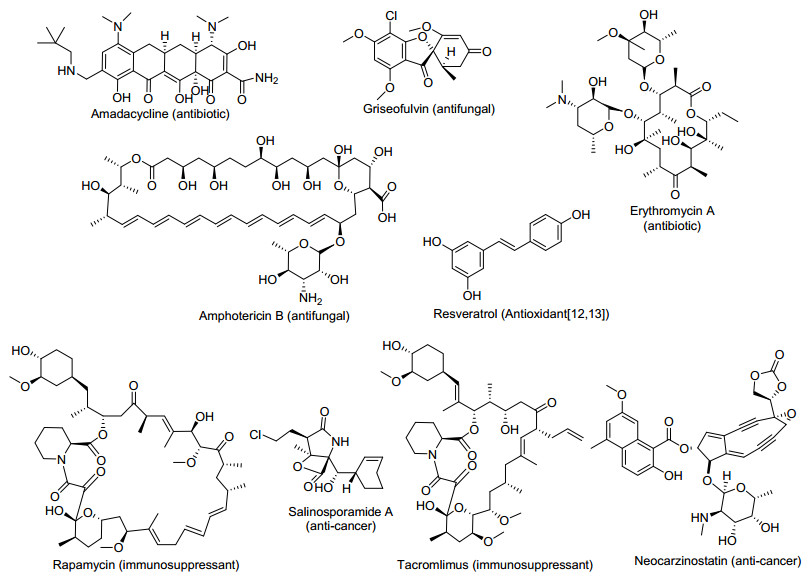
图 1.
聚酮化合物的结构和活性功能
Figure 1.
Structure and bio-activity of polyketides

青霉素是人类文明历史上第一个用于治疗人类感染疾病的抗生素, 它的发现挽救了无数人的生命, 并激发越来越多的科学家挖掘天然药物的热情.迄今为止, 约15000多种次级代谢天然产物被挖掘出来, 根据其结构成分可分为聚酮类化合物、非核糖体多肽、萜类和甾醇类化合物等, 其中绝大多数属于聚酮类化合物[1, 2].聚酮化合物是一类由细菌﹑真菌和植物将低级羧酸通过连续的缩合反应, 再经修饰形成的天然产物, 它包括很多的药用活性化合物, 如具有抗细菌活性的tetrono-mycin[3]和红霉素A[4]、抗真菌活性的两性霉素B[5]和安丝菌素[6]、抗癌活性的salinosporamide A[7]和neocarzino-statin[8, 9]、作为免疫抑制剂的他克莫司(FK506)[10]和雷帕霉素[11]等(图 1).
聚酮化合物主要是由聚酮合酶(PKSs)催化产生的[2], 根据结构和反应机制的不同, 可分为Ⅰ、Ⅱ和Ⅲ型. Ⅰ型PKS包括Ⅰ型模块PKS和Ⅰ型迭代PKS, 其中典型的Ⅰ型模块PKS是以模块形式存在的多功能酶.模块中的催化功能域包含β-酮酰基硫酯合酶(KS)结构域、酰基转移酶(AT)结构域和酰基受体蛋白(ACP)结构域, 这三个结构域组成最小的催化模块或PKS.其中, KS结构域的功能是催化碳-碳键的形成增长主链, AT结构域能够转运不同功能的酰基基团渗入主链, ACP结构域的功能是接受和传递AT结构域转移的酰基部分.除了三个必需结构域外, Ⅰ型PKS模块中还存在其它结构域, 如对主链延伸的碳原子进行功能修饰的β-酮基还原酶(KR)、脱水酶(DH)和烯酰还原酶(ER)和水解终产物且具有纠错功能的硫酯酶(TE)[2, 14].而Ⅰ型迭代PKS是以重复使用单模块合酶来合成聚酮化合物, 单模块中的功能结构域与典型Ⅰ型PKS中的结构域相同. Ⅱ型PKS是以多酶复合体的迭代形式行使合成功能, 包含由2个酮酰合酶单元合成的异源二聚体(KSα和KSβ)、ACP结构域和其它结构域如KR、环化酶(CYC)和芳香化酶(ARO)等, 主要催化蒽环类和四环素类化合物的生物合成[15]. Ⅲ型PKS主要是查尔酮合成酶, 是一类可重复使用的同源双亚基自主式合酶, 其上只含有一个KS结构域, 能直接催化酰基-CoA间的缩合, 形成单环或双环等芳香类聚酮化合物[2].
随着聚酮化合物的广泛使用, 人们发现其具有品种局限性、产量低、易产生耐药性及药物毒副作用等问题, 寻找活性强、毒性弱的聚酮化合物变得越来越困难.基于上述原因, 越来越多的科学家们开始探索合成聚酮化合物PKS内在奥秘, 并借助PKS线性模块和结构域的工程——AT结构域的改造, 寻求产生活性更好的新型聚酮化合物.总结了近年Ⅰ型PKS中AT结构域的催化机制与底物选择性相关研究, 并介绍通过AT结构域的替换、点突变和trans-AT结构域互补的基因工程改造得到新型聚酮化合物的范例. PKS中AT结构域的改造是获取聚酮化合物生物多样性的手段, 也是新药开发的重要策略.
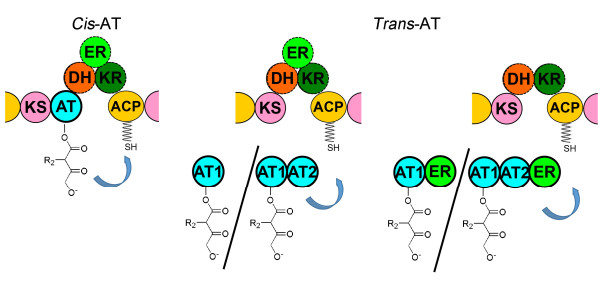
Ⅰ型PKS中AT根据其所处的位置主要分为cis-AT和trans-AT结构域. cis-AT结构域位于PKS模块中, 执行转运底物功能[16].大部分cis-AT结构域发现于放线菌、粘细菌和蓝细菌的PKS中, 其进化来源于基因复制和分化[1, 16]. Trans-AT结构域位于Trans-AT PKS(又称AT-less PKS)模块外, 一般情况下可同时转移n (n≥1)个底物到n (n≥1)个ACP(s)结构域上[17, 18].例如在首个被生化和体内表征的AT-less PKS——leinamycin PKS中, trans-ATLnmG转运6个丙二酰(M)单元到PKS的6个模块中[19, 20].大部分trans-AT结构域存在于变形杆菌、粘细菌和芽孢杆菌的PKS中.与cis-AT结构域进化来源不同, trans-AT结构域的进化源自于基因水平转移[1, 16].另外, trans-AT结构域根据其在PKS中串联方式的差异可细分为四种.第一种, 在合成黄色霉素的PKS中, 有一个单独AT结构域KirCⅡ[21](图 2). Williams课题组[17]利用KirCⅡ与叠氮化物/炔基官能团修饰的延伸单元进行共价连接, 通过十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)蛋白电泳和荧光定量检测叠氮化物/炔基官能团的荧光强度差异, 从而监测KirCⅡ结合底物的活性.结果显示KirCⅡ特异性转运乙基丙二酰(EM)单元到特定ACP5结构域, 从而将EM单元渗入到黄色霉素主链中.这是有史以来首次通过生化实验证明trans-AT结构域转运延伸单元到特定模块中.第二种, 变绿黏球菌中合成myxovirescin的PKS中的TaV含有2个串联AT结构域[21](图 2), 即TaV-AT1和TaV-AT2. Calderone课题组通过体外生化实验分析发现, 虽然TaV-AT1和TaV-AT2都含有高度保守的催化活性中心, 却只有TaV-AT2能特异性转运M单元到多个ACP, 包括独立结构的holo-TaB/TaE和Tal PKS模块中holo-ACP5/6/7上, 但TaV-AT1结构域无此功能[22]; 第三种, 在2-硝基丙烷双加氧酶家族中多元不饱和脂肪酸的合成途径中存在一个C末端与额外的ER结构域相连的AT结构域(图 2)[23].这两个串联的结构域独立行使还原和转运延伸单元的功能; 第四种, 在黄色霉素合成途径中, KirCI串联了两个AT结构域与ER结构域(图 2). Yadav课题组[24]发现KirCI-AT2结构域上含有识别M单元的氨基酸模序, 而在KirCI-AT1结构域中, 其高度保守的His91和Arg117分别被Ala和Glu取代进而失去活性.
AT结构域是Ⅰ型PKS的必需功能域.不同AT结构域转运不同的底物, 而底物决定了聚酮产物的骨架结构, 这是聚酮化合物之所以种类繁多的最主要原因[1].按照转运底物可分为选择性引入的起始单元和选择性上载的主链延伸单元. AT结构域可引入的起始底物种类繁多(图 3), 主要是对α-碳羧酸CoA的识别, 其中M单元和甲基丙二酰(MM)单元是目前报道最多的起始底物[1], 如两性霉素B[5]和盐霉素[25]以丙二酰单元为起始底物, 阿雷西霉素[26]和黄色霉素[17, 21]以甲基丙二酰单元为起始底物.同时, 有些AT结构域以ACP携带的酰基基团作为转运起始底物, 如以丁基丙二酰-ACP (BM-ACP)为起始底物合成阿诺霉素[27]和以己基丙二酰-ACP (HM-ACP)为起始底物合成苯那他丁[28].有趣的是, AT结构域还可直接以多种化合物作为起始底物, 如以4, 5-二羟基-1-烯-环己酸为起始单元合成FK506[29]和雷帕霉素[30].
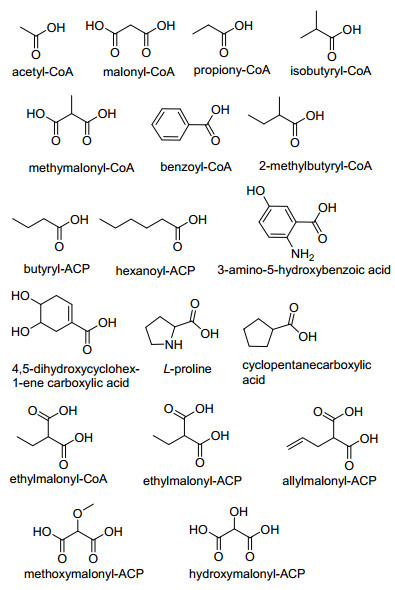
The substrates of AT domains are in three forms as carboxylic acid, acyl-CoA and acyl-ACP. Their structures are shown in the form of free carboxylic acid in Figure 3
主链的延伸也需要AT结构域特异性地引入延伸底物(图 3).与AT结构域能转运多种类型的起始单元相比, AT结构域所识别的延伸底物较为单一, 主要由CoA和ACP结构域所携带[1].根据产生来源的不同, 延伸底物可分为初级代谢和次级代谢产生的延伸底物.初级代谢过程中产生的延伸底物如M和MM参与绝大部分聚酮产物主链的延伸.次级代谢过程中基因簇内相关亚基因簇指导合成且由ACP结构域携带的延伸底物如烯丙基丙二酰ACP (AM-ACP)[31]和甲氧基丙二酰ACP (MeO-ACP)[32]参与此簇合成的产物主链的延伸. Yoon课题组[31]研究发现FK506合成基因簇中tcsa~tcsd亚基因簇催化合成的延伸底物AM-ACP. AM-ACP渗入到FK506主链中, 成为主链的一个罕见的烯丙基侧链. Floss课题组[32]发现安丝菌素基因簇中的asm13~asm17亚基因簇催化合成延伸单元MeO-ACP.另外, 还存在初级代谢和次级代谢过程中都能产生的延伸底物如EM-CoA.产生次级代谢主产物FK506的筑波链霉菌在营养生长过程中, 高效液相色谱(HPLC)监测其体内M-, MM-, EM-和丙酰(Pr)-CoA的含量, 发现检测到EM-CoA的存在[33], 这说明EM-CoA可以在初级代谢过程中产生. Petkovic课题组[34]在子囊霉素的合成基因簇中发现fkbg~fkbk亚基因簇负责合成EM-CoA.这说明EM-CoA可以在次级代谢过程中产生, 且它参与组成子囊霉素主链的乙基侧链.
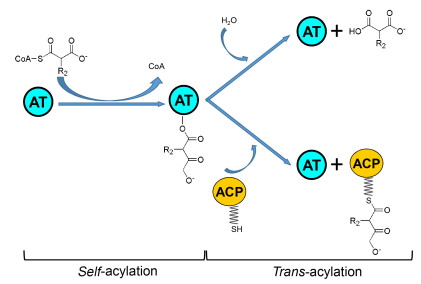
AT结构域转运底物的催化机制包括自酰化反应和转酰化反应.自酰化反应是指CoA或ACP结构域所携带的酯酰基与AT结构域形成AT-酰基中间体的过程; 转酰化反应是指AT结构域转运结合自身的酰基部分到相应ACP结构域的磷酸泛酸巯基乙胺臂上的过程(图 4)[35].根据AT结构域的两步催化过程来探讨哪一步决定AT底物专一性的相关内容. Leadlay课题组[36]发现菌株Saccharopolyspora erythraea发酵主产物红霉素的6-脱氧红霉素B合酶(DEBS)中的AT结构域只结合(2S)-MM单元形成中间体, 不识别(2R)-MM单元.并且DEBS AT3在结合M和MM单元酶活动力曲线的实验结果表明:中间体MM-O-AT3的形成速率显著高于M-O-AT3的形成, 且是M-O-AT3形成速率的200倍[35].这说明AT结构域的自酰化过程决定底物的专一性.尽管上述文献报道AT的自酰化对底物有选择性, 但也有研究表明AT结构域的转酰化过程决定底物专一性.苦霉素PKS中的AT6PikAIV在自酰化反应中可结合MM和EM单元形成MM-O-AT6PikAIV和EM-O-AT6PikAIV两种中间体, 但在转酰化反应中, 只产生MM-ACP6PikAIV[37]. Weber课题组[38]发现菌株Streptomyces collinus的次级代谢主产物黄色霉素PKS中KirCI-AT2结构域转运M单元, 而不是MM或EM单元到ACP5Kir上.我们实验室[39]也发现筑波链霉菌YN06的FK506 PKS模块中AT10FkbA在自酰化反应中可结合M和MM单元形成M-O-AT10FkbA和MM-O-AT10FkbA两种中间体, 但在转酰化反应中只形成M-ACP10FkbA.除上述两种情况之外, AT结构域在这两步反应中对底物都具有专一性作用. FK506 PKS的AT4FkbB在自酰化反应中可结合AM、EM和MM单元形成3种中间体, 但不能与M形成中间体; 随后在转酰化反应中转运与自身结合的AM和EM单元到相应ACP4FkbB上, 但不转运MM单元[40].有趣的是, 也有研究表明, 除AT结构域外, ACP结构域也参与协助决定底物专一性.如Williams课题组[17]发现黄色霉素PKS中的KirCⅡ只转运EM单元到ACP5Kir上, 这一过程是由KirCⅡ和ACP5Kir协同决定的.
The spring structure of ACP domain was the phosphopantetheine arm
AT结构域转运底物的催化过程虽已解释.但AT结构域上哪些氨基酸对底物专一性起重要作用还未了解, 这促使人们深入解析AT结构域、AT结构域与小分子复合物、AT与其他结构域的蛋白质晶体结构.查阅AT结构域蛋白质晶体的相关报道, 发现AT结构域在立体结构上较为保守, 它在自然形态下以二聚体的形式存在.单体的AT是由2个亚结构域组成.核心亚结构域为α/β-水解酶, 其上含有保守的催化模序(GHSxL)和底物结合模序(xxxH). GHSxL模序中的Ser和xxxH模序中的His是AT结构域中高度保守的氨基酸, 两者组成了AT结构域的催化活性中心(图 5A).另一个附属亚结构域为传递电子的类铁氧还蛋白结构(图 5A)[4, 18, 41~43].科学家解析AT-酰基中间体的蛋白质晶体结构发现AT结合酰基底物的自酰化过程如下: AT结构域的催化模序GHSxL中Ser的羟基以硫酯键的形式结合CoA上水解下来的脂酰基单元形成AT-酰基中间体(图 5B)[18, 41], 从而完成自酰化反应.这为酰基渗入主链的合成奠定基础.同时, AT结构域中的底物结合模序xxxH中高度保守的His在AT-酰基中间体的形成中起辅助稳定中间态作用(图 5B)[44].经过大量AT结构域的氨基酸序列比对, 底物结合模序xxxH绝大多数情况下识别M单元为HAFH, 而识别MM单元为YASH[1]等.根据底物结合模序xxxH来推测AT结构域识别的延伸底物, 这为PKS产生聚酮化合物的推测提供方法.但也存在个别例子如两性霉素B PKS上的AT3识别MM单元, 其模序为YAGH, 而不是YASH[45].因此, AT结构域中其他氨基酸在AT自酰化过程中也具有协同稳定中间态的作用, 如Enediyne PKS DynE8与酰基底物结合的晶体结构解析中发现, 除上述提到催化活性中心Ser和His外, AT核心亚结构域α/β-水解酶结构上存在模序PGQGXQ与酰基基团直接形成氧负离子穴来稳定AT-酰基中间体结构的状态(图 5B). AT结构域催化口袋附近的高度保守氨基酸Arg676不仅决定了催化口袋的深度, 而且与M单元的羧基形成盐桥来稳定M-ATDynE8中间体的形成(图 5B)[41].
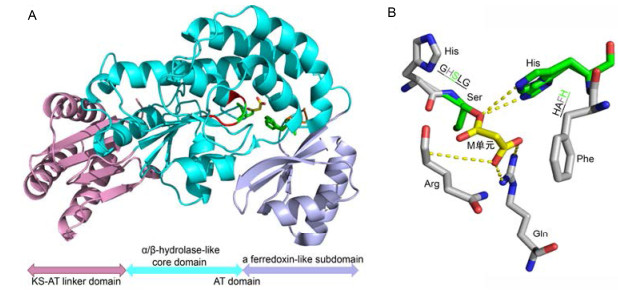
The data was from protein data bank (PDB, ID: 4AMP). (A) Overall structure of ATDynE8 with malonate. The motifs GHSLG and HAFH in α/β-hydrolase-like core domain were colored red and orange. (B) The catalytic pocket of ATDynE8 domain with malonate. Ser in motif GHSLG and His in motif HAFH, malonate, oxygen and hydrogen atoms were colored green, yellow, red and blue. Hydrogen bonds were shown by the yellow dotted lines
AT结构域的转酰化过程, 需要AT与ACP结构域的参与, 但迄今为止, 因AT与ACP结构域之间的相互作用力较小而无法得到AT与ACP的蛋白晶体复合物[18].研究人员通过解析晶体或同源建模得到AT与ACP的结构,并利用docking将这两个蛋白进行对接模拟,研究AT与ACP结构域之间的相互作用, 从而分析AT结构域的转酰化过程[18, 41]. AT的晶体结构在上面已详细介绍,这里主要介绍ACP的晶体结构: ACP结构有4个α螺旋和其间3个无规则卷曲区域组成, 它的活性催化中心为Ser, 位于α螺旋上. AT结构域的催化中心Ser与ACP结构域催化中心Ser相距约20 Å, 即伸展磷酸泛酸巯基乙胺臂的长度, 以满足转酰化反应的进行, 这是对接模拟的前提条件[18, 41].根据对接模拟数据发现, 这两个结构域之间以静电互补作用相互接触[4, 18, 41]. Liew课题组通过AT与ACP结构域的对接模拟, 发现KS与AT结构域之间的连接区域与ACPDYN的第一、第二无规则卷曲部分有静电作用. KS与AT结构域之间的连接区域上的氨基酸Asp477, Arg534和Arg562与ACPDYN上的氨基酸Arg956, Asp960和Glu945形成盐桥.
ATDYN大亚基上的一些无规则卷曲部分与ACPDYN之间也存在静电作用.这些静电互补作用为这两个蛋白之间的接触和传递底物提供环境等作用.并且, Williams课题组在对预测得到AT与ACP结构域表面接触的氨基酸进行点突变后发现:当AT结构域原带有负电的氨基酸突变为中性氨基酸, AT结构域的活性受到影响, 只具有较小的活性; 原氨基酸突变为相反电荷的氨基酸, 即带正点的氨基酸, 则AT结构域的活性减少为原来活性的1/2倍.这进一步说明AT与ACP结构域之间存在静电作用.
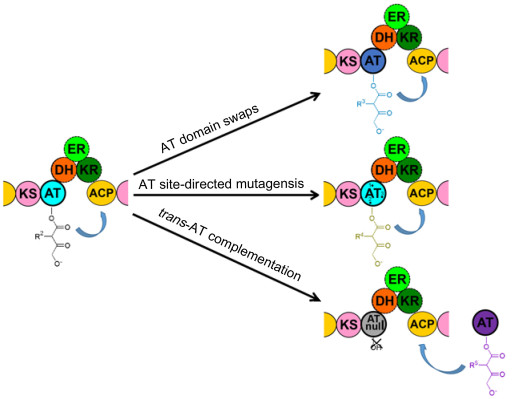
组合生物合成是利用基因工程技术, 有目的地改变天然产物的生物合成途径以期得到新结构产物, 即PKS系统中各个模块和结构域均有一定活性, 可通过人工设计替换或重组等方法在模块和结构域这两个水平上改造, 从而产生新聚酮化合物[46]. PKS中AT结构域因能转运各种起始和延伸单元, 与PKS中其它结构域相比专一性差, 根据底物结合位点易于预测其转运的延伸单元等特点而被人们关注并对此结构域进行组合生物合成研究.目前对AT结构域进行的组合生物合成技术有AT替换、AT点突变和trans-AT互补.本文总结近年来这三种技术应用的案例, 分析和阐述这三种技术的优缺点和面临的问题.
AT结构域替换, 顾名思义是两者氨基酸序列和结构同源但来源不同的AT结构域进行替换, 进而得到杂合模块或PKS, 从而渗入新起始或延伸单元到主链中产生新型聚酮化合物.大部分AT结构域替换的主要操作是利用异源AT结构域两侧保守位点的限制性内切酶酶切并克隆异源片段, 随后将克隆得到的异源片段替换本身AT结构域, 从而建立杂合模块的过程[47](图 6).尽管目前关于AT组合生物合成成功改造获得目的产物的报道较少, 但AT结构域的替换是这三种方法中最大程度上改变AT结构域底物选择性并且也是获得新产物成功率较高的方法.
首次成功利用AT结构域替换得到新型“人工天然产物”的是Leadlay课题组. Leadlay等[48]尝试将DEBS1-TE PKS中转运MM单元的AT1替换为雷帕霉素PKS转运M单元的AT2.突变株的发酵产物中, 除检测到两种本身次级代谢主产物外, 还检测到比主产物缺少1个甲基的两种衍生物.这一技术的利用和新颖聚酮化合物的产生为之后AT结构域成功替换得到“人工天然产物”奠定了理论基础和技术方法.
随后, 关于AT结构域替换技术的文章相继被报道.如将DEBS模块4中转运MM单元的AT替换为尼达霉素PKS模块5中转运EM单元的AT.突变株的发酵产物除原主次级代谢产物红霉素A外, 还存在少量红霉素A的衍生物6-去甲基-6-乙基红霉素A, 并且新衍生物具有抗真菌和细菌的活性[49].将格尔德霉素PKS中转运MM/MeO单元的AT结构域成功替换为雷帕霉素PKS中转运M的AT结构域, 这两者结构域替换产生的少量格尔德霉素同系物不仅对蛋白药物靶点Hsp90具有原有抗生素的亲和力, 且是原有抗生素亲和力的4倍[50].将泰乐菌素PKS转运MM单元的AT6替换为DEBS中转运相同单元的AT1. HPLC检测突变菌株的发酵产物, 发现其内仍含有泰乐菌素[51].上述利用AT结构域替换产生新化合物的过程中, 还伴随产生本身化合物的例子说明底物选择性不仅受同源替换AT结构域的影响, 也受到PKS中其他结构域或因素的影响.利用雷帕霉素中转运M的AT结构域替换DEBS转运MM单元的AT结构域的方法产生的突变菌株能产生少量的次级代谢去甲基聚酮化合物同系物——去甲基红霉素B或去甲基-6-脱氧红霉素B[52], 说明这两类新化合物的产生受到线性PKS中AT结构域所处的位置影响. Keasling和Yuzawa课题组[53]通过单个埃博霉素PKS AT4结构域、AT4及其携带单侧连接部分、AT4及其携带两侧连接部分来替换DEBS AT6的酶活动力学实验, 分析表明埃博霉素PKS AT4结构域是替换DEBS AT6的最佳选择片段.这说明AT结构域的成功替换取决于其结构域边界的确定和该结构域与如KS, ACP等其他结构域之间足够长的连接部分.
尽管上述很多例子证实AT结构域的替换可改变底物选择性, 从而产生新衍生物.但大部分案例是以降低自身发酵主产物的产量或减少主效价来得到少量的新型聚酮化合物.同时很多文献也报道有的AT结构域替换非常困难.为探寻或解决AT结构域替换的瓶颈问题, Khosla课题组[54]分析野生型和AT结构域替换后的杂合PKS系统中AT酰化和KS介导缩合的动力学反应.酶活动力学实验结果发现杂合模块中的缩合反应速率是野生型缩合反应速率的1/10, 并且Khosla课题组利用纯化和限制性蛋白酶水解野生型和杂合PKS蛋白的方法, 发现与野生型PKS蛋白相比, 杂合PKS蛋白的表达量低, 且杂合PKS水解后的蛋白条带与野生型蛋白条带不一致.这说明杂合PKS的蛋白稳定性差, 且具有不同的蛋白构型.上述结果推测在杂合体系中, AT结构域本身氨基酸之间和KS、ACP结构域间形成的分子间作用力不同于野生型分子间作用力来维持自身稳定的二聚体形式和与其他结构域间的相互作用, 而是转移了本身氨基酸之间和其他结构域之间相互作用力, 从而改变蛋白质稳定的方向.总的来说, AT结构域的替换不仅需要考虑单个AT结构域的边界及其与其他结构域之间的连接区域, 而且需要考虑其他结构域乃至整个PKS对底物选择性的影响.
尽管AT结构域的替换有很多成功例子, 但有更多杂合PKS体系不产生预期产物或明显降低产物活性的例子.这严重限制了非天然或其他延伸单元的渗入.同时, 随着DEBS中[KS3][AT3][42]和[KS5][AT5][43]3D蛋白晶体结构的首次解析, 比对转运M-或MM-CoA的绝大多数AT结构域的同源序列, 人们发现了AT结构域的催化位点(GHSxL)及其附近的氨基酸序列和底物结合位点(xxxH)对底物的选择性起不同程度的作用.这为之后出现的AT点突变技术指明了方向. AT点突变技术不仅对AT结构域的影响最小, 既保留AT结构域自身环境, 且排除了AT结构域替换中结构域之间上下游传递的相互作用和结构域之间相互作用等因素[1, 47, 55].接着, 越来越多转运其他延伸单元的底物结合位点模序被发现, 如FK506 PKS中转运AM单元的氨基酸模序CPTH[40]和转运MeO单元的HAGH[1]; epothilone PKS中同时转运M和MM的AT模序为HASH[56].这扩大了AT结构域点突变的组合生物合成范围.
AT点突变是通过AT点突变饱和突变库的发酵筛选, 或通过催化位点及其附近氨基酸和结合底物模序的改变和发酵筛选得到新化合物的技术[1, 47](图 6). DEBS中特异性识别MM单元的AT1, AT4和AT6上的结合底物模序YASH突变为特异性识别M单元的氨基酸模序HAFH.结果显示突变后AT结构域的底物选择性宽泛, 可同时识别M和MM单元, 但识别M单元的能力较低[57~59].这说明除了结合底物位点之外, AT结构域上的其他氨基酸序列也对底物的选择性起作用. Klopries课题组[60]通过量子力学/标准分子力学(QM/MM)的动力学模拟发现, DEBS AT6催化位点结合底物口袋处的氨基酸Val295能控制延伸单元侧链的大小. AT6结构域中氨基酸Val295成功突变为Ala的菌株发酵产生少量含有非天然延伸单元2-丙基丙二酰的2-炔丙基红霉素A[59, 60]. V295A能转运非天然延伸单元这一结果推测可能不是增强AT6对非天然延伸单元的选择性, 而是突变为Ala来降低AT6转运自身底物的活性, 来增强转运其他底物的可能性[59].
因为不同底物的含量是不可控的, 且目前体内技术还不能描述AT结构域底物选择性的相关动力学参数, 所以原位AT定点突变株转运底物原因存在不确定性.再加上AT结构域底物专一性的机制还不够明朗.因此随着蛋白质晶体技术的发展以及监测体内物质技术的加强, AT点突变的研究仍存在着很大的发展空间.
相关研究发现有的AT结构域替换非常困难, AT决定底物专一性的关键氨基酸不明确, 这都为trans-AT互补技术的产生埋下伏笔.同时像disorazole和黄色霉素的PKS一样, 其模块中缺少cis-AT结构域, 只存在独立的trans-AT结构域[16].尽管cis-AT和trans-AT的来源和进化方式不同, 氨基酸序列同源性较低, 但这两者的晶体立体结构较为保守.它们都含有α/β-水解酶的核心亚结构域和类铁氧还蛋白的附属亚结构域.它们的催化机制都以催化活性中心Ser-His的机制来转运底物[4, 18, 41].这也成为trans-AT结构域补充cis-AT结构域功能组合生物合成的前提.且与cis-AT结构域的功能相比, trans-AT可转运多个底物, 如黄色霉素中转运EM单元的trans-AT (KirCⅡ)也可转运丙基和烯丙基单元[61].这都为trans-AT互补技术产生新型聚酮化合物指明方向.
Trans-AT互补技术是将独立trans-AT结构域整合到无活性cis-AT结构域(AT0)的PKS中, 以trans-AT转运底物的功能补充无活性AT的功能, 从而渗入不同单元得到新聚酮化合物[1, 47](图 6). AT0是通过将其催化位点Ser突变为Ala得到的. Khosla课题组[62]通过体外生化实验发现disorazole PKS中的trans-ATDszs能补充DEBS1AT0的功能.随后体内实验也证实在缺少cis-AT结构域功能的DEBS1中, DszsAT可转运氟化丙二酰单元到ACP上, 从而合成氟化红霉素衍生物.并且可利用trans-ATDszs能同时转运延伸单位到多个ACP上的功能, 如Chang课题组[63]将trans-ATDszs整合到AT20-AT30双模块都缺失cis-AT结构域功能的DEBS中, 并筛选突变株, 进行发酵, 检测发现发酵产物中含有二氟化TKL. Wesener课题组[64]发现菌株Chromobacterium violaceum 968所产生次级代谢主产物罗米地辛(FK228)的PKS (DepBC)中缺失AT结构域, 随后将大肠中的酰基转移酶FabD1和FabD2转入菌株中, 结果显示发酵产物中含有FK228, 且其产量为0.4 mg/L.同时与野生型菌株相比, 改造后菌株对厌氧条件下的适应性更强.苔藓虫素PKS中的trans-AT能补充DEBS AT60无活性的功能[65].虽然上述内容介绍了trans-AT结构域互补技术成功的一些例子, 但关于trans-AT成功替换的作用机制还未了解, 此技术的应用研究也仍处于起步阶段.
Ⅰ型PKS的线性模块及其结构域行使功能的特点, 是生物合成聚酮化合物的内在奥秘, 其产物生物活性多样性赋予巨大的新药开发潜力和商业价值.特别是Ⅰ型AT结构域基因工程改造可以人工设计新型聚酮药物, 与传统技术筛选有价值聚酮化合物的过程相比, AT结构域的改造方法可以大幅减少繁重筛选工作、提高效率; 和化学合成技术相比, AT结构域的改造可替代化学无法合成步骤.采用AT结构域的改造产生了很多新型聚酮类化合物, 如雷帕霉素PKS的AT2替换了DEBS中AT2, AT4和AT10的突变菌株发酵检测有红霉素B同系物10-脱甲基-、4-脱甲基-和2-脱甲基-红霉素B的存在, 且这些化合物对金黄色葡萄球菌、表皮葡萄球菌、松骨链球菌和粪肠球菌具有较好的抗性[57~59]; 与格尔德霉素相比, 格尔德霉素PKS中转运MM/MeO单元的AT替换为雷帕霉素PKS中转运M单元AT结构域, 突变菌株所产生的格尔德霉素同系物对蛋白药物靶点Hsp90具有较强的亲和力[50].
尽管解析了一些AT结构域的晶体结构, 也明确了AT的催化机制与底物选择性相关的一些氨基酸, 但对AT序列中其它相对保守氨基酸的功能还未精确了解; 已报道了一些AT结构域改造成功的案例, 但AT结构域的底物选择性不仅受AT结构域自身上的氨基酸调控, 也受KS结构域、ACP结构域及与AT连接区域的影响; 另外, 包含AT、KS和ACP等结构域的整个PKS是结构复杂的生物大分子, 各个结构域间的相互作用机制十分复杂, 其上蛋白结构的微小变化可能导致催化过程发生改变或停止.以上因素阻碍了AT基因工程广泛应用于新型药物的开发, 因此对Ⅰ型PKS复合酶的催化机制需要更深入的解析.
AT结构域的基因改造技术虽然处于初级阶段, 但随着对AT结构域的序列、结构、机制等深入的理解, 以及PKS的基因操作、蛋白表达和分析等技术的利用, 有望成为新药发现的又一途径, 有利于PKS合成生物学技术的发展, 并为人类缓解微生物天然药物的耐药性作出贡献.
Dunn, B. J.; Khosla, C. J. R. Soc., Interface 2013, 10, 20130297. doi: 10.1098/rsif.2013.0297
Shen, B. Curr. Opin. Chem. Biol. 2003, 7, 285. doi: 10.1016/S1367-5931(03)00020-6
Demydchuk, Y.; Sun, Y.; Hong, H.; Staunton, J.; Spencer, J. B.; Leadlay, P. F. ChemBioChem 2008, 9, 1136. doi: 10.1002/(ISSN)1439-7633
Khosla, C.; Tang, Y.; Chen, A. Y.; Schnarr, N. A.; Cane, D. E. Annu. Rev. Biochem. 2007, 76, 195. doi: 10.1146/annurev.biochem.76.053105.093515
Bailey, C. B.; Pasman, M. E.; Keatinge-Clay, A. T. Chem. Commun. (Camb) 2016, 52, 792. doi: 10.1039/C5CC07315D
Li, S.; Lu, C.; Chang, X.; Shen, Y. Appl. Microbiol. Biotechnol. 2016, 100, 2641. doi: 10.1007/s00253-015-7127-7
Luhavaya, H.; Williams, S. R.; Hong, H.; Gonzaga de Oliveira, L.; Leadlay, P. F. ChemBioChem 2014, 15, 2081. doi: 10.1002/cbic.201402300
Liang, Z. X. Nat. Prod. Rep. 2010, 27, 499. doi: 10.1039/b908165h
Lanen, S. G. V.; Shen, B. Curr. Top. Med. Chem. 2008, 8, 448. doi: 10.2174/156802608783955656
Fu, L. F.; Tao, Y.; Jin, M. Y.; Jiang, H. Biotechnol. Lett. 2016, 38, 2015. doi: 10.1007/s10529-016-2202-4
Taguchi, C.; Taura, F.; Tamada, T.; Shoyama, Y.; Tanaka, H.; Shoyama, Y.; Kuroki, R.; Morimoto, S. Acta Crystallogr., Sect. F 2008, 64, 217. doi: 10.1107/S1744309108003795
Yu, D. Y.; Xu, F. C.; Zeng, J.; Zhan, J. X. IUBMB Life 2012, 64, 285. doi: 10.1002/iub.v64.4
Fang, W. J.; Wang, C. J.; He, Y.; Zhou, Y. L.; Peng, X. D.; Liu, S. K. Acta Pharmacol. Sin. 2018, 39, 59. doi: 10.1038/aps.2017.50
Fischbach, M. A.; Walsh, C. T. Chem. Rev. 2006, 106, 3468. doi: 10.1021/cr0503097
Kotowska, M.; Pawlik, K.; Smulczyk-Krawczyszyn, A.; Bar-tosz-Bechowski, H.; Kuczek, K. Appl. Environ. Microbiol. 2009, 75, 887. doi: 10.1128/AEM.01371-08
Musiol, E. M.; Weber, T. Med. Chem. Commun. 2012, 3, 871. doi: 10.1039/c2md20048a
Ye, Z.; Musiol, E. M.; Weber, T.; Williams, G. J. Chem. Biol. 2014, 21, 636. doi: 10.1016/j.chembiol.2014.02.019
Wong, F. T.; Jin, X.; Mathews, I. I.; Cane, D. E.; Khosla, C. Biochemistry 2011, 50, 6539. doi: 10.1021/bi200632j
Cheng, Y. Q.; Tang, G. L.; Shen, B. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3149. doi: 10.1073/pnas.0537286100
Pan, G. H.; Xu, Z. R.; Guo, Z. K.; Hindra; Ma, M.; Yang, D.; Zhou, H.; Gansemans, Y.; Zhu, X. C.; Huang, Y.; Zhao, L. X.; Jiang, Y.; Cheng, J. H.; Nieuwerburgh F. V.; Suh, J. W.; Duan, Y. W.; Shen, B. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E11131. doi: 10.1073/pnas.1716245115
Helfrich, E. J. N.; Piel, J. Nat. Prod. Rep. 2016, 33, 231. doi: 10.1039/C5NP00125K
Calderone, C. T.; Iwig, D. F.; Dorrestein, P. C.; Kelleher, N. L.; Walsh, C. T. Chem. Biol. 2007, 14, 835. doi: 10.1016/j.chembiol.2007.06.008
Butcher, R. A.; Schroeder, F. C.; Fischbach, M. A.; Straight, P. D.; Kolter, R.; Walsh, C. T.; Clardy, J. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 1506. doi: 10.1073/pnas.0610503104
Yadav, G.; Gokhale, R. S.; Mohanty, D. J. Mol. Biol. 2003, 328, 335. doi: 10.1016/S0022-2836(03)00232-8
Jiang, C.; Qi, Z.; Kang, Q.; Liu, J.; Jiang, M.; Bai, L. Angew. Chem., Int. Ed. 2015, 54, 9097. doi: 10.1002/anie.201503561
Luzhetskyy, A.; Mayer, A.; Hoffmann, J.; Pelzer, S.; Holzenkamper, M.; Schmitt, B.; Wohlert, S. E.; Vente, A.; Bechthold, A. ChemBio-Chem 2007, 8, 599. doi: 10.1002/(ISSN)1439-7633
Blauenburg, B.; Oja, T.; Klika, K. D.; Metsa-Ketela, M. ACS Chem. Biol. 2013, 8, 2377. doi: 10.1021/cb400384c
Xu, Z.; Schenk, A.; Hertweck, C. J. Am. Chem. Soc. 2007, 129, 6022. doi: 10.1021/ja069045b
Lowden, P. A.; Wilkinson, B.; Böhm, G. A.; Handa, S.; Floss, H. G.; Leadlay, P. F.; Staunton, J. Angew. Chem., Int. Ed. 2001, 40, 777. doi: 10.1002/1521-3773(20010216)40:4<>1.0.CO;2-X
Paiva, N. L.; Roberts, M. F.; Demain, A. L. J. Ind. Microbiol. 1993, 12, 423. doi: 10.1007/BF01569676
Mo, S.; Kim, D. H.; Lee, J. H.; Park, J. W.; Basnet, D. B.; Ban, Y. H.; Yoo, Y. J.; Chen, S. W.; Park, S. R.; Choi, E. A.; Kim, E.; Jin, Y. Y.; Lee, S. K.; Park, J. Y.; Liu, Y.; Lee, M. O.; Lee, K. S.; Kim, S. J.; Kim, D.; Park, B. C.; Lee, S. G.; Kwon, H. J.; Suh, J. W.; Moore, B. S.; Lim, S. K.; Yoon, Y. J. J. Am. Chem. Soc. 2011, 133, 976. doi: 10.1021/ja108399b
Kato, Y.; Bai, L. Q.; Xue, Q.; Revill, W. P.; Yu, T. W.; Floss, H. G. J. Am. Chem. Soc. 2002, 124, 5268. doi: 10.1021/ja0127483
Xia, M. L.; Huang, D.; Li, S. S.; Wen, J. P.; Jia, X. Q.; Chen Y. Biotechnol. Bioeng. 2013, 110, 2717. doi: 10.1002/bit.24941
Goranovic, D.; Kosec, G.; Mrak, P.; Fujs, S.; Horvat, J.; Kuscer, E.; Kopitar, G.; Petkovic, H. J. Biol. Chem. 2010, 285, 14292. doi: 10.1074/jbc.M109.059600
Dunn, B. J.; Cane, D. E.; Khosla, C. Biochemistry 2013, 52, 1839. doi: 10.1021/bi400185v
Marsden, A. F.; Caffrey, P.; Aparicio, J. F.; Loughran, M. S.; Staunton, J.; Leadlay, P. F. Science 1994, 263, 378. doi: 10.1126/science.8278811
Tsai, S. C.; Lu, H.; Cane, D. E.; Khosla, C.; Stroud, R. M. Bio-chemistry 2002, 41, 12598. http://www.ncbi.nlm.nih.gov/pubmed/12379102
Reeves, C. D.; Murli, S.; Ashley, G. W.; Piagentini, M.; Hutchinson, C. R.; McDaniel, R. Biochemistry 2001, 40, 15464. doi: 10.1021/bi015864r
Wang, Y. Y.; Bai, L. F.; Ran, X. X.; Jiang, X. H.; Wu, H.; Zhang, W.; Jin, M. Y.; Li, Y. Q.; Jiang, H. Protein Pept. Lett. 2015, 22, 2.
Jiang, H.; Wang, Y. Y.; Guo, Y. Y.; Shen, J. J.; Zhang, X. S.; Luo, H. D.; Ren, X. X.; Jiang, X. H.; Li, Y. Q. FEBS J. 2015, 282, 2527. doi: 10.1111/febs.13296
Liew, C. W.; Nilsson, M.; Chen, M. W.; Sun, H. H.; Cornvik, T.; Liang, Z. X.; Lescar, J. J. Biol. Chem. 2012, 287, 23203. doi: 10.1074/jbc.M112.362210
Tang, Y.; Kim, C. Y.; Mathews, I. I.; Cane, D. E.; Khosla, C. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11124. doi: 10.1073/pnas.0601924103
Tang, Y.; Chen, A. Y.; Kim, C. Y.; Cane, D. E.; Khosla, C. Chem Biol. 2007, 14, 931. doi: 10.1016/j.chembiol.2007.07.012
Li, Y.; Zhang, W.; Zhang, H.; Tian, W. Y.; Wu, L.; Wang, S. W.; Zheng, M. M.; Zhang, J. R.; Sun, C. H.; Deng, Z. X.; Sun, Y. H.; Qu, X. H.; Zhou, J. H. Angew. Chem., Int. Ed. 2018, 10.1002/anie.201802805. doi: 10.1002/anie.201802805
Caffrey, P.; Lynch, S.; Flood, E.; Finnan, S.; Oliynyk, M. Chem. Biol. 2001, 8, 713. doi: 10.1016/S1074-5521(01)00046-1
冯建飞, 周日成, 郭兴庭, 张扬, 现代农业科技, 2011, 3, 24. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=ahny201103005Feng, J. F.; Zhou, R. C.; Guo, X. T.; Zhang, Y. Mod. Agric. Sci. Technol. 2011, 3, 24(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=ahny201103005
Barajas, J. F.; Blake-Hedges, J. M.; Bailey, C. B.; Curran, S.; Keasling, J. D. Synth. Syst. Biotechnol. 2017, 2, 147. doi: 10.1016/j.synbio.2017.08.005
Oliynyk, M.; Brown, M. J.; Cortes, J.; Staunton, J.; Leadlay, P. F. Chem. Biol. 1996, 3, 833. doi: 10.1016/S1074-5521(96)90069-1
Stassi, D. L.; Kakavas, S. J.; Reynolds, K. A.; Gunawardana, G.; Swanson, S.; Zeidner, D.; Jackson, M.; Liu, H.; Buko, A.; Katz, L. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7305. doi: 10.1073/pnas.95.13.7305
Patel, K.; Piagentini, M.; Rascher, A.; Tian, Z. Q.; Buchanan, G. O.; Regentin, R.; Hu, Z.; Hutchinson, C. R.; McDaniel, R. Chem. Biol. 2004, 11, 1625. doi: 10.1016/j.chembiol.2004.09.012
Wong, F. T.; Jin, X.; Mathews, I. I.; Cane, D. E.; Khosla, C. Biochemistry 2011, 50, 6539. doi: 10.1021/bi200632j
Mcdaniel, R.; Thamchaipenet, A.; Gustafsson, C.; Fu, H.; Betlach, M.; Betlach, M.; Ashley, G. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 1846. doi: 10.1073/pnas.96.5.1846
Yuzawa, S.; Deng, K.; Wang, G.; Baidoo, E. E.; Northen, T. R.; Adams, P. D.; Katz, L.; Keasling, J. D. ACS Synth. Biol. 2017, 6, 139. doi: 10.1021/acssynbio.6b00176
Hans, M.; Hornung, A.; Dziarnowski, A.; Cane, D. E.; Khosla, C. J. Am. Chem. Soc. 2003, 125, 5366. doi: 10.1021/ja029539i
Koryakina, I.; Kasey, C.; McArthur, J. B.; Lowell, A. N.; Chemler, J. A.; Hansen, D. A.; Sherman, D. H.; Williams, G. ACS Chem. Biol. 2017, 12, 114. doi: 10.1021/acschembio.6b00732
Petković, H.; Sandmann, A.; Challis, I. R.; Hecht, H. J.; Silakowski, B.; Low, L.; Beeston, N.; Kuščer, E.; Gar-cia-Bernardo, J.; Leadlay, P. F.; Kendrew, S. G.; Wilkinson, B.; Müller R. Org. Biomol. Chem. 2008, 6, 500. doi: 10.1039/B714804F
Vecchio, F. D.; Petkovic, H.; Kendrew, S. G.; Low, L.; Wilkinson, B.; Lill, R.; Cortés, J.; Rudd, B. A. M.; Staunton, J.; Leadlay, P. F. J. Ind. Microbiol. Biotechnol. 2003, 30, 489. doi: 10.1007/s10295-003-0062-0
Ruan, X. A.; Pereda, A.; Stassi, D.; Zeidner, D.; Summers, R. G.; Jackson, M.; Shivakumar, A.; Kakavas, S.; Staver, M. J.; Donadio, S.; Katz, L. J. Bacteriol. 1997, 179, 6416. doi: 10.1128/jb.179.20.6416-6425.1997
Sundermann U.; Bravo-Rodriguez, K.; Klopries, S.; Kushnir, S.; Gomez, H.; Sanchez-Garcia, E.; Schulz, F. ACS. Chem. Biol. 2013, 8, 443. doi: 10.1021/cb300505w
Klopries, S.; Sundermann U.; Schulz, F. Beilstein J. Org. Chem. 2013, 9, 664. doi: 10.3762/bjoc.9.75
Koryakina, I.; McArthur, J.; Randall, S.; Draelos, M. M.; Musiol, E. M.; Muddiman, D. C.; Weber, T.; Williams, G. J. ACS Chem. Biol. 2013, 8, 200. doi: 10.1021/cb3003489
Dunn, B. J.; Watts, K. R.; Robbins. T.; Cane, D. E.; Khosla, C. Biochemistry 2014, 53, 379. http://www.ncbi.nlm.nih.gov/pubmed/24871074
Ad, O.; Thuronyi, B. W.; Chang, M. C. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 660. doi: 10.1073/pnas.1614196114
Wesener, S. R.; Potharla, V. Y.; Cheng, Y. Q. Appl. Environ. Microbiol. 2011, 77, 1501. doi: 10.1128/AEM.01513-10
Lopanik, N. B.; Shields, J. A.; Buchholz, T. J.; Rath, C. M.; Hothersall, J.; Haygood, M. G.; Hakansson, K.; Thomas, C. M.; Sherman, D. H. Chem. Biol. 2008, 15, 1175. doi: 10.1016/j.chembiol.2008.09.013

图 2 cis-AT和trans-AT在整个模块中的位置及其所处模块的结构域划分
Figure 2 Locations of cis-AT and trans-AT in module and their domain architectures

图 3 AT结构域转运的起始和延伸底物单元
Figure 3 Transfer of start and extender units of AT domains
The substrates of AT domains are in three forms as carboxylic acid, acyl-CoA and acyl-ACP. Their structures are shown in the form of free carboxylic acid in Figure 3

图 4 AT结构域的催化机制
Figure 4 Catalytic mechanism of AT domain
The spring structure of ACP domain was the phosphopantetheine arm

图 5 Enediyne PKS DynE8结构域与M单元复合物的晶体结构
Figure 5 Three-dimensional structure of the Enediyne PKS DynE8 covalent complex with malonate
The data was from protein data bank (PDB, ID: 4AMP). (A) Overall structure of ATDynE8 with malonate. The motifs GHSLG and HAFH in α/β-hydrolase-like core domain were colored red and orange. (B) The catalytic pocket of ATDynE8 domain with malonate. Ser in motif GHSLG and His in motif HAFH, malonate, oxygen and hydrogen atoms were colored green, yellow, red and blue. Hydrogen bonds were shown by the yellow dotted lines

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

 下载:
下载: