
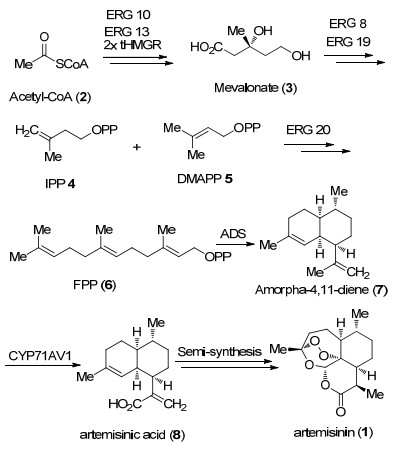
图式 1.
青蒿酸的生物发酵合成途径
Scheme 1.
Biological fermentation pathway for the synthesis of artemisinic acid

天然产物是药物先导化合物的重要来源, 据统计在1981~2014年间上市的小分子药物中, 超过一半的小分子药物直接或间接来源于天然产物及其类似物[1], 因此具有重要生物活性的天然产物的全合成及功能研究具有重要意义.天然产物全合成主要包括化学合成与生物合成.化学合成的历史可以追溯到1828年Wöhler实现尿素的合成[2], 随后的一个多世纪时间里, 天然产物全合成取得了巨大的进展, 化学家们不断挑战结构复杂的天然产物, 完成了包括维生素B12、海葵毒素等一系列具有里程碑意义的全合成工作[3]. 1992年Kishi课题组[4]首次完成了软海绵素(Halichondrin B)的全合成, 随后的药物化学研究中, 通过对合成中间体进行结构改造成功研发出用于治疗乳腺癌的药物艾瑞布林.近年来, 随着分子生物学技术的发展, 天然产物生物合成也取得了长足的进展.例如, 在抗癌药物紫杉醇[5]的合成中, Stephanopoulos等[6]报道了通过多元模块化方法利用大肠杆菌发酵合成紫杉二烯, 该化合物也是紫杉醇合成的重要中间体.阿片类药物是西药中治疗疼痛的主要药物[7], 其主要来源于罂粟. 2015年Smolke等[8]利用酵母来生产阿片类药物, 联合酶发现、酶工程、生物途径和菌株优化等在酵母中实现了阿片类药物的合成, 该方法有可能成为阿片类药物的另一种来源.本文将选取一些重要的天然产物, 从天然产物化学合成与生物合成, 以及两者结合的角度, 介绍此策略的重要性.
青蒿素是二十世纪六七十年代经中国科学家分离并用于治疗疟疾的有效成分, 其对脑型疟疾和抗氯喹疟疾具有速效和低毒的特点, 以青蒿素为先导化合物衍生得到的蒿甲醚和青蒿琥酯等青蒿素类药物是治疗疟疾唯一有效的药物, 并成为世界卫生组织推荐的药品[9].青蒿素的抗疟机理与其它类型抗疟药不同, 它主要通过干扰疟原虫的表膜2线粒体功能, 导致虫体结构瓦解[10, 11].青蒿素主要来源于植物黄花蒿的叶和花蕾, 目前市售青蒿素主要来自于植物黄花蒿的提取, 而黄花蒿从育种种植到企业收购及提取销售需3年左右时间, 这也导致青蒿素市场价格波动较大, 因此青蒿素的化学合成及工业生产也引起了化学家们的关注[12].
|
|
青蒿素是一种倍半萜内酯化合物, 结构上最特殊的是具有过氧桥环, 这也使其合成具有很大的挑战[13].经过30多年的研究, 有关青蒿素人工合成的相关文献报道较多[14], 我国合成化学家在此领域也做出了突出的贡献[15], 相关工作已经被多次总结[16].张万斌课题组[17]最近也系统回顾了青蒿素的化学合成及可工业合成路线等.此部分主要从化学合成与生物合成结合的角度, 简单介绍青蒿素合成的最新进展.
自20世纪80年代开始, 植物学家就对植物体内青蒿素(1)的生物合成途径进行了研究, 研究发现青蒿酸(2)是黄花蒿植物体内形成青蒿素(1)的重要中间体[18]. 2006年Keasling和Sarpong等[19]联合报道了使用酿酒酵母发酵生产青蒿酸的方法, 该方法经优化后产量可达到25 g/L.作者通过三个步骤实现发酵法合成青蒿酸.首先, 作者改变法尼基焦磷酸(6, FPP)的生物合成途径以增加FPP产量并降低其用于固醇类化合物的合成.其次, 将来自A. annuain的紫穗槐二烯合成酶的基因(ADS)引入酿酒酵母中, 并顺利将FPP转化为紫穗槐- 4, 11-二烯(7).最后通过克隆得到一种新型的细胞色素P450单加氧酶(CYP71AV1), 其可以使青蒿二烯7发生三步氧化转化成青蒿酸8.在完成青蒿酸的合成后, 接下来通过半合成就可以完成青蒿素(1)的全合成[20] (Scheme 1).
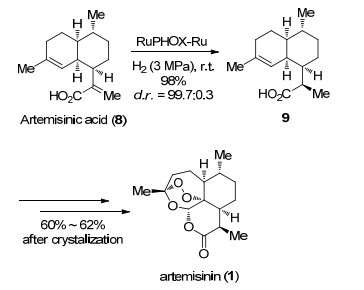
张万斌课题组[21]经过多年努力, 成功发展了一种无光照高效合成青蒿素(1)的方法(Scheme 2).作者以发酵制备的青蒿酸8为原料, 将他们课题组发展的面手性催化剂RuPHOX-Ru用于青蒿酸8的不对称氢化, 能够高收率高选择性地完成二氢青蒿酸9的合成.随后以过氧化氢为氧化剂利用他们课题组发展的过渡金属催化剂顺利完成过氧桥环及内酯化, 经重结晶后以60%的收率完成青蒿素(1)的合成.该反应目前已完成300 L规模的中试研究, 成果已完成技术转让, 相关企业正在进行产业化研究.
随着青蒿酸生物发酵技术的发展, 由青蒿酸制备青蒿素的半合成工艺将会趋于完善, 而由生物发酵与化学合成相结合的方式规模化制备青蒿素将有可能取代黄花蒿提取, 成为青蒿素类抗疟药物的主要原料来源.
Spinosyn A (10)是一种重要的有机和天然杀虫剂, 其作为多杀菌素的主要组成部分, 在世界农业中广泛使用. Spinosyns A (10)和D (11)是从刺糖多孢菌的发酵液中分离得到, 二者比例为17:3.生物学家证实Spinosyn A (10)具有新颖的作用模式, 它们主要靶向昆虫神经系统的烟碱乙酰胆碱受体, 同时也可以作用于γ-氨基丁酸受体, 其整体效果是使昆虫过度兴奋, 最终导致死亡.此外, Spinosyn A (10)除了高效和广谱的杀虫作用之外, 它具有非常低的哺乳动物毒性以及良好的环境兼容性[22].
|
|
结构上, Spinosyn A以独特的5, 6, 5, 12-四并环结构作为基本骨架, 并通过氧糖苷键分别与两个糖基片段D-脱氧氨基葡萄糖和2, 3, 4-三甲氧基-L-鼠李糖连接, 其中D环为大环内酯环, 整个分子中含有9个手性中心. Spinosyn A (10)的合成具有很大的挑战性, 主要难点是在于四环骨架的高效立体选择性构建, 此外选择性地引入D-脱氧氨基葡萄糖和2, 3, 4-三甲氧基-L-鼠李糖也是其合成难点.
1993年, Evans课题组[23]报道了(+)-Spinosyn A的首次全合成工作.作者以噁唑烷酮诱导的不对称aldol反应[24]及大环内酯化为关键反应首先构建D环[25], 随后利用Stille偶联[26]和分子内Diels-Alder反应构建A, B环[27], 最后利用分子内aldol反应构建C环完成四环骨架的构建, 接下来分别通过酸催化和银盐促进的糖苷化引入糖基片段完成(+)-Spinosyn A的首次全合成.
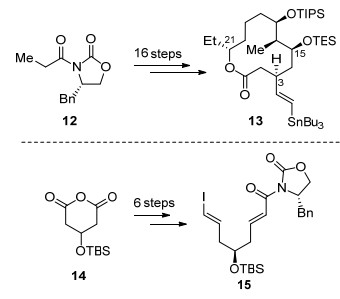
作者首先利用辅基诱导的不对称aldol反应、Mukaiyama aldol反应及Yamaguchi大环内酯化作为关键反应, 从化合物12出发经16步转化首先完成D环的构筑得到化合物13, 该化合物中已经完成4个手性中心的构建.在完成D环的构建后, 作者随后将注意力转向烯基侧链的合成, 从化合物14出发利用去对称化反应、Swern氧化、Takai烯基化及Wittig反应为关键反应经6步转化得到烯基碘化合物15, 完成烯基侧链的合成(Scheme 3).
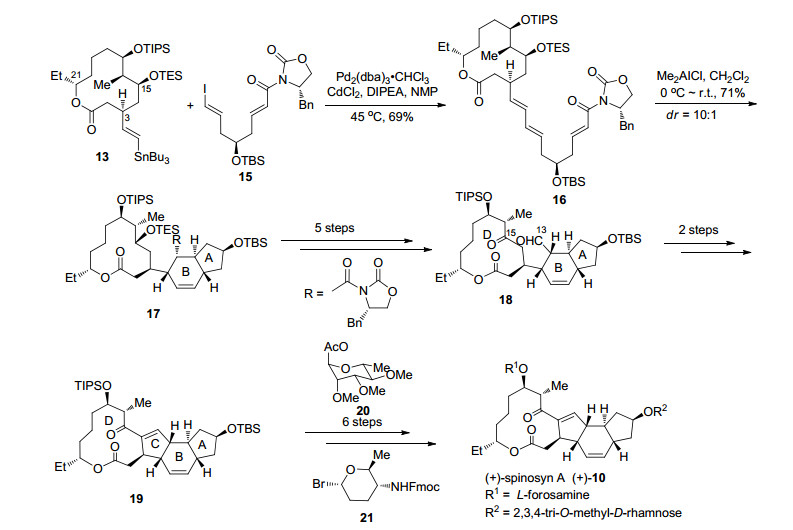
在完成D环和烯基侧链的合成后, 作者利用Stille偶联将两片段连接得到化合物16, 随后作者希望利用分子内Diels-Alder反应顺利构建A, B环, 在二甲基氯化铝的作用下化合物16可以顺利发生Diels-Alder反应得到化合物17, 其dr值为10:1, 在该反应中, 作者提前引入手性辅基可以很好地控制Diels-Alder反应的endo/exo选择性.化合物17经6步转化调整C(15)位氧化态并对C(13)位进行官能团转化得到化合物18, 在碱性条件发生分子内aldol反应、脱水即可完成四环骨架的构建.对于两个糖片段的合成, 作者从D-(+)-甘露糖出发经6步简单转化即可完成D-鼠李糖21的合成, 而氨基葡萄糖20由已知化合物经8步转化得到.接下来化合物19分步脱除C(9)位及C(17)位保护基后, 分别在银盐和高氯酸盐作用下与D-鼠李糖21及氨基葡萄糖20构建氧糖苷键, 经6步转化完成了(+)-Spinosyn A的首次全合成(Scheme 4).
Evans课题组对(+)-Spinosyn A [(+)-10]的全合成确认了天然产物的绝对构型, 该合成中使用辅基参与的不对称aldol反应及Diels-Alder反应很好地控制了反应的立体选择性, 为后续的研究提供了重要参考.
1998年Paquette课题组[28]利用负离子参与的氧杂Cope重排为关键反应首先构建A, B, C三环, 经多步转化引入侧链后, 通过大环内酯化构建D环完成基本骨架的构建, 最后通过氧糖苷键连接两个糖片段完成Spinosyn A (10)的全合成(Scheme 5).
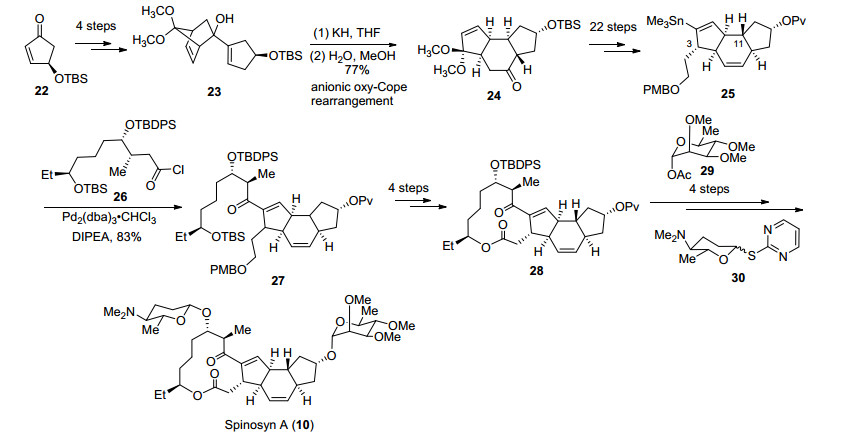
对于四环骨架的构建, 作者采取先构建A, B, C三环再构建D环的顺序.化合物22经四步转化得到重排前体化合物23, 在氢化钾作用下发生负离子促进的氧杂Cope重排, 利用甲醇捕获相应的中间体可以立体选择性地构建ABC三环.针对D环的合成, 作者希望在C环引入侧链后进行大环内酯化来构建.从化合物24出发以HWE反应为关键步骤经过22步转化在C(3)位引入侧链, 同时对BC环的氧化态和C(11)位立体化学进行调整得到化合物25.接下来通过Stille偶联引入C(4)位侧链, 利用Yamaguchi大环内酯化完成D环的构建.最后化合物28经过4步转化与L-鼠李糖29和氨基葡萄糖30作用顺利构建C(9)及C(17)位氧糖苷键完成Spinosyn A (10)的全合成[29].
Paquette课题组采用汇聚式路线完成Spinosyn A (10)的全合成, 但由于C(3)侧链的引入、氧化态及立体化学的调整导致整体的合成效率不高.
2004年Roush课题组[30]采取先构建A, B环再构建C, D环的构环顺序, 利用跨环Diels-Alder反应和MBH反应作为关键策略完成了Spinosyn A (10)的全合成.
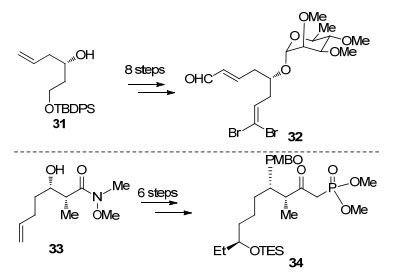
作者希望通过两次HWE反应制备Diels-Alder反应前体, 从手性化合物31出发, 经过8步转化引入L-鼠李糖片段并延长碳链得到醛32, 而磷酸酯片段34可以从化合物33出发经过6步转化引入手性乙基及磷酸酯得到(Scheme 6).
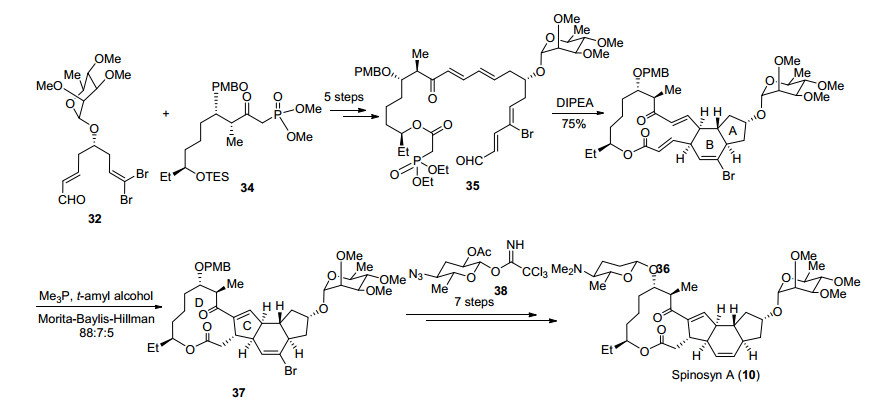
在完成片段的合成后, 作者希望将两片段连接, 醛32与磷酸酯34经HWE反应、Suzuki偶联及氧化态的调整得到化合物35, 在二异丙基乙基胺的作用下发生分子内Horner-Wadsworth-Emmons (HWE)反应构建大环内酯, 同时发生分子内跨环Diels-Alder反应完成AB环的构建, 该反应可以一步构建三环体系, 作者认为提前引入C(9)位L-鼠里糖可以很好地控制Diels-Alder反应的立体选择性[31].化合物36在三甲基膦活化下顺利发生MBH反应可以完成C, D环的构建[32], 脱除C(17)位对甲氧基苄基保护, 在Lewis酸活化下与38作用构建C(17)位氧糖苷键, 最后利用Barton去氧化及氮甲基化和脱溴反应完成Spinosyn A (10)的全合成(Scheme 7).
Roush课题组通过HWE反应和跨环Diels-Alder反应及MBH反应高效构建四环骨架, 最终以31步3.1%的总收率完成Spinosyn A (10)的全合成, 作者通过提前引入L-鼠里糖可以很好地控制Diels-Alder反应的立体选择性, 相比于前人的工作已经取得了较大进步.
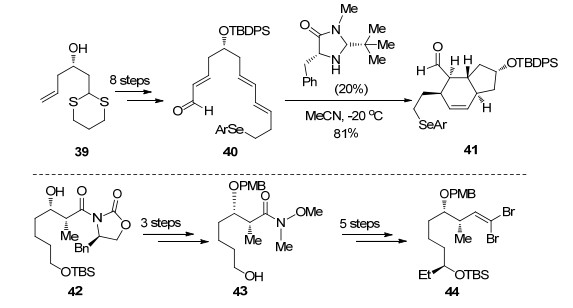
2016年Dai课题组[33]报道了最新的Spinosyn A (10)的合成方案, 作者以手性胺催化的不对称Diels-Alder反应及钯催化分子内Heck串联插羰环化作为关键策略完成四环骨架的构建, 最后利用俞飚课题组发展的金催化糖苷化反应完成Spinosyn A (10)的全合成.
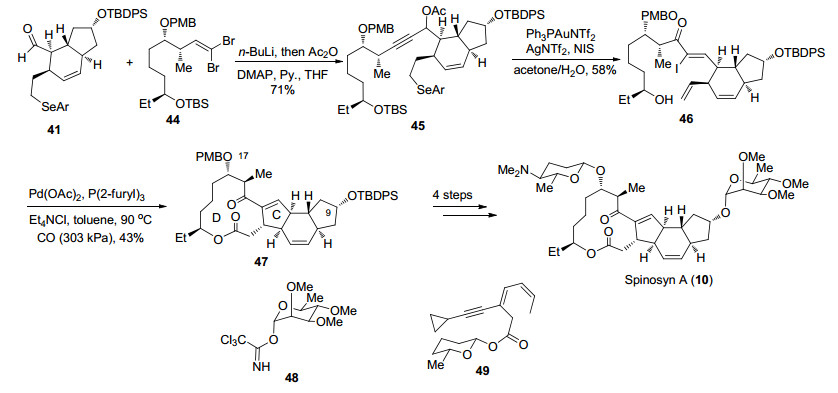
作者从化合物39出发, 以烯烃复分解反应、Takai烯基化及Stille偶联为关键反应经8步转化得到Diels- Alder反应前体40, 随后在MacMillan催化剂作用下发生分子内Diels-Alder反应完成AB环的构建[34], 该反应可以很好地控制其立体选择性得到单一产物41.另一片段则从化合物42出发经过3步转化脱除辅基并调整保护基得到43, 随后经过5步转化引入乙基, 将Weinreb酰胺还原得到醛后发生二溴烯基化完成片段44的合成(Scheme 8).
化合物41和44在正丁基锂作用下发生Corey-Fuchs反应得到炔化合物45, 随后利用金催化炔丙醇乙酸酯重排得到烯基碘化合物46.接下来作者希望利用钯催化的Heck串联插羰环化反应一步构建C, D环.钯催化的插羰反应被广泛应用于复杂天然产物全合成中[35], 然而Heck串联插羰环化反应用于构建大环内酯尚未见报道, 作者在他们小组工作的基础上, 通过条件优化发现, 在醋酸钯催化下化合物46可以顺利完成5-exo-trig类型环化, 同时发生插羰被分子内的羟基捕获后完成四环骨架的高效构筑.脱除C(9)和C(17)位保护基后, 在Lewis酸和金催化下分别与48和49作用构建氧糖苷键[36]实现Spinosyn A (10)的全合成(Scheme 9).
Dai课题组以15步的线性步骤完成了Spinosyn A (10)的全合成, 其中作者发展的Heck串联插羰环化非常高效地实现四环骨架的快速构建, 该方法也为大环内酯的合成提供了一种新的可能. Dai课题组的合成路线简洁高效, 也为Spinosyn A (10)类似物的快速合成提供了可能.
2014年Liu课题组[37]报道利用化学合成和生物合成结合的策略, 实现了Spinosyn A (10)的首次酶催化全合成.作者采取新的构环策略, 先构建多烯大环内酯化合物, 随后利用酶催化的连续分子内环化完成基本骨架的构建, 最后阶段引入氨基葡萄糖实现Spinosyn A (10)的全合成.
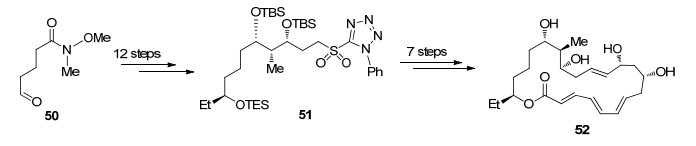
对于多烯大环内酯化合物的合成, 作者从化合物50出发通过不对称乙基化和aldol反应等5步转化得到化合物51, 随后发生Stille偶联、Julia-Kocienski烯基化及Yamaguchi大环内酯化, 经7步转化完成酶催化前体52的构建(Scheme 10).
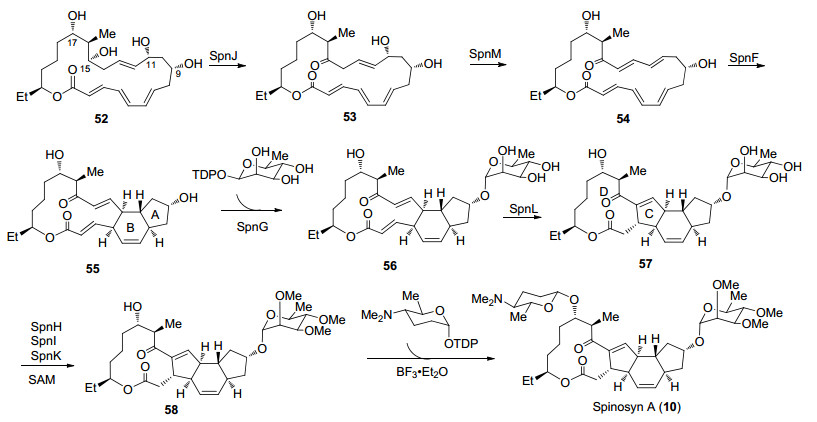
作者通过对刺糖多孢菌中的聚酮合成酶进行修饰表达分别得到SpnJ, SpnM, SpnF, SpnG, SpnL, SpnH, SpnI, SpnK, 研究发现这些酶可以用于Spinosyn A (10)的合成中(Scheme 11).化合物52首先在SpnJ催化下将C(15)位选择性脱氢得到羰基化合物53[38], SpnM催化脱除C(11)位羟基得到多烯化合物54, SpnF可以催化54发生跨环Diels-Alder反应完成AB环的构建[39], 而酶催化的Diels-Alder反应可以选择性地得到单一目标产物.接下来在SpnG催化下可以选择性引入C(9)位L-鼠李糖得到化合物56, SpnL可以催化56发生Morita-Baylis-Hillman (MBH)类型的环化完成C, D环的构建, 化合物57在三个酶SpnH, SpnI, SpnK催化下选择性甲基化及氧糖苷化可以完成化合物58的合成[40], 最后在Lewis酸活化下引入C(17)位氨基葡萄糖实现Spinosyn A (10)的全合成.
Liu课题组完成了Spinosyn A (10)的首次酶催化全合成, 该合成策略通过多步化学转化完成酶催化前体52的合成, 在8个酶作用下实现一锅法合成化合物58, 证实了酶催化转化的高效性和专一性, 同时也说明了化学与生物合成结合的策略在复杂聚酮类天然产物合成中的重要价值.而相对于Spinosyn A的化学全合成, Spinosyn A的酶催化合成在由于其酶催化底物的特殊性, 导致其特定底物的制备步骤冗长, 这也是目前酶催化反应的局限之一.
Myceliothernophin E (59)是由Wu课题组[41]在2007年从嗜热真菌Myceliophthora thermophila的次级代谢产物中分离得到的聚酮类天然产物, 具有很好的抗肿瘤活性, 其对肝癌细胞、肺癌细胞及乳腺癌细胞的IC50值仅为0.26, 0.25, 0.28 μg/mL.从结构来看, Myceliothernophin E (59)具有反式氢化萘环骨架, C环则是tetramic acid环, 整个分子中含有5个手性中心, 合成上也具有一定的挑战性.
|
|
2012年Uchiro课题组[42]利用分子内Diels-Alder反应为关键步骤首次完成了Myceliothernophin E (59)的全合成.作者从(R)-香茅醛(60)出发, 以Corey-Fuchs炔基化和Suzuki-Miyaura偶联作为关键反应首先合成多烯化合物61, 在三氟化硼乙醚作用下发生分子内Diels-Alder反应顺利完成反式氢化萘环骨架的构建, 该反应可以发生完全endo选择性环化得到目标产物62, 同时也完成了5个手性中心的构筑. C环的构建则是利用化合物63在二异丙基氨基锂(LDA)作用下与62发生aldol反应, 经Swern氧化后得到三环化合物64, 接下来经过两步转化在C环引入双键, 脱除保护基并消除C(21)位羟基完成Myceliothernophin E (59)的全合成. Uchiro课题组通过对Myceliothernophin E (59)全合成验证了天然产物的绝对构型, 该策略也有助于对Myceliothernophin E (59)展开生物活性方面的研究(Scheme 12).
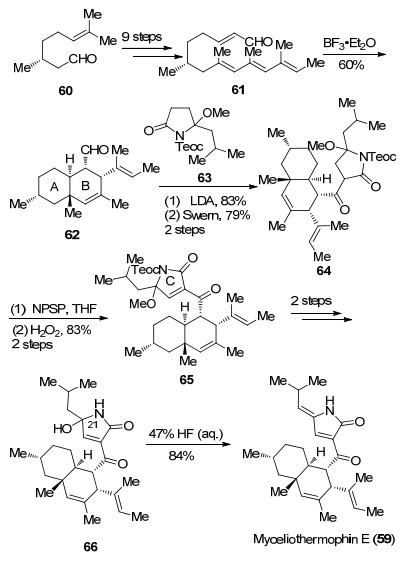
2014年Nicolaou课题组[43]利用他们发展的串联环化反应作为关键步骤实现了Myceliothernophin E (59)的全合成.作者从香茅醛衍生物67出发, 通过3步转化延长碳链得到化合物68, 随后在对甲苯磺酸作用下发生分子内Michael加成及aldol缩合串联环化反应, 可以高效构建反式氢化萘环骨架得到化合物69, 该方法也为反式氢化萘环骨架的构建提供了新的选择.接下来作者通过8步转化分别在C(3)和C(4)位引入侧链, 并对C(1)位氧化得到化合物70, 值得注意的是, 该化合物已经具备了完整的5个手性中心, 化合物70经7步转化可以得到高级中间体62, 随后与63在碱性条件再次发生aldol反应并氧化得到64, 最后通过四步简单转化调整C环的氧化态从而完成Myceliothernophin E (59)的全合成(Scheme 13).
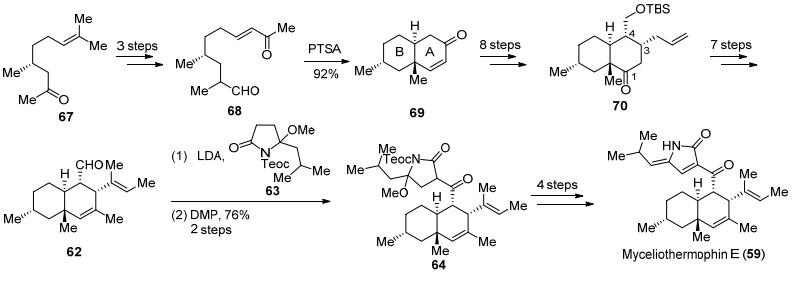
2016年Tang课题组[44]报道了Myceliothernophin E (59)的首次酶催化合成.作者首先从嗜热真菌M. thermophile ATCC 42462中分离得到三种聚酮合成酶-非核糖体多肽合成酶(PKS-NRPS), 分别是MycA, MycB和MycC, 研究发现这三种酶可能参与了Myceliothermophin E (59)的生物合成.为了验证这一设想, 作者首先在MycA和MycC的作用下合成化合物71, 随后化合物71经Knoevenagel缩合构建tetramic acid环并完成化合物72的合成.作者认为化合物72可能经历两种途径的反应历程, 在MycB催化下72经历Path a历程直接发生Diels-Alder反应, 完成ABC三环的构建, 随后发生C(18)位羰基的烯醇化, 在氧气的作用下发生氧化得到Myceliothernophin E (59).而Path b路径中, 化合物72首先发生C(18)位烯醇化, 首先在氧气作用下氧化得到tetramic acids环, 随后在MycB催化下发生Diels-Alder反应完成Myceliothernophin E (59)的合成(Scheme 14).
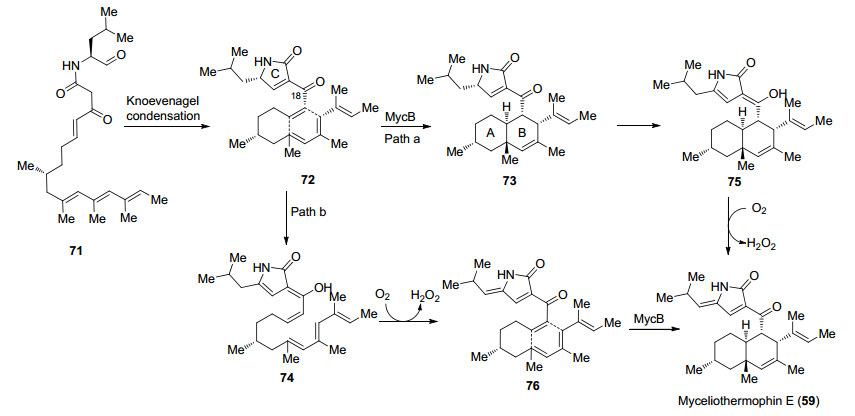
Tang课题组发展的酶催化合成可以实现从简单原料出发完成Myceliothernophin E (59)的全合成, 通过对反应机理研究证实了MycB是一种Diels-Alder酶, 同时也为反式氢化萘环的合成提供了新的方法, 与传统化学合成相比, 酶催化合成在合成效率以及步骤经济性上有了很大提高.
Equisetin (77)是从Fusarium equiseti真菌中分离得到的聚酮类天然产物, 其具有优秀的生物活性, 包括HIV抑制活性和一定的细胞毒性.结构上来看, Equisetin (77)具有反式氢化萘环骨架, 含有6个手性中心, 其中C16位为季碳手性中心, C环为tetramic acids环[45].
|
|
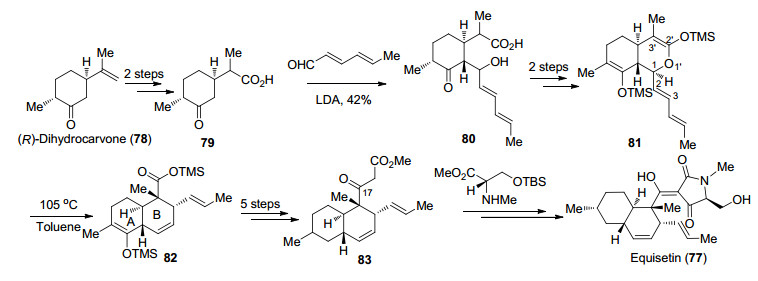
1989年Danishefsky课题组[46]首次完成了Equisetin (77)的全合成.作者采用分步构建反式氢化萘环骨架的策略, 利用(R)-二氢香芹酮(78)为原料, 对烯烃连续氧化得到羧酸化合物79, 在LDA作用下与2, 4-己二烯醛发生aldol反应得到化合物80, 随后在缩合剂的作用下得到内酯81.接下来作者利用Claisen重排反应可以顺利构建B环及C(16)位季碳手性中心.对于tetramic acids环的构建, 化合物82经过5步转化调整保护基并在C(17)位延长碳链得到83, 随后与N-甲基-L-丝氨酸衍生物缩合, 脱保护后发生Dieckmann缩合即可实现Equisetin (77)的全合成.在该部分工作中, Danishefsky课题组利用aldol反应和Claisen重排反应分步的策略完成了反式氢化萘环骨架的构建(Scheme 15).
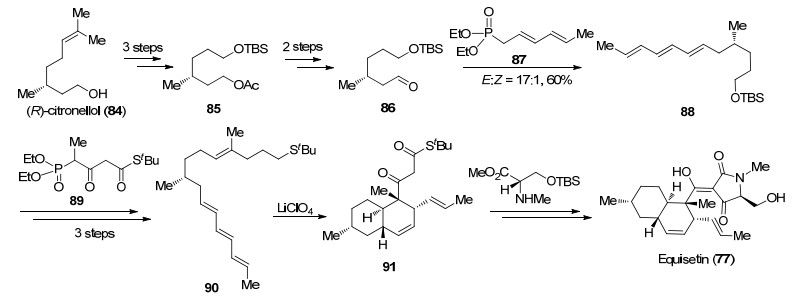
2000年Dixon课题组[47]利用分子内Diels-Alder反应作为关键策略完成了Equisetin (77)的全合成(Scheme 16).针对多烯化合物的合成, 作者从(R)-香茅醇(84)出发, 经乙酰基保护、臭氧化切断及还原保护得到化合物85, 将乙酰基脱除并氧化得到醛86, 碱性条件下与磷酸酯87发生HWE反应, 以17:1的E:Z比得到三烯化合物88.接下来脱除88中的硅基保护基并氧化得到醛, 再与化合物89作用发生HWE反应可以得到多烯化合物90.随后作者尝试了不同Lewis酸促进的Diels-Alder反应, 发现在高氯酸锂作用下能以70%的收率及90%的de值得到目标产物91, 该反应可以一步构建反式氢化萘环骨架, 同时引入5个手性中心.最后在三氟乙酸银作用下与N-甲基-L-丝氨酸衍生物缩合, 脱除保护基后发生Dieckmann缩合完成Equisetin (77)的全合成.
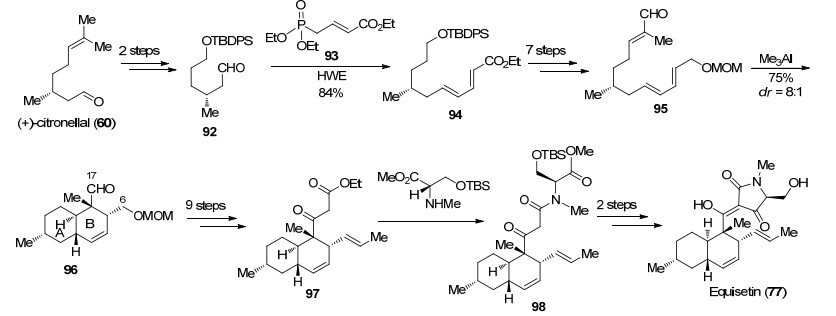
2001年Shishido课题组[48]同样以分子内Diels-Alder反应作为关键策略实现了Equisetin (77)的全合成.对于多烯化合物的合成, 作者选择以(R)-香茅醛(60)为原料, 经两步转化得到醛92, 与磷酸酯93发生HWE反应得到二烯化合物94, 随后以Wittig反应为关键反应经7步转化延长碳链得到化合物95.通过对不同条件下分子内Diels-Alder反应的条件考察, 作者发现化合物95在三甲基铝的作用下顺利完成反式氢化萘环骨架的构建, 能够以75%的收率和8:1的dr值得到化合物96.接下来作者以Reformatsky反应和Swern氧化为关键反应经9步转化分别在C(6)及C(17)位延长碳链, 最后采用类似的策略, 在三氟乙酸银作用下与N-甲基-L-丝氨酸衍生物缩合, 脱除硅基保护发生Dieckmann缩合完成Equisetin (77)的全合成(Scheme 17). Shishido课题组利用三甲基铝促进的分子内Diels-Alder反应很好地控制该反应的非对映选择性, 但后续官能团转化较为繁琐, 使得整体路线冗长.
高栓虎课题组[49]在2012年报道了Equisetin (77)的全合成, 在此工作基础上, 该课题组提出了Equisetin (77)的生源合成途径[50](Scheme 18).认为多烯氨基酸化合物102可能是(-)-Equisetin (77)的生源合成前体, 化合物99与化合物100和87作用发生连续的HWE反应延长碳链, 得到的化合物101在三氟乙酸银作用下与N-甲基-L-丝氨酸缩合完成前体化合物102的合成.随后作者对其生源合成途径进行了研究, 希望通过先构建tetramic acids环再构筑反式氢化萘环的策略完成Equisetin (77)的合成.多烯氨基酸化合物102在甲醇钠作用下发生Dieckmann缩合顺利得到多烯tetramic acid化合物103, 接下来作者考察了化合物103的Diels-Alder反应, 在常用的Lewis酸及加热条件下该反应的非对映选择性(endo/exo)较差, 在甲苯中加热时仅以56%的收率和2:1的endo/exo选择性得到目标产物.近年来, 酶催化Diels-Alder反应也引起了合成化学家们的关注[51], 作者通过与刘文课题组[52]合作探索酶催化的Diels-Alder反应.基于Osada课题组[53]的报道, 将fsa2基因序列克隆到pET-28a载体中, 在大肠杆菌中异源表达得到具有N-末端His6标记的fsa2蛋白并对其纯化.将纯化的酶fsa2与多烯tetramic acid化合物104在缓冲溶液[Tris-HCl buffer (pH=7.0)]中作用, 发现103可以经历完全endo选择性环化得到Equisetin (77).
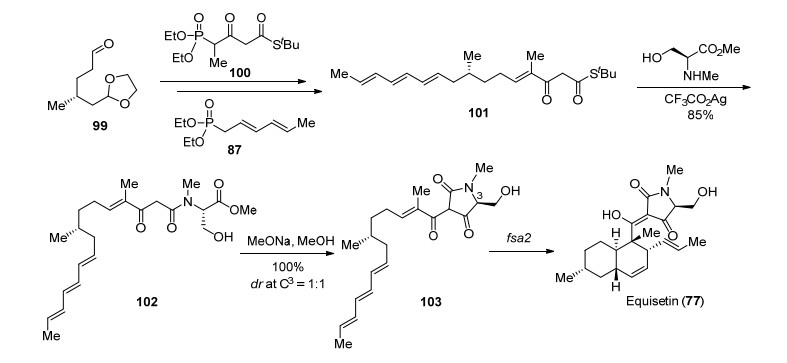
高栓虎课题组与刘文课题组合作实现了Equisetin (77)的首次酶催化合成[54], 与传统的化学合成相比, 利用酶催化Diels-Alder反应构建反式氢化萘环也体现了酶催化反应的高效性和专一性.
主要从化学合成和生物合成结合的角度, 对复杂天然产物的合成研究进行了综述.化学合成已有100多年的历史, 合成方法学及新反应和新试剂的发展极大促进了其在天然产物中的应用.而生物合成则是近年来发展的新兴技术, 随着分子生物学技术的发展, 生物合成已经可以实现在细菌或酵母中合成天然产物, 并有不少成功的例子报道.但目前生物合成发展仍然受到一定的限制, 这可能与酶的种类及酶促反应的识别有关, 另外科学家对酶的作用机制研究将进一步促进未来生物合成研究领域的发展.
Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2016, 79, 629. doi: 10.1021/acs.jnatprod.5b01055
Wöhler, F.; Ueber K. B.; Des, H. Ann. Phys. 1828, 88, 253. doi: 10.1002/(ISSN)1521-3889
(a) Nicolaou, K. C.; Vourloumis, D.; Winssinger, N.; Baran, P. S. Angew. Chem., Int. Ed. 2000, 39, 44.
(b) Nicolaou, K. C. J. Org. Chem. 2009, 74, 951.
(c) Hoffmann, R. W. Angew. Chem., Int. Ed. 2013, 52, 123.
(d) Keasling, J. D.; Mendoza, A.; Baran, P. S. Nature 2012, 492, 188.
(a) Aicher, T. D.; Buszek, K. R.; Fang, F. G.; Forsyth, C. J.; Jung, S. H.; Kishi, Y.; Matelich, M. C.; Scola, P. M.; Spero, D. M.; Yoon, S. K. J. Am. Chem. Soc. 1992, 114, 3162.
(b) Duan, J. J.-W.; Kishi, Y. Tetrahedron Lett. 1993, 34, 7541.
(c) Stamos, D. P.; Kishi, Y. Tetrahedron Lett. 1996, 37, 8643.
(d) Stamos, D. P.; Sean, S. C.; Kishi, Y. J. Org. Chem. 1997, 62, 7552.
(e) Namba, K.; Kishi, Y. J. Am. Chem. Soc. 2005, 127, 15382.
(f) Dong, C.-G.; Henderson, J. A.; Kaburagi, Y.; Sasaki, T.; Kim, D.-S.; Kim, J. T.; Urabe, D.; Guo, H.; Kishi, Y. J. Am. Chem. Soc. 2009, 131, 15642.
(g) Yang, Y.-R.; Kim, D.-S.; Kishi, Y. Org. Lett. 2009, 20, 4516.
(h) Liu, L.; Henderson, J. A.; Yamamoto, A.; Bremond, P.; Kishi, Y. Org. Lett. 2012, 14, 2262.
(i) Shan, M.; Kishi, Y. Org. Lett. 2012, 14, 660.
(j) Lee, J. H.; Li, Z.; Osawa, A.; Kishi, Y. J. Am. Chem. Soc. 2016, 138, 16248.
(a) Kingston, D. G. Phytochemistry 2007, 68, 1844.
(b) Wani, M. C.; Taylor, H. L.; Wall, M. E.; Coggon, P.; McPhail, A. T. J. Am. Chem. Soc. 1971, 93, 2325.
Ajikumar, P. K.; Xiao, W.-H.; Tyo, K. E. J.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T. H.; Pfeifer, B.; Stephanopoulo, G. Science 2010, 330, 70. doi: 10.1126/science.1191652
(a) World Health Organization 18th WHO Essential Medicines List, Geneva, Switzerland, 2013.
(b) Seya, M. J.; Gelders, S. F.; Achara, O. U.; Milani, B.; Scholten, W. K. J. Pain Palliat. Care Pharmacother. 2011, 25, 6.
(c) International Narcotics Control Board (INCB) Narcotic Drugs: Estimated World Requirements for 2015——Statistics for 2013, New York, 2014.
Galanie, S.; Thodey, K.; Trenchard, I. J.; Interrante, M. F.; Smolke, C. D. Science 2015, 349, 1095. doi: 10.1126/science.aac9373
Klayman, D. L. Science 1985, 228, 104.
Davies, E. E. Ann. Trop. Med. Parasitol. 1975, 69, 147-154. doi: 10.1080/00034983.1975.11686996
Theakston, R. D. G. Life Sci. 1969, 8, 521-529. doi: 10.1016/0024-3205(69)90251-3
刘春朝, 王玉春, 欧阳藩, 化学进展, 1999, 11, 41. doi: 10.3321/j.issn:1005-281X.1999.01.006Liu, C. C.; Wang, Y. C.; Ouyang, F. Prog. Chem. 1999, 11, 41. doi: 10.3321/j.issn:1005-281X.1999.01.006
(a) Research Group of Artemisinin Structure Chin. Sci. Bull. 1977, 22, 142 (in Chinese).
(青蒿素结构研究协作组, 科学通报, 1977, 22, 142.)
(b) Liu, J. M.; Ni, M. Y.; Fan, J. F.; Tu, Y. Y.; Wu, Z. H.; Wu, Y. L.; Zhou, W. S. Acta Chim. Sinica 1979, 37, 129 (in Chinese).
(刘静明, 倪幕云, 樊菊芬, 屠呦呦, 吴照华, 吴毓林, 周维善, 化学学报, 1979, 37, 129.)
(c) Research Group of Artemisinin Structure Chin. Pharm. Bull. 1979, 14, 49 (in Chinese).
(青蒿素结构研究协作组, 药学通报, 1979, 14, 49.)
(a) Schmid, G.; Hofheinz, W. J. Am. Chem. Soc. 1983, 105, 624.
(b) Yadav, J. S.; Babu, R. S.; Sabitha, G. Tetrahedron Lett. 2003, 44, 387.
(c) Yadav, J. S.; Thirupathaiah, B.; Srihari, P. Tetrahedron 2010, 66, 2005.
(d) Zhu, C.; Cook, S. P. J. Am. Chem. Soc. 2012, 134, 13577.
(a) Xu, X. X.; Zhu, J.; Huang, D. Z.; Zhou, W. S. Acta Chim. Sinica 1983, 41, 574 (in Chinese).
(许杏祥, 朱杰, 黄大中, 周维善, 化学学报, 1983, 41, 574.)
(b) Xu, X. X.; Zhu, J.; Huang, D. Z.; Zhou, W. S. Acta Chim. Sinica 1984, 42, 940 (in Chinese).
(许杏祥, 朱杰, 黄大中, 周维善, 化学学报, 1984, 42, 940.)
(c) Xu, X. X.; Zhu, J.; Huang, D. Z.; Zhou, W. S. Tetrahedron 1986, 42, 819.
(d) Zhou, W. S.; Xu, X. X. Acc. Chem. Res. 1994, 27, 211.
(e) Ye, B.; Wu, Y. L. J. Chem. Soc., Chem. Commun. 1990, 10, 726.
(f) Hao, H. D.; Li, Y.; Han, W. B.; Wu, Y. K. Org. Lett. 2011, 13, 4212.
(g) Chen, H. J.; Han, W. B.; Hao, H. D.; Wu, Y. K. Tetrahedron 2013, 69, 1112.
(a) Wang, Z. L.; Yang, L. Y.; Yang, X. W.; Zhang, X. H. Synth. Commun. 2014, 44, 1987.
(b) Fu, Y. H.; Zheng, J.; Luo, S. Q.; Hou, Q. W.; Wang, G. C.; Zhan, M. Z. Chin. Pharm. J. 2014, 49, 795 (in Chinese).
(付彦辉, 钟俊, 罗素琴, 侯庆伟, 王国成, 战明哲, 中国药学杂志, 2014, 49, 795.)
(c) Corsello, M. A.; Garg, N. K. Nat. Prod. Rep. 2015, 32, 359.
刘德龙, 张万斌, 科学通报, 2017, 62, 1997. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=KXTB201718014&dbname=CJFD&dbcode=CJFQLiu, D.; Zhang, W. Chin. Sci. Bull. 2017, 62, 1997(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=KXTB201718014&dbname=CJFD&dbcode=CJFQ
(a) Wang, Y.; Xia, Z. Q.; Zhou, F. Y. Acta Chim. Sinica 1988, 46, 1152 (in Chinese).
(汪猷, 夏志强, 周凤仪, 化学学报, 1988, 46, 1152.)
(b) Wallaart, T. E.; Pras, N.; Quax, W. J. J. Nat. Prod. 1999, 62, 1161.
(c) Chappell, J.; Wolf, F.; Proulx, J. Plant Physiol. 1995, 109, 1337.
(a) Martin, V. J. J.; Pitera, D. J.; Withers, S. T.; Newman, J. D.; Keasling, J. D. Nat. Biotechnol. 2003, 21, 796.
(b) Brown, G. D. Molecules 2010, 15, 7603.
(c) Tsuruta, H.; Paddon, C. J.; Eng, D.; Lenihan, J. R.; Horning, T.; Anthony, L. C.; Regentin, R.; Keasling, J. D.; Renninger, N. S.; Newman, J. D. PLoS One 2009, 4, e4489.
(d) Ro, D.-K.; Paradise, E. M.; Ouellet, M.; Fisher, K. J.; Newman, K. L.; Ndungu, J. M.; Ho, K. A.; Eachus, R. A.; Ham, T. S.; Kirby, J.; Chang, M. C. Y.; Withers, S. T.; Shiba, Y.; Sarpong, R.; Keasling, J. D. Nature 2006, 440, 940.
(e) Westfall, P. J.; Pitera, D. J.; Lenihan, J. R.; Eng, D.; Woolard, F. X.; Regentin, R.; Horning, T.; Tsuruta, H.; Melis, D. J.; Owens, A.; Fickes, S.; Diola, D.; Benjamin, K. R.; Keasling, J. D.; Leavell, M. D.; McPhee, D. J.; Renninger, N. S.; Newman, J. D.; Paddon, C. J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E111.
(f) Bouwmeester, H. J.; Wallaart, T. E.; Janssen, M. H. A.; Loo, B. V.; Jansen, B. J. M.; Posthumus, M. A.; Schmidt, C. O.; De Kraker, J.-W.; Kö nig, W. A.; Franssen, M. C. R. Phytochemistry 1999, 52, 843.
(g) Mercke, P.; Bengtsson, M.; Bouwmeester, H. J.; Posthumus, M. A.; Brodelius, P. E. Arch. Biochem. Biophys. 2000, 381, 173.
(h) Teoh, K. H.; Polichuk, D. R.; Reed, D. W.; Covello, P. S. Botany 2009, 87, 635.
(i) Paddon, C. J.; Westfall, P. J.; Pitera, D. J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M. D.; Tai, A.; Main, A.; Eng, D.; Polichuk, D. R.; Teoh, K. H.; Reed, D. W.; Treynor, T.; Lenihan, J.; Jiang, H.; Fleck, M.; Bajad, S.; Dang, G.; Dengrove, D.; Diola, D.; Dorin, G.; Ellens, K. W.; Fickes, S.; Galazzo, J.; Gaucher, S. P.; Geistlinger, T.; Henry, R.; Hepp, M.; Horning, T.; Iqbal, T.; Kizer, L.; Lieu, B.; Melis, D.; Moss, N.; Regentin, R.; Secrest, S.; Tsuruta, H.; Vazquez, R.; Westblade, L. F.; Xu, L.; Yu, M.; Zhang, Y.; Zhao, L.; Lievense, J.; Covello, P. S.; Keasling, J. D.; Reiling, K. K.; Renninger, N. S.; Newman, J. D. Nature 2013, 496, 528.
(a) Acton, N.; Roth, R. J. J. Nat. Prod. 1989, 52, 1183.
(b) Acton, N.; Roth, R. J. J. Org. Chem. 1992, 57, 3610.
(c) Haynes, R. K.; Vonwiller, S. C. J. Chem. Soc., Chem. Commun. 1990, 451.
(d) Sy, L.-K.; Brown, G. D. Tetrahedron 2002, 58, 897.
(a) Zhang, W.; Liu, D.; Yuan, Q. CN 102718773, 2012.
(b) Li, J.; Shen, J.; Xia, C.; Wang, Y.; Liu, D.; Zhang, W. Org. Lett. 2016, 18, 2122.
(a) Sparks, T. C.; Crouse, G. D.; Durst, G. Pest Manage. Sci. 2001, 57, 896.
(b) Kirst, H. A. J. Antibiot. 2010, 63, 101.
(c) Kirst, H. A.; Michel, K. H.; Martin, J. W.; Creemer, L. C.; Chio, E. H.; Yao, R. C.; Nakatsukasa, W. M.; Boeck, L. D.; Occolowitz, J. L.; Paschal, J. W.; Deeter, J. B.; Jones, N. D.; Thompson, G. D. Tetrahedron Lett. 1991, 32, 4839.
Evans, D. A.; Black, W. C. J. Am. Chem. Soc. 1993, 115, 4497. doi: 10.1021/ja00064a011
(a) Evans, D. A.; Bartroli, J.; Shih, T. L. J. Am. Chem. Soc. 1981, 103, 2127.
(b) Evans, D. A.; Gage, J. R. Org. Synth. 1989, 68, 83.
(a) Hikota, M.; Tone, H.; Horita, K.; Yonemitsu, O. J. Org. Chem. 1990, 55, 7.
(b) Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc. Jpn. 1979, 52, 1989.
(a) Stille, J. K.; Groh, B. L. J. Am. Chem. Soc. 1987, 109, 813.
(b) Stille, J. K.; Sweet, M. P. Tetrahedron Lett. 1989, 30, 3645.
Evans, D. A.; Chapman, K. T.; Bisaha, J. J. Am. Chem. Soc. 1988, 110, 1238. doi: 10.1021/ja00212a037
(a) Paquette, L. A.; Gao, Z.; Ni, Z.; Smith, G. F. J. Am. Chem. Soc. 1998, 120, 2543.
(b) Paquette, L. A.; Collado, I.; Purdie, M. J. Am. Chem. Soc. 1998, 120, 2553.
(a) Mukaiyama, T.; Kobayashi, S.; Shoda, S.-I. Chem. Lett. 1984, 907.
(b) Evans, D. A.; Kaldor, S. W.; Jones, T. K.; Clardy, J.; Stout, T. J. J. Am. Chem. Soc. 1990, 112, 7001.
(a) Mergott, D. J.; Frank, S. A.; Roush, W. R. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 11955.
(b) Winbush, S. M.; Mergott, D. J.; Roush, W. R. J. Org. Chem. 2008, 73, 1818.
(a) Boeckman, R. K. Jr.; Barta, T. E. J. Org. Chem. 1985, 50, 3421.
(b) Roush, W. R.; Kageyama, M. Tetrahedron Lett. 1985, 26, 4327.
(c) Roush, W. R.; Kageyama, M.; Riva, R.; Brown, B. B.; Warmus, J. S.; Moriarty, K. J. J. Org. Chem. 1991, 56, 1192.
(a) Wang, L. C.; Luis, A. L.; Agaplou, K.; Jang, H. Y.; Krische, M. J. J. Am. Chem. Soc. 2002, 124, 2402.
(b) Frank, S. A.; Mergott, D. J.; Roush, W. R. J. Am. Chem. Soc. 2002, 124, 2404.
Bai, Y.; Shen, X. Y.; Li, Y.; Dai, M. J. J. Am. Chem. Soc. 2016, 138, 10838. doi: 10.1021/jacs.6b07585
(a) Wilson, R. M.; Jen, W. S.; MacMillan, D. W. C. J. Am. Chem. Soc. 2005, 127, 11616.
(b) Lambert, T. H.; Danishefsky, S. J. J. Am. Chem. Soc. 2006, 128, 426.
(a) Davis, D. C.; Walker, K. L.; Hu, C.; Zare, R. N.; Waymouth, R. M.; Dai, M. J. J. Am. Chem. Soc. 2016, 138, 10693.
(b) Bai, Y.; Davis, D. C.; Dai, M. J. Angew. Chem., Int. Ed. 2014, 53, 6519.
(c) Gehrtz, P. H.; Hirschbeck, V.; Ciszek, B.; Fleischer, I. Synthesis 2016, 48, 1573.
(d) Wu, X. F.; Neumann, H.; Beller, M. Chem. Rev. 2013, 113, 1.
(e) Wu, X.-F.; Neumann, H.; Beller, M. Chem. Rev. 2013, 113, 1.
(f) Bai, Y.; Davis, D. C.; Dai, M. J. J. Org. Chem. 2017, 82, 2319.
(a) Li, Y.; Yang, Y.; Yu, B. Tetrahedron Lett. 2008, 49, 3604.
(b) Yang, Y.; Li, Y.; Yu, B. J. Am. Chem. Soc. 2009, 131, 12076.
(c) Yu, B.; Sun, J.; Yang, X. Acc. Chem. Res. 2012, 45, 1227.
(d) Nie, S.; Li, W.; Yu, B. J. Am. Chem. Soc. 2014, 136, 4157.
Kim, H. J.; Choi, S.-H.; Jeon, B.-S.; Kim, N.; Pongdee, R.; Wu, Q.; Liu, H.-W. Angew. Chem., Int. Ed. 2014, 53, 13553. doi: 10.1002/anie.201407806
Kim, H. J.; Pongdee, R.; Wu, Q.; Hong, L.; Liu, H.-W. J. Am. Chem. Soc. 2007, 129, 14582. doi: 10.1021/ja076580i
(a) Kim, H. J.; Ruszczycky, M. W.; Choi, S.-H.; Liu, Y.-N.; Liu, H.-W. Nature 2011, 473, 109.
(b) Kim, H. J.; Ruszczycky, M. W.; Liu, H.-W. Curr. Opin. Chem. Biol. 2012, 16, 124.
(a) Oikawa, H. Bull. Chem. Soc. Jpn. 2005, 78, 537.
(a) Chen, Y.-L.; Chen, Y.-H.; Lin, Y.-C.; Tsai, K.-C.; Chiu, H.-T. J. Biol. Chem. 2009, 284, 7352.
(b) Kim, H. J.; White-Phillip, J. A.; Ogasawara, Y.; Shin, N.; Isiorho, E. A.; Liu, H.-W. J. Am. Chem. Soc. 2010, 132, 2901.
(c) Isiorho, E. A.; Liu, H.-w.; Keatinge-Clay, A. T. Biochemistry 2012, 51, 1213.
Yang, Y. L.; Lu, C. P.; Chen, M. Y.; Chen, K. Y.; Wu, Y. C.; Wu, S. H. Chem.-Eur. J. 2007, 13, 6985. doi: 10.1002/(ISSN)1521-3765
Shionozaki, N.; Yamaguchi, T.; Kitano, H.; Tomizawa, M.; Makino, K.; Uchiro, H. Tetrahedron Lett. 2012, 53, 5167. doi: 10.1016/j.tetlet.2012.07.058
Nicolaou, K.; Shi, L.; Lu, M.; Pattanayak, M. R.; Shah, A. A.; Ioannidou, H. A.; Lamani, M. Angew. Chem., Int. Ed. 2014, 53, 10970. doi: 10.1002/anie.201406815
Li, L.; Yu, P.; Tang, M.-C.; Zou, Y.; Gao, S-S.; Hung, Y.-S.; Zhao, M.; Watanabe, K.; Houk, K. N.; Tang, Y. J. Am. Chem. Soc. 2016, 138, 15837. doi: 10.1021/jacs.6b10452
Burmeister, H. R.; Bennet, G. A.; Vesonder, R. F.; Hesseltine, C. W. Antimicrob. Agents Chemother. 1974, 5, 634. doi: 10.1128/AAC.5.6.634
Turos, E.; Audia, J. E.; Danishefsky, S. J. J. Am. Chem. Soc. 1989, 111, 8231. doi: 10.1021/ja00203a026
(a) Burke, L. T.; Dixon, D. J.; Ley, S. V.; Rodríguez, F. Org. Lett. 2000, 2, 3611.
(b) Burke, L. T.; Dixon, D. J.; Ley, S. V.; Rodríguez, F. Org. Biomol. Chem. 2005, 3, 274.
Kumiko, Y. K.; Shindo, M.; Shishido, K. Tetrahedron Lett. 2001, 42, 2517. doi: 10.1016/S0040-4039(01)00217-9
Yin, J.; Wang, C.; Kong, L.; Cai, S.; Gao, S. Angew. Chem., Int. Ed. 2012, 51, 7786. doi: 10.1002/anie.201202455
(a) Yin, J.; Kong, L.; Wang, C.; Shi, Y.; Cai, S.; Gao, S. Chem.- Eur. J. 2013, 19, 13040.
(b) Kong, L.; Rao, M.; Ou, J.; Yin, J.; Lu, W.; Liu, M.; Pang, X.; Gao, S. Org. Biomol. Chem. 2014, 12, 7591.
(c) Yin J.; Gao, S. Synlett 2014, 25, 1.
(d) Yin, J.; Kong, L.; Gao, S. Chin. J. Org. Chem. 2013, 33, 259 (in Chinese).
(尹军, 孔丽丽, 高栓虎, 有机化学, 2013, 33, 259.)
(a) Oikawa, H. Cell Chem. Biol. 2016, 23, 429.
(b) Oikawa, H.; Tokiwano, T. Nat. Prod. Rep. 2004, 21, 321.
(c) Klas, K.; Tsukamoto, S.; Sherman, D. H.; Williams, R. M. J. Org. Chem. 2015, 80, 11672.
(d) Minami, A.; Oikawa, H. J. Antibiot. 2016, 1.
(e) Oikawa, H. Bull. Chem. Soc. 2005, 78, 537.
(f) Jeon, B.-S.; Wang, S.-A.; Ruszczycky, M. W.; Liu, H.-W. Chem. Rev. 2017, 117, 5367.
(a) Wu, Q.; Wu, Z.; Qu, X.; Liu, W. J. Am. Chem. Soc. 2012, 134, 17342.
(b) Tian, Z.; Sun, P.; Yan, Y.; Wu, Z.; Zheng, Q.; Zhou, X.; Zhang, H.; Yu, F.; Jia, X.; Chen, D.; Mandi, A.; Kurtan T.; Liu, W. Nat. Chem. Biol. 2015, 11, 259.
(c) Pang, B.; Zhong, G.; Tang, Z.; Liu, W. Methods Enzymol. 2016, 575, 39.
(d) Zheng, Q.; Tian, Z.; Liu, W. Curr. Opin. Chem. Biol. 2016, 31, 95.
(e) Zheng, Q.; Guo, Y.; Yang, L.; Zhao, Z.; Wu, Z.; Zhang, H.; Liu, J.; Cheng, X.; Wu, J.; Yang, H.; Jiang, H.; Pan, L.; Liu, W. Cell Chem. Biol. 2016, 23, 352.
(f) Zheng, Q.; Gong, Y.; Guo, Y.; Zhao, Z.; Wu, Z.; Zhou, Z.; Chen, D.; Pan, L.; Liu, W. Cell Chem. Biol. 2018, 25, 718.
Kato, N.; Nogawa, T.; Hirota, H.; Jang, J. H.; Takahashi, S.; Ahn, J. S.; Osada, H. Biochem. Biophys. Res. Commun. 2015, 460, 210. doi: 10.1016/j.bbrc.2015.03.011
Li, X.; Zheng, Q.; Yin, J.; Liu, W.; Gao, S. Chem. Commun. 2017, 53, 4695. doi: 10.1039/C7CC01929G

图式 1 青蒿酸的生物发酵合成途径
Scheme 1 Biological fermentation pathway for the synthesis of artemisinic acid

图式 5 Paquette课题组对Spinosyn A (10)的全合成
Scheme 5 Paquette's total synthesis of spinosyn A (10)

图式 12 Uchiro课题组关于Myceliothernophin E (50)的全合成
Scheme 12 Total synthesis of myceliothernophin E (50) by Uchiro group

图式 13 Nicolaou课题组关于Myceliothernophin E (59)的全合成
Scheme 13 Total synthesis of Myceliothernophin E (59) by Nicolaou group

图式 14 Tang课题组关于Myceliothernophin E (59)的酶催化合成
Scheme 14 Enzymatic synthesis of myceliothernophin E (59) by Tang group

图式 15 Danishefsky课题组关于Equisetin (77)的全合成
Scheme 15 Total synthesis of equisetin (77) by Danishefsky group

图式 16 Dixon课题组关于Equisetin (77)的全合成
Scheme 16 Total synthesis of equisetin (77) by Dixon group

图式 17 Shishido课题组关于Equisetin (77)的全合成
Scheme 17 Total synthesis of equisetin (77) by Shishido group

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:










 下载:
下载: