于烷基溴不如碘化物活泼, 反应速率相对较慢, 生成大量的芳烃偶联副产物.可通过添加催化量的NaI将溴化物转化成活泼的烷基碘[16], 反应以4, 4'-二甲氧基联吡啶(L3)或菲啰啉(L8)作配体.另外, 添加Zn粉作还原剂比Mn作还原剂效果要好, 这极可能与反应生成的MX2 (ZnX2, MnX2)有关, 已有相关文献报道金属卤化物在偶联反应中有着非常重要的作用[17].活泼的氯代芳烃如4-氰基氯苯在不添加NaI情况下能与溴代烷烃以很高的收率进行偶联.若加入NaI, 反应反而不能发生, 可能是因为生成的碘代烷烃中间体与氯代芳烃的活性差异太大, 导致溴代烷烃快速生成其它副产物, 而无还原偶联产物生成.卤代杂环如2-氯吡啶等也可以在类似的条件下进行偶联, 反应以4, 7-二苯基-1, 10-菲啰啉(L9)作配体[18].除联吡啶、菲啰啉系列配体外, 其它含N杂环如吡啶甲脒类配体(L10, L11)也能很好地应用于镍催化还原偶联反应[19].
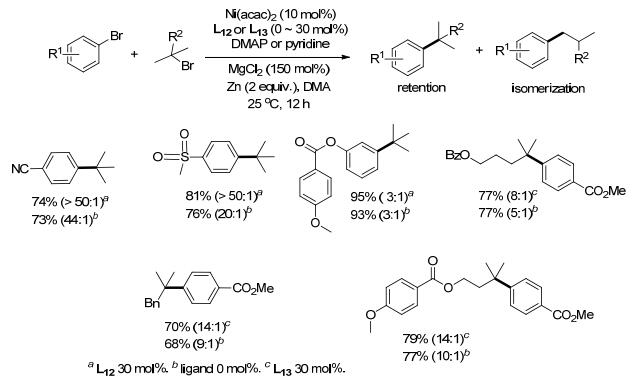
叔卤代烷烃由于空间位阻更容易发生脱卤或β-H消除副反应, 在与卤代芳烃偶联时相比伯/仲卤代烃更具有挑战性. 2015年, Gong课题组[20]报道了叔卤代烷烃与Ar—X偶联, 通过添加二甲氨基吡啶(DMAP)(或吡啶)以及无机盐MgCl2, 可取得很好的收率.添加剂在这个反应中有着极其重要的作用, 没有添加剂反应很难发生, 前者可能有着配体的作用, 后者可能加速镍的还原.配体L12, L13对偶联反应的影响相对较小, 是否添加配体收率相差无几(Scheme 9).芳环上有吸电子基如酯基、甲磺酰基、三氟甲基、氰基等取代的溴代芳烃反应时一般以DMAP作添加剂, 主要生成烷基结构保持的产物, 重排产物较少.含弱供电子基如对甲苯甲酰氧基、邻苯二甲酰亚胺基的溴代芳烃, 以DMAP作添加剂难以进行偶联反应, 若以吡啶代替DMAP收率明显提高, 但烷基结构保持的产物与重排产物比例明显降低.含强供电子基的卤代芳烃难以进行偶联反应. DMAP/吡啶对反应活性的影响及产物异构化的影响机制目前尚不明确.该反应主要用于底物α, α-二甲基取代的叔卤代烷烃的还原偶联, 而位阻更大的叔卤代烷烃则难以反应, 且主要生成重排产物.
亲电试剂的还原偶联反应通常需要使用过量的Mn、Zn等金属还原剂, 以便将NiII转化为活性的Ni0并且保证整个催化循环的进行.除此之外, 采用电化学方法[21]或者光催化方法[22]作为还原体系也能实现Ni催化的还原偶联. 2016年, MacMillan[22c]与雷爱文课题组[22b]先后报道了在光催化剂Ir作用下, 通过IrII还原NiII至Ni0, 再对卤代芳烃氧化加成. MacMillan课题组通过实验推测光催化氧化溴负离子成溴自由基, 后者立即从H供体(TMS)3SiH拔H, 形成稳定的自由基中间体(TMS)3Si.在R—X与Ar—X同时存在下, 三(三甲基硅基)硅自由基会选择性地从更弱的烷烃C—X键拔溴, 生成活泼的烷基自由基中间体与(TMS)3SiBr, 后续反应历程与Scheme 8类似.而不加(TMS)3SiH或改变Si—H试剂无产物生成或收率降低, 进一步验证了烷基自由基的形成与Si—H键有关联.电中性与含供电子基、吸电子基的溴代芳烃, 卤代的六元杂环(吡嗪、嘧啶、哒嗪), 五元杂环(N-甲基咪唑/吡唑)等都能反应, 邻位有取代基的卤代芳烃也能以很好的收率得到产物.未保护的芳香胺可直接参与反应, 氨基不受影响.伯/仲/叔卤代烷烃也都能很好地参与偶联(Scheme 10).
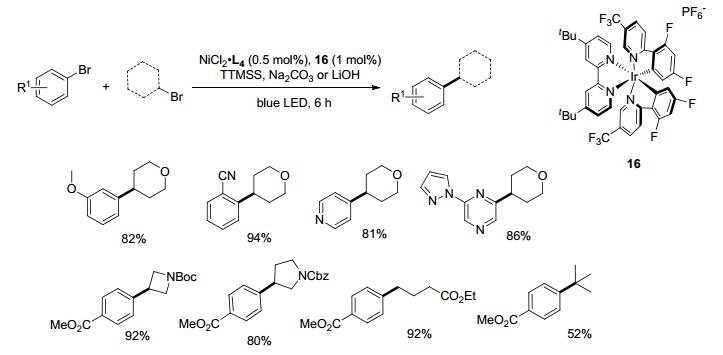
Lei课题组[22b]采用的光催化剂为Ir(ppy)2(dtbbpy)- PF6, 烷基自由基的形成可能经由Ni0与R—X反应或通过光催化条件下单电子转移机理.另外, 反应也需添加MgCl2与配体4, 4'-二叔丁基联吡啶(L4), 且二者对反应很关键, 无MgCl2产率急剧下降, 不加配体则只有痕量产物生成(Eq. 3).
|
|
(3) |
其它能生成烷基自由基的底物也能与卤代芳烃进行还原偶联.例如, 在光催化剂16与NiCl2•glyme共同作用下, Boc保护的α-氨基酸可以脱羧形成烷基自由基, 吸电子、供电子基取代的卤代芳烃都能与自由基中间体偶联(Eq. 4)[23].若采用N-羟基邻苯二甲酰亚胺(NHP)酯, 在(dtbbpy)NiBr2催化下, 无需光照与光催化剂, 也可脱羧形成烷基自由基进行偶联反应, 反应过程中, 羧基、硼酸酯等官能团不受影响(Eq. 5)[24a].草酸酯在光催化下通过脱两分子二氧化碳也能得到烷基自由基, 草酸酯可直接由醇与草酰氯反应得到, 不需进一步纯化便能直接参与后续还原偶联反应.在该条件下, 伯仲醇都能以中等的收率与卤代芳烃偶联, 供电子基取代的卤代芳烃收率较低, 有吸电子基的卤代芳烃更容易反应(Eq. 6)[24b].尽管草酸酯作烷基化底物收率相比卤代烃、NHP酯等更低, 但由于醇在自然界中广泛存在, 能直接购买得到的醇比卤代烷烃更多, 且醇羟基很容易转化成其它形式的离去基团, 因此醇及其衍生物的还原偶联反应更值得化学家们深入研究.
|
|
(4) |
|
|
(5) |
|
|
(6) |
芳甲基在药物分子中广泛存在, 在芳环上引入甲基可以改变化合物的生物活性.通过还原偶联方法可以在芳环上引入烷基, 然而采用同样的方法引入甲基却难以实现, 主要由于卤代甲烷容易快速二聚或形成MeZnI[25a]. 2017年, Gong课题组[25b]通过采用对甲苯磺酸甲酯, 成功实现了芳环的甲基化(Scheme 11).反应有很好的官能团耐受性, 硼酸酯、酰胺基、烯基等在反应过程中不受影响, 卤代杂环化合物也能很好地进行反应.二溴芳烃通过加倍催化剂、配体、对甲苯磺酸甲酯的用量, 可以成功引入两个甲基.该偶联反应还可以采用Ar—OTs作甲基化的底物, 反应条件与前者有所不同, Ni催化下以dppf或PPh3作配体, 反应需加入大量的添加剂和还原剂, 与前者相比, 产率相对较低.但由于酚类在天然产物和药物分子中广泛存在, 且Ar—OH很容易转化成Ar—OTs并进行后续反应, 因此Ar—OTs在偶联反应中的用途不容小觑.若采用其它不活泼的对甲苯磺酸烷基酯作烷基化试剂, 可以通过双金属催化体系, 如加入钴催化剂活化R—OTs, 镍活化卤代芳烃, 最终实现芳烃的烷基化[25c].
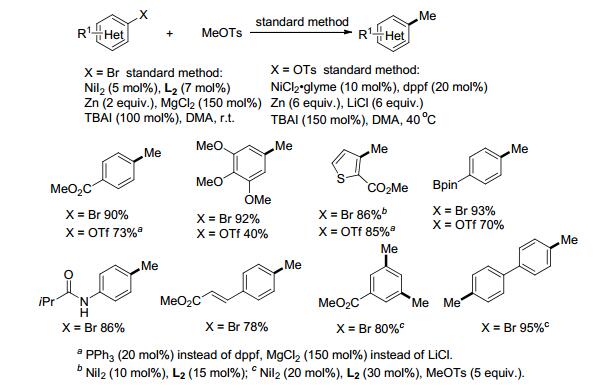
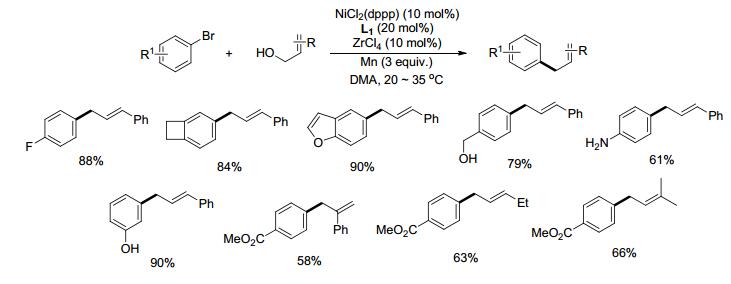
对于苄基与芳基的偶联, 若采用苄基卤化物, 由于其比卤代芳烃活泼, 很容易与镍催化剂、金属还原剂反应最终形成苄基二聚的副产物.为保证镍与卤代芳烃能进行氧化加成形成Ar—NiII—X, 可以采用有机还原剂四(二甲胺基)乙烯(TDAE)[26]或选择活性相对较低的甲磺酸酯(OMs)[27a]、特戊酸酯(OPiv)[27b]等.芳烃的烯丙基化可以通过卤代芳烃与烯丙基醋酸酯的还原偶联来实现[28].实现活性差异较大的两类亲电试剂的偶联反应一直以来备受挑战. 2018年, Shu课题组[29]发展了Ni与Lewis酸共催化的催化体系, 实现了芳基溴代物与惰性亲电试剂烯丙醇的交叉偶联反应.其中, 催化量的ZrCl4作为Lewis酸起到了活化烯丙醇的作用.反应具有很好的区域选择性和立体选择性, 最终生成直链的E型烯丙基化产物.吸电子与供电子取代的芳烃都能很好地进行还原偶联, 芳环上的亲核基团如酚羟基、醇羟基、氨基、吲哚等不受影响(Scheme 12).部分烷基取代的烯丙醇底物如2-丁烯- 1-醇和1-戊烯-3-醇等偶联反应时区域选择性和立体选择性较差, 若需高选择性地合成相应的烯丙基化产物, 可以参考后续章节介绍的烯基-烷基还原偶联方法.
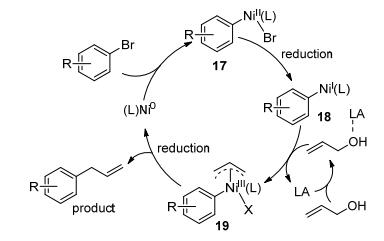
溴苯与烯丙醇均可与镍发生氧化加成, 生成Ar—NiII—Br与Ally—NiII—X, 为确定哪种镍中间体优先形成, 作者对溴苯与烯丙醇对Ni0的氧化加成能力进行了对比研究.实验结果证明:在该反应体系中, 溴苯对Ni0的氧化加成能力明显强于烯丙醇.这说明反应很可能起始于溴苯与Ni0反应生成Ar—NiII—Br中间体, 之后再与烯丙醇反应得到目标产物.为进一步验证Ar—NiII(bpy)—Br为反应可能的中间体, 作者通过Ar—Br与(bpy)Ni0(cod)反应制备得到了该化合物.通过比较Ar—NiII(bpy)—Br与前体催化剂(bpy)Ni0(cod)分别催化Ar—Br与肉桂醇的偶联反应, 发现前者产物的生成速度明显快于后者, 进一步说明Ar—NiII(bpy)—Br很可能是反应的关键中间体. 17与肉桂醇反应, 有还原剂Mn存在下有产物生成, 无还原剂则无偶联产物, 推测17还原成18才能与肉桂醇继续反应.加入自由基捕捉剂后偶联反应仍能进行, 说明烯丙醇并不以烯丙基自由基中间体进行反应, 而是Ar—NiI对Lewis酸活化后的烯丙醇氧化加成, 形成的中间体19还原消除得到烯丙基-芳基偶联产物(Scheme 13).
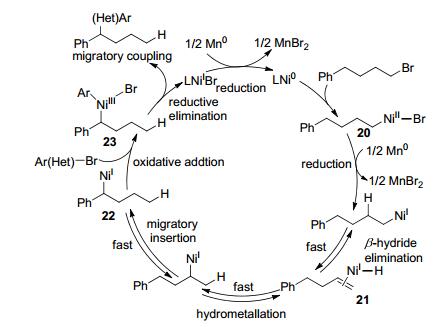
镍催化下, 卤代烷烃容易发生β-H消除, 从而得到异构化的副产物, 通过控制反应条件, 可以得到单一的重排偶联产物. 2017年, Zhu课题组[30]报道了在Ni(ClO4)2/6, 6'-二甲基联吡啶(L7)催化下, 卤代芳烃与卤代烷烃可以得到重排的偶联产物.联吡啶配体连接的取代基对反应有很大的影响, 采用联吡啶(L1)或4, 4'-二甲氧基联吡啶(L3)得到正常偶联产物, 无重排; 以6, 6'-二甲基联吡啶配体(L7)作配体则得到重排产物.若添加1-溴丙烷为H供体, 可提高收率, 1-溴丙烷自身并不与卤代芳烃反应.吸电子基和供电子基取代的卤代芳烃都能发生反应, 芳环上含醇羟基、酚羟基、氨基以及硼酸酯等官能团在反应过程中均不受影响, 伯/仲卤代烷都能得到迁移重排的偶联产物(Scheme 14).
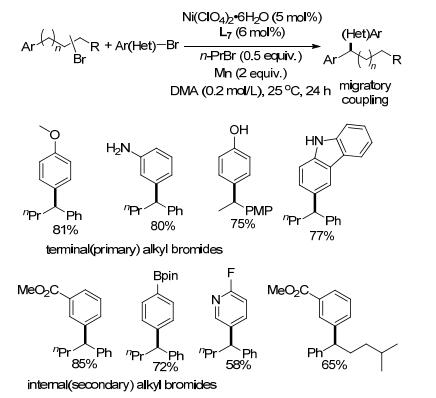
可能的反应历程为卤代烷烃与镍氧化加成形成NiII 20, 经Mn还原成NiI再经过β-H消除形成烯烃21与NiI—H, 之后NiI—H与烯烃中间体经过多次插入与β-H消除反应, 双键位置逐步迁移, 最终得到的苄基型NiI中间体22, 再与卤代芳烃偶联(Scheme 15).直接采用烯烃作底物, 在此条件下若添加1-溴丙烷, 也能与溴代芳烃偶联得到相应的重排产物, 每个催化循环开始都需通过对1-溴丙烷氧化加成以及β-H消除形成NiI—H中间体, Z/E烯烃且无论双键位置如何都能反应.通过机理实验证明, 烯烃与镍的双键迁移反应优先于与卤代芳烃的氧化加成, 在没有卤代芳烃的情况下, 烯烃的双键迁移反应也能进行.另外, 通过溴苯与碘苯参与反应得到的重排产物与其它异构体比例不同, 也可以推测烯烃迁移的过程将优先于与卤代芳烃的氧化加成.若先与卤代芳烃氧化加成, 则所得产物比例应只有微小差别.若烯烃的α位或溴的β位无H, 双键的迁移将会受阻碍, 无迁移产物生成.中间体脱β-H生成的烯烃与NiI—H中间体会经历不断的解离与重新络合的过程, 由此可解释为何反应体系中同时存在等量的结构类似的卤代烷烃与卤代烯烃时, 所得到的对应产物比例相当.
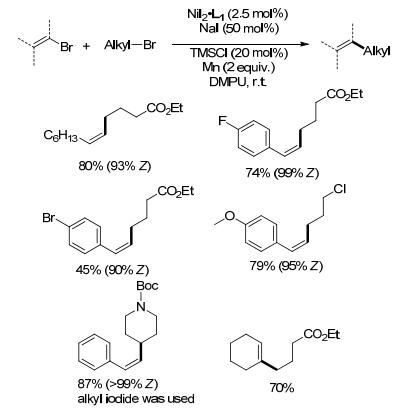
卤代烯烃在镍催化下可与卤代烷烃进行还原偶联生成烷基取代的烯烃[16a, 31], 反应以NiI2(bpy)作催化剂, 芳基或烷基取代的卤代烯烃都能与溴代烷烃进行偶联反应.当采用仲溴代烷烃时, 与NaI形成碘代烷烃中间体的速度相比伯溴代烷烃更慢, 易形成烯烃二聚的副产物, 直接采用R—I则可取得很好的收率.苯乙烯芳环上的氟、氯取代基不受影响.若为芳基溴, 则会与烯基溴竞争反应(Scheme 16).反应表现出很好的立体选择性.小部分(Z)-溴代烯烃还原偶联反应后会生成少量的E型产物, 可能是由于反应过程中底物烯基溴或烯基镍中间体发生了异构[32a].
(E)-溴代烯烃空间位阻比Z型小, 镍催化下更容易发生烯烃的二聚副反应[32a], 可以通过降低镍催化剂的用量或改变配体来实现(E)-溴代烯烃与卤代烷烃的还原偶联.例如, Gong课题组[32b]以吡啶代替联吡啶类的双齿配体, 反应表现出很好的立体专一性, Z/E型的溴代烯烃都分别得到对应的Z/E构型的产物.烯基镍中间体并不会发生构型翻转.该还原偶联条件下, 仲溴代烷烃能很好地参与反应, 伯卤代烷烃反应较差(Eq. 7).除卤代烷烃外, 羧酸[32c]、烷基硅酸盐[32d]也可以作为sp3碳源, 通过在光催化条件形成烷基自由基再与卤代烯烃偶联, 反应同样有极好的立体专一性, Z/E构型的卤代烯烃反应后构型保持.前者一般需要羧基α位存在N、O等杂原子稳定形成的烷基自由基中间体, 后者在伯碳与烯烃的偶联上可以取得很好的收率.尽管底物硅酸盐的制备比卤代烷烃繁琐, 却可以弥补卤代烷烃与卤代烯烃偶联的底物局限性(Eq. 8), 两种方法相结合可以成功实现一系列伯仲烷基与烯基的偶联.
|
|
(7) |
|
|
(8) |
酰卤与卤代烷烃在NiCl2(dme)/4, 4'-二叔丁基联吡啶(L4)催化下可进行还原偶联反应生成酮[33](Eq. 9).由于卤代甲烷容易进行二聚或脱卤等副反应, 如需制备甲基酮, 采用酰卤与卤代甲烷的还原偶联方法则难以实现.若采用Me—OTs作甲基化试剂, Ni(acac)2催化下, 以5, 5'-二甲基联吡啶(L6)作配体, 可以中等收率得到甲基酮(Eq. 10)[25a].
|
|
(9) |
|
|
(10) |
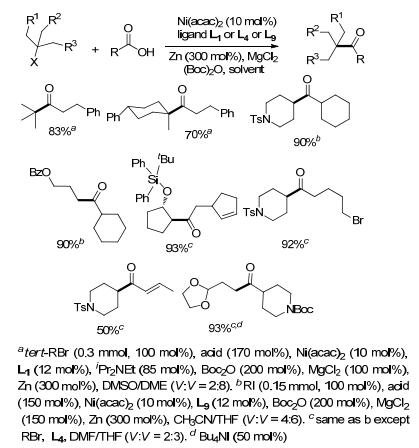
酰卤非常活泼, 很容易发生其它副反应, 因此可采用其它温和稳定的羧酸衍生物, 如酰胺[34]、2-吡啶硫酯[33]和酸酐[35]等.通过在反应体系中加入酸酐, 如羧酸在酸酐(Boc)2O存在下, 先生成酸酐中间体, 再与卤代烷烃进行还原偶联反应生成酮(Scheme 17)[36].向反应体系中加入MgCl2可以加速NiII的还原过程.在条件Scheme 17a下, 以联吡啶(L1)为配体, 脂肪族羧酸与叔卤代烷烃偶联生成酮, 芳香族羧酸如苯甲酸或苯甲酸酐则难以进行偶联反应.仲溴代烷烃在该条件下也难以进行偶联, 将配体换成L4或采用碘代仲烷烃以4, 7-二苯基-1, 10-菲罗啉(L9)作配体则可取得很好的收率.伯卤代烷在该偶联反应中不够活泼, 需加入Bu4NI作添加剂.反应历程与卤代芳烃和卤代烷烃的偶联反应类似, 首先Ni0对RC(O)—OBoc氧化加成, 形成RC(O)—NiII—OBoc中间体, 后续过程与Ar—NiII—X与卤代烷烃的偶联反应历程一致[36, 37].通过卤代芳烃与Ni氧化加成再插入羰基, 可将Ar—NiII—X转化成ArC(O)—NiII—X, 再与卤代烷烃偶联也能生成酮.
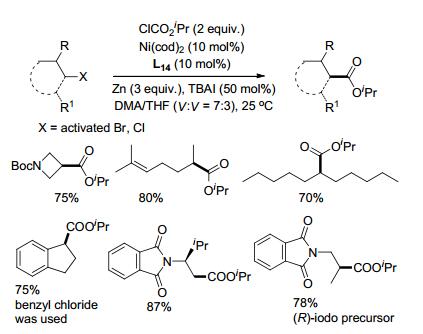
通过卤代烷烃与酰基化合物的还原偶联还可以制备羧酸酯[38].例如, 碘代烷烃与氯甲酸酯在镍催化下, 以联吡啶(L1)、菲啰啉(L8)或2-(二氢咪唑)吡啶(L14)作配体, 都能以很好的收率得到羧酸酯.伯仲碘代烷烃均能以很好的收率进行羧基化反应.在该条件下, 苄氯如1-氯茚满及其衍生物可以中等收率得到羧基化产物.邻苯二甲酰保护的α/β位卤代的胺在该条件下可以用于制备氨基酸酯.卤代烷在该还原偶联反应中同样以自由基中间体进行, 手性的卤碳原子参与反应后最终得到外消旋的产物(Scheme 18).
杂环结构广泛存在于药物分子中, 杂环化合物参与的不对称还原偶联反应制备手性化合物非常具有挑战性. 2015年, Reisman课题组[39]报道了α-氯代腈与碘代杂环芳烃还原偶联制备手性的α-杂环取代的腈(Scheme 19).通过筛选一系列的双噁唑啉(BOX)与膦噁唑啉(PhOX)配体, 发现NiCl2(dme)催化下, 在配体DMMB-PhOX (L15)作用下, 反应可以取得很好的收率与对映选择性.若用还原剂Zn代替Mn, 收率和ee值都大大降低.反应若采用α-溴代腈, 则更容易进行脱卤素和消除等副反应.一系列C(2)有取代基的碘代杂环芳烃可以进行还原偶联反应, C(2)无取代基的底物反应难以进行, 可能是无取代基的碘代杂环有更强的Lewis碱性而不利于反应.对于底物α-氯代腈, 空间位阻对其有着较大的影响, 位阻较小的底物可以取得很好的收率与对映选择性, 位阻较大的底物收率相对较低.该还原偶联对底物α-氯代腈也有很好的官能团耐受性, 烷基碳链上连有的氨基甲酸酯、酯基及氯等在反应中不受影响.
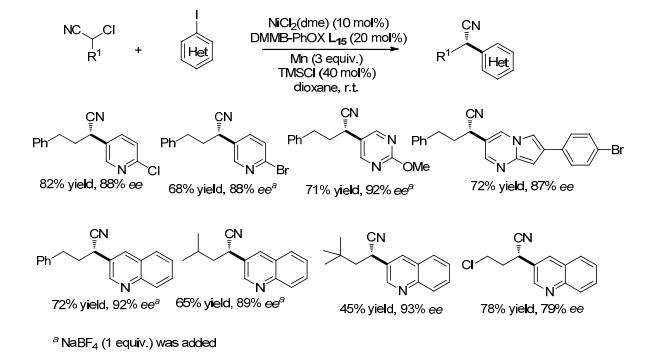
2017年, 他们课题组[40]尝试将上述不对称催化体系应用于卤代杂环与α-烷基取代苄氯的还原偶联反应, 然而并没有取得目标产物.通过筛选双噁唑啉配体(BOX), 发现增加BOX烷基取代基的碳链长度可以取得很好的收率, 对映选择性最高可达95% (Eq. 11).当采用溴代杂环代替碘代杂环, 收率和对映选择性略有降低, 而采用苄溴则主要生成苄基自身偶联的副产物. C(2)取代的碘代嘧啶、吡啶等以及碘代芳烃都能很好地进行偶联反应.当烷基碳链上有多个氯, 如α-(3-氯丙基)苄氯, 苄位的氯参与偶联反应, 端位的氯不受影响.
|
|
(11) |
烯丙位芳基取代的手性化合物非常有应用价值.它的合成一般通过烯丙基亲电试剂与芳基金属试剂的偶联来实现[41a, 41b].通过还原偶联方法也能高对映选择性地合成α-芳基取代的烯丙基化合物.例如, 溴代烯烃与α-烷基取代的苄氯, 在NiCl2(dme)催化下, 以二氢茚基取代的噁唑啉作配体, 可以取得很好的收率与优秀的对映选择性(Scheme 20)[41c].反应添加三氟乙酸(TFA)或三甲基氯硅烷(TMSCl), 此类酸性添加剂可以活化金属的表面, 收率反而降低.添加NaI反应活性增大, 可能是NaI加速了Mn与Ni之间的电子转移或者反应过程中促进烷基碘化物中间体的形成.反应过程中, 底物上连接的醇羟基、酚羟基和硼酸酯等不受影响.自由基机理下很容易进行环化反应的底物α-(4-戊烯基)苄氯在偶联反应中仅得到了链状的产物. β-烷基取代苄氯及非共轭体系的溴代烯烃都能进行还原偶联, 而二取代的溴代烯烃却难以进行反应. Z型的烯烃也难以进行反应, 且双键会发生异构化, 最终以很低的收率得到E型产物.该还原偶联体系, 添加酞菁钴(CoPc)作共催化剂, 可应用于溴代烯烃与α-三甲基硅基苄氯的不对称还原偶联制备烯丙基硅烷(Eq. 12)[41d].
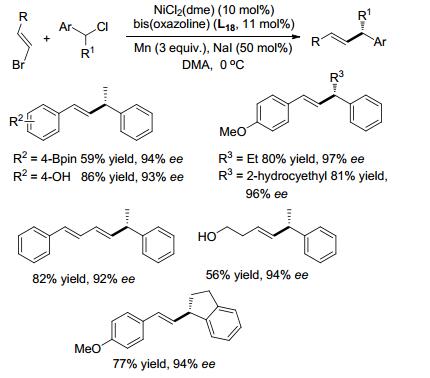
|
|
(12) |
N-羟基邻苯二甲酰亚胺(NHP)酯在偶联反应中广泛用作sp3碳源, 用NHP酯可以代替氯化物进行还原偶联反应[42], 尤其是在氯化物难以制备或不稳定的情况下.比如α-甲氧基苄氯, 该底物极不稳定, 难以进行偶联反应, 若采用NHP酯则可以不错的收率得到α-甲氧基烯丙基化合物(Eq. 13). NHP酯与氯化物分别作sp3碳源的这两种方法可以相互补充, 进一步扩大该不对称还原偶联的应用范围.
|
|
(13) |
Ni与噁唑啉不对称催化体系还可以应用于苄氯与酰卤的还原偶联(Eq. 14)[43].配体上连接噁唑啉基团非常关键, 以亚甲基或2, 6-亚吡啶基连接噁唑啉, 反应没有对映选择性, 而换成亚异丙基L17, 则可以中等收率得到α, α-二取代的酮, 对映选择性最高可达94%.改变噁唑啉环上的取代基, 收率和对映选择性也会大大降低.
|
|
(14) |
镍催化下, 亲电试剂的还原偶联反应已经得到了一定的发展, 为构筑C(sp2)—C(sp2)和C(sp2)—C(sp3)键提供了非常重要的手段.镍催化下, 通过调整配体、添加剂, 或引入双金属催化, 可对化学性质相近的C(sp2)—X及C(sp2)—OTf选择性地氧化加成, 伯仲叔卤代烷烃都能作sp3碳源参与还原偶联.通过引入噁唑啉类配体, 还能催化C(sp2)—X与C(sp3)—X高对映选择性地构建C(sp2)—C(sp3)键.尽管亲电试剂的还原偶联已经取得了长足的发展, 但底物的普适性仍需进一步深入研究, 如叔卤代烷在还原偶联反应中的应用.反应的机理及各种添加剂的作用也有待进一步明确.在未来, 还可以进一步探索弱的亲电试剂如醇和酯等在还原偶联反应中的应用价值.设计新型的还原体系代替金属还原剂也是一个有待发展的重要领域.
(a) Moragas, T.; Correa, A.; Martin, R. Chem.-Eur. J. 2014, 20, 8242.
(b) Knappke, C. E. I.; Grupe, S.; Gä rtner, D.; Corpet, M.; Gosmini, C.; Jacobi von Wangelin, A. Chem.-Eur. J. 2014, 20, 6828.
(c) Everson, D. A.; Weix, D. J. J. Org. Chem. 2014, 79, 4793.
Wurtz, A. Ann. Chem. Pharm. 1855, 96, 364. doi: 10.1002/(ISSN)1099-0690
Ullmann, F.; Bielecki, J. Chem. Ber. 1901, 34, 2174. doi: 10.1002/(ISSN)1099-0682
(a) Hanna, L. E.; Jarvo, E. R. Angew. Chem., Int. Ed. 2015, 54, 15618.
(b) Gu, J.; Wang, X.; Xue, W.; Gong, H. Org. Chem. Front. 2015, 2, 1411.
(c) Weix, D. J. Acc. Chem. Res. 2015, 48, 1767.
Amatore, M.; Gosmini, C. Angew. Chem., Int. Ed. 2008, 47, 2089. doi: 10.1002/(ISSN)1521-3773
(a) Takashi, K.; Hayashi, T. Tetrahedron Lett. 1997, 38, 7087.
(b) Littke, A. F.; Dai, C.; Fu, G. C. J. Am. Chem. Soc. 2000, 122, 4020.
(c) Schoenebeck, F.; Houk, K. N. J. Am. Chem. Soc. 2010, 132, 2496.
(d) Ackerman, L. K. G.; Lovell, M. M.; Weix, D. J. Nature 2015, 524, 454.
Olivares, A. M.; Weix, D. J. J. Am. Chem. Soc. 2018, 140, 2446.
(a) Fillon, H.; Gosmini, C.; Périchon, J. J. Am. Chem. Soc. 2003, 125, 3867.
(b) Sibille, S.; Ratovelomanana, V.; Périchon, J. J. Chem. Soc., Chem. Commun. 1992, 283.
(a) Denmark, S. E.; Butler, C. R. Chem. Commun. 2009, 20.
(b) Riviere, C.; Pawlus, A. D.; Merillon, J.-M. Nat. Prod. Rep. 2012, 29, 1317.
(c) Zhang, Z.; Qin, Y. Macromolecules 2016, 49, 3318.
(a) Moncomble, A.; Le Floch, P.; Lledos, A.; Gosmini, C. J. Org. Chem. 2012, 77, 5056.
(b) Amatore, M.; Gosmini, C.; Périchon, J. Eur. J. Org. Chem. 2005, 2005, 989.
(c) Gomes, P.; Gosmini, C.; Périchon, J. Tetrahedron 2003, 59, 2999.
Liu, J.; Ren, Q.; Zhang, X.; Gong, H. Angew. Chem., Int. Ed. 2016, 55, 15544. doi: 10.1002/anie.201607959
(a) Ariafard, A.; Lin, Z. Organometallics 2006, 25, 4030.
(b) Cristian, M.; Maria, B.; Feliu, M.; Gregorio, A.; Mercedes, M. S. Chem.-Eur. J. 2010, 16, 13390.
(c) Frisch, A. C.; Beller, M. Angew. Chem., Int. Ed. 2005, 44, 674.
(a) Jones, G. D.; McFarland, C.; Anderson, T. J.; Vicic, D. A. Chem. Commun. 2005, 4211.
(b) González-Bobes, F.; Fu, G. C. J. Am. Chem. Soc. 2006, 128, 5360.
Everson, D. A.; Shrestha, R.; Weix, D. J. J. Am. Chem. Soc. 2010, 132, 920. doi: 10.1021/ja9093956
Biswas, S.; Weix, D. J. J. Am. Chem. Soc. 2013, 135, 16192. doi: 10.1021/ja407589e
(a) Everson, D. A.; Jones, B. A.; Weix, D. J. J. Am. Chem. Soc. 2012, 134, 6146.
(b) Prinsell, M. R.; Everson, D. A.; Weix, D. J. Chem. Commun. 2010, 46, 5743.
(c) Takahashi, H.; Inagaki, S.; Nishihara, Y.; Shibata, T.; Takagi, K. Org. Lett. 2006, 8, 3037.
McCann, L. C.; Organ, M. G. Angew. Chem., Int. Ed. 2014, 53, 4386. doi: 10.1002/anie.201400459
Everson, D. A.; Buonomo, J. A.; Weix, D. J. Synlett 2014, 25, 233.
Hansen, E. C.; Pedro, D. J.; Wotal, A. C.; Gower, N. J.; Nelson, J. D.; Caron, S.; Weix, D. J. Nat. Chem. 2016, 8, 1126. doi: 10.1038/nchem.2587
Wang, X.; Wang, S.; Xue, W.; Gong, H. J. Am. Chem. Soc. 2015, 137, 11562. doi: 10.1021/jacs.5b06255
(a) Rollin, Y.; Troupel, M.; Tuck, D. G.; Perichon, J. J. Organomet. Chem. 1986, 303, 131.
(b) Meyer, G.; Rollin, Y.; Perichon, J. J. Organomet. Chem. 1987, 333, 263.
(c) Gosmini, C.; Lasry, S.; Nedelec, J.-Y.; Perichon, J. Tetrahedron 1998, 54, 1289.
(d) Sengmany, S.; Vitu-Thiebaud, A.; Le Gall, E.; Condon, S.; Leonel, E.; Thobie-Gautier, C.; Pipelier, M.; Lebreton, J.; Dubreuil, D. J. Org. Chem. 2013, 78, 370.
(e) Perkins, R. J.; Pedro, D. J.; Hansen, E. C. Org. Lett. 2017, 19, 3755.
(a) Pratsch, G.; Overman, L. E. J. Org. Chem. 2015, 80, 11388.
(b) Duan, Z.; Li, W.; Lei, A. Org. Lett. 2016, 18, 4012.
(c) Zhang, P.; Le, C. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 8084.
(a) Zuo, Z.; Ahneman, D. T.; Chu, L.; Terrett, J. A.; Doyle, A. G.; MacMillan, D. W. C. Science 2014, 345, 437.
(b) Zuo, Z.; Cong, H.; Li, W.; Choi, J.; Fu, G. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 1832.
(a) Huihui, K. M. M.; Caputo, J. A.; Melchor, Z.; Olivares, A. M.; Spiewak, A. M.; Johnson, K. A.; DiBenedetto, T. A.; Kim, S.; Ackerman, L. K. G.; Weix, D. J. J. Am. Chem. Soc. 2016, 138, 5016.
(b) Zhang, X.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 13862.
(a) Liang, Z.; Xue, W.; . Lin, K.; Gong, H. Org. Lett. 2014, 16, 5620.
(b) Wang, J.; Zhao, J.; Gong, H. Chem. Commun. 2017, 53, 10180.
(c) Komeyama, K.; Ohata, R.; Kiguchi, S.; Osaka, I. Chem. Commun. 2017, 53, 6401.
Anka-Lufford, L. L.; Huihui, K. M. M.; Gower, N. J.; Ackerman, L. K. G.; Weix, D. J. Chem.-Eur. J. 2016, 22, 11564. doi: 10.1002/chem.201602668
(a) Ackerman, L. K. G.; Anka-Lufford, L. L.; Naodovic, M.; Weix, D. J. Chem. Sci. 2015, 6, 1115.
(b) Konev, M. O.; Hanna, L. E.; Jarvo, E. R. Angew. Chem., Int. Ed. 2016, 55, 6730.
(a) Anka-Lufford, L. L.; Prinsell, M. R.; Weix, D. J. J. Org. Chem. 2012, 77, 9989.
(b) Wang, S.; Qian, Q.; Gong, H. Org. Lett. 2012, 14, 3352.
(c) Gomes, P.; Gosmini, C.; Périchon, J. Org. Lett. 2003, 5, 1043.
Jia, X.-G.; Guo, P.; Duan, J.; Shu, X.-Z. Chem. Sci. 2018, 9, 640. doi: 10.1039/C7SC03140H
Chen, F.; Chen, K.; Zhang, Y.; He, Y.; Wang, Y.-M.; Zhu, S. J. Am. Chem. Soc. 2017, 139, 13929. doi: 10.1021/jacs.7b08064
(a) Cannes, C.; Condon, S.; Durandetti, M.; Périchon, J.; Nédélec, J.-Y. J. Org. Chem. 2000, 65, 4575.
(b) Qiu, C.; Yao, K.; Zhang, X.; Gong, H. Org. Biomol. Chem. 2016, 14, 11332.
(a) Johnson, K. A.; Biswas, S.; Weix, D. J. Chem.-Eur. J. 2016, 22, 7399.
(b) Gu, J.; Qiu, C.; Lu, W.; Qian, Q.; Lin, K.; Gong, H. Synthesis 2017, 49, 1867.
(c) Noble, A.; McCarver, S. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2015, 137, 624.
(d) Patel, N. R.; Kelly, C. B.; Jouffroy, M.; Molander, G. A. Org. Lett. 2016, 18, 764.
Wotal, A. C.; Weix, D. J. Org. Lett. 2012, 14, 1476. doi: 10.1021/ol300217x
Ni, S.; Zhang, W.; Mei, H.; Han, J.; Pan, Y. Org. Lett. 2017, 19, 2536. doi: 10.1021/acs.orglett.7b00831
Yin, H.; Zhao, C.; You, H.; Lin, K.; Gong, H. Chem. Commun. 2012, 48, 7034. doi: 10.1039/c2cc33232a
Zhao, C.; Jia, X.; Wang, X.; Gong, H. J. Am. Chem. Soc. 2014, 136, 17645. doi: 10.1021/ja510653n
Wotal, A. C.; Ribson, R. D.; Weix, D. J. Organometallics 2014, 33, 5874. doi: 10.1021/om5004682
Zheng, M.; Xue W.; Xue, T.; Gong, H. Org. Lett. 2016, 18, 6152. doi: 10.1021/acs.orglett.6b03158
Kadunce, N. T.; Reisman, S. E. J. Am. Chem. Soc. 2015, 137, 10480. doi: 10.1021/jacs.5b06466
Poremba, K. E.; Kadunce, N. T.; Suzuki, N.; Cherney, A. H.; Reisman, S. E. J. Am. Chem. Soc. 2017, 139, 5684. doi: 10.1021/jacs.7b01705
(a) Evans, P. A.; Uraguchi, D. J. Am. Chem. Soc. 2003, 125, 7158.
(b) Alexakis, A.; El Hajjaji, S.; Polet, D.; Rathgeb, X. Org. Lett. 2007, 9, 3393.
(c) Cherney, A. H.; Reisman, S. E. J. Am. Chem. Soc. 2014, 136, 14365.
(d) Hofstra, J. L.; Cherney, A. H.; Ordner, C. M.; Reisman, S. E. J. Am. Chem. Soc. 2018, 140, 139.
Suzuki, N.; Hofstra, J. L.; Poremba, K. E.; Reisman, S. E. Org. Lett. 2017, 19, 2150. doi: 10.1021/acs.orglett.7b00793
Cherney, A. H.; Kadunce, N. T.; Reisman, S. E. J. Am. Chem. Soc. 2013, 135, 7442. doi: 10.1021/ja402922w

图式 1 Ar—X/OTf选择性氧化加成
Scheme 1 Selective oxidative addition of aryl halides and triflates

图式 4 卤代烯烃与烯基三氟甲基磺酸酯的偶联
Scheme 4 Cross-electrophile coupling of vinyl bromides with vinyl triflates

图式 8 卤代芳烃与卤代烷烃偶联反应机理
Scheme 8 Proposed mechanism for the cross-coupling of aryl halides with alkyl halides

图式 9 卤代芳烃与叔卤代烷烃的偶联
Scheme 9 Cross coupling of aryl halides with tertiary alkyl halides

图式 10 光催化卤代芳烃与卤代烷烃的偶联
Scheme 10 Photoredox-enabled cross coupling of aryl halides with alkyl halides

图式 11 Ar—X/OTs与Me—OTs偶联
Scheme 11 Cross coupling of aryl halides/tosylates with methyl tosylate

图式 13 卤代芳烃与烯丙醇偶联反应机理
Scheme 13 Proposed mechanism for the cross-coupling of aryl halides with allylic alcohols

图式 14 卤代烷烃与卤代芳烃的重排偶联
Scheme 14 Remote migratory cross coupling of aryl bromides with alkyl bromides

图式 19 α-氯代腈与碘代杂环的还原偶联
Scheme 19 Representative scope of cross coupling of α-chloronitriles with heteroaryl iodides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:














 下载:
下载:












 下载:
下载: