引用本文:
李遥, 罗三中. 基于实验数据和反应机理的手性催化剂理性设计[J]. 有机化学,
2018, 38(9): 2363-2376.
doi:
10.6023/cjoc201806013 Citation:
Li Yao, Luo Sanzhong. Rational Design of Chiral Catalysts Based on Experimental Data and Reaction Mechanism[J]. Chinese Journal of Organic Chemistry,
2018, 38(9): 2363-2376.
doi:
10.6023/cjoc201806013
Received Date:
09 June 2018 Revised Date:
10 July 2018 Available Online:
01 September 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21672217, 21390400)
Abstract:
Asymmetric catalysis is the most efficient chiral synthesis strategy. Chemists have already developed a variety of catalysts to achieve many asymmetric transformations. However, most of the deveoped chiral catalysts and the asymmetric catalytic reactions were developed on the basis of trios-errors approaches involving massive random screening. How to effectively obtain catalysts with higher activity and selectivity is still a challenge. In recent years, the rapid development of physical organic chemistry and computational chemistry has greatly facilitated the study of the reaction mechanism and the origin of selectivity, setting basis for rational catalyst design and evolution. This review will briefly introduce some representative works on the rational design of chiral catalysts in recent years, including rational design based on structure-activity relationship analysis, rational design based on reaction mechanism research, and computational design of enzymes.
(a) Hutt, A. J. In Smith and Williams' Introduction to the Principles of Drug Design and Action, Ed.: Smith, H. J.; CRC Press, Taylor & Francis, Boca Raton, FL, 2006. (b) Garrison, A. W. Environ. Sci. Technol. 2006, 40, 16. (c) Kitzerow, H.-S.; Bahr, C. Chirality in Liquid Crystals, Springer-Verlag, New York, 2001.
[2]
(a) Hembury, G. A.; Borovkov, V. V.; Inoue, Y. Chem. Rev. 2008, 108, 1. (b) Tsukamoto, M.; Kagan, H. B. Adv. Synth. Catal. 2002, 344, 453. (c) Okamoto, Y.; Yashima, E. Angew. Chem., Int. Ed. 1998, 37, 1020. (d) Knochel, P.; Singer, R. D. Chem. Rev. 1993, 93, 2117. (e) Zhou, Q.-L. Privileged Chiral Ligands and Catalysts, Wiley-VCH, Weinheim, Germany, 2011.
[3]
丁奎岭, 范青华, 不对称催化新概念与新方法, 化学工业出版社, 北京, 2009.Ding, K. L.; Fan, Q. H. Asymmetric Catalysis:New Concepts and Methods, Chemical Industry Press, Beijing, 2009(in Chinese).
[4]
(a) Chen, Y.; Yekta, S.; Yudin, A. K. Chem. Rev. 2003, 103, 3155. (b) Doyle, A. G.; Jacobsen E. N. Chem. Rev. 2007, 107, 5713. (c) Ooi, T.; Maruoka, K. Angew. Chem., Int. Ed. 2007, 46, 4222. (d) Hargaden, G. C.; Guiry, P. J. Chem. Rev. 2009, 109, 2505. (f) Ward T. R. Acc. Chem. Res. 2011, 44, 47. (g) Yu, Y.-N.; Xu, M.-H. Acta Chim. Sinica2017, 75, 655(in Chinese). (于月娜, 徐明华, 化学学报, 2017, 75, 655.)
[5]
(a) Harper, K. C.; Sigman, M. S. J. Org. Chem. 2013, 78, 2813. (b) Houk, K. N.; Cheong, P. H.-Y. Nature2008, 455, 309.
(a) Knowles, R. R.; Jacobsen, E. N. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20678. (b) Zuend, S. J.; Jacobsen, E. N. J. Am. Chem. Soc. 2009, 131, 15358.
[19]
Jones, R. N.; Forbes W. F.; Mueller, W. A. Can. J. Chem. 1957, 35, 504. doi: 10.1139/v57-071
[20]
Milo, A.; Bess, E. N.; Sigman, M. S. Nature 2014, 507, 210. doi: 10.1038/nature13019
[21]
(a) Knowles, R. R.; Jacobsen, E. N. Proc. Natl. Acad. Sci. U. S. A.2010, 107, 20678. (b) Neel, A. J.; Hilton, M. J.; Sigman, M. S.; Toste, F. D. Nature2017, 543, 637. (c) Toste, F. D.; Sigman, M. S.; Miller, S. J. Acc. Chem. Res. 2017, 50, 609.
[22]
Wheeler, S. E.; Houk, K. N. J. Am. Chem. Soc. 2008, 130, 10854. doi: 10.1021/ja802849j
[23]
Orlandi, M.; Coelho, J. A. S.; Hilton, M. J.; Toste, F. D.; Sigman, M. S. J. Am. Chem. Soc. 2017, 139, 6803. doi: 10.1021/jacs.7b02311
[24]
Orlandi, M.; Hilton, M. J.; Yamamoto, E.; Toste, F. D.; Sigman, M. S. J. Am. Chem. Soc. 2017, 139, 12688. doi: 10.1021/jacs.7b06917
Yang, C.; Wang, J.; Liu, Y.; Ni, X.; Li, X.; Cheng, J. P. Chem.-Eur. J. 2017, 23, 5488. doi: 10.1002/chem.v23.23
[33]
Belokon, Y. N.; Green, B.; Ikonnikov, N. S.; Larichev, V. S.; Lokshin, B. V.; Moscalenko, M. A.; North, M.; Orizu, C.; Peregudov, A. S; . Timofeeva, G. I. Eur. J. Org. Chem. 2000, 2655. doi: 10.1002/1099-0690(200007)2000:14<2655::AID-EJOC2655>3.0.CO;2-O
[34]
Belokon, Y. N.; Blacker, A. J.; Carta, P.; Clutterbuck, L. A.; North, M. Tetrahedron 2004, 60, 10433. doi: 10.1016/j.tet.2004.07.098
DiRocco, D. A.; Ji, Y.; Sherer, E. C.; Klapars, A.; Reibarkh, M.; Dropinski, J.; Mathew, R.; Maligres, P.; Hyde, A. M.; Limanto, J.; Brunskill, A.; Ruck, R. T.; Campeau, L.-C.; Davies, I. W. Science 2017, 356, 426. doi: 10.1126/science.aam7936
[37]
Mitsumori, S.; Zhang, H.; Cheong, P. H.-Y.; Houk, K. N.; Tanaka, F.; Barbas, C. F. J. Am. Chem. Soc. 2006, 128, 1040. doi: 10.1021/ja056984f
[38]
Cheong, P. H.-Y.; Zhang, H.; Thayumanavan, R.; Tanaka, F.; Houk, K. N.; Barbas, C. F. Org. Lett. 2006, 8, 811. doi: 10.1021/ol052861o
[39]
Jang, K. P.; Hutson, G. E.; Johnston, R. C.; McCusker, E. O.; Cheong, P. H.-Y.; Scheidt, K. A. J. Am. Chem. Soc. 2014, 136, 76. doi: 10.1021/ja410932t
[40]
Rooks, B. J.; Haas, M. R.; Sepúlveda, D.; Lu, T.; Wheeler, S. E. ACS Catal. 2015, 5, 272. doi: 10.1021/cs5012553
[41]
Doney, A. C.; Rooks, B. J.; Lu, T.; Wheeler, S. E. ACS Catal. 2016, 6, 7948. doi: 10.1021/acscatal.6b02366
[42]
Guan, Y.; Wheeler, S. E. Angew. Chem., Int. Ed. 2017, 56, 9101. doi: 10.1002/anie.201704663
[43]
Gerosa, G. G.; Spanevello, R. A.; Suárez, A. G.; Sarotti, A. M. J. Org. Chem. 2015, 80, 7626. doi: 10.1021/acs.joc.5b01214
[44]
Straker, R. N.; Peng, Q.; Mekareeya, A.; Paton, R. S.; Anderson, E. A. Nat. Commun. 2016, 7, 10109. doi: 10.1038/ncomms10109
[45]
Burrows, L. C.; Jesikiewicz, L. T.; Lu, G.; Geib, S. J.; Liu, P.; Brummond, K. M. J. Am. Chem. Soc. 2017, 139, 15022. doi: 10.1021/jacs.7b07121
[46]
Daubignard, J.; Detz, R. J.; Jans, A. C. H.; Bruin, B. de; Reek, J. N. H. Angew. Chem., Int. Ed. 2017, 56, 13056. doi: 10.1002/anie.201707670
Siegel, J. B.; Zanghellini, A.; Lovick, H. M.; Kiss, G.; Lambert, A. R.; Clair, J. L. S.; Gallaher, J. L.; Hilvert, D.; Gelb, M. H.; Stoddard, B. L.; Houk, K. N.; Michael, F. E.; Baker, D. Science 2010, 329, 309. doi: 10.1126/science.1190239
[58]
Segler, M. H. S.; Preuss, M.; Waller, M. P. Nature 2018, 555, 604. doi: 10.1038/nature25978
[59]
Ahneman, D. T.; Estrada, J. G.; Lin, S.; Dreher, S. D.; Doyle, A. G. Science 2018, 360, 186. doi: 10.1126/science.aar5169
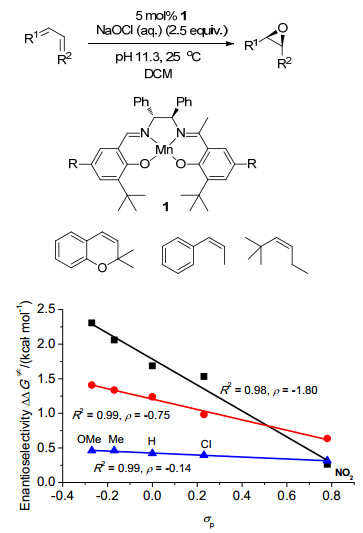
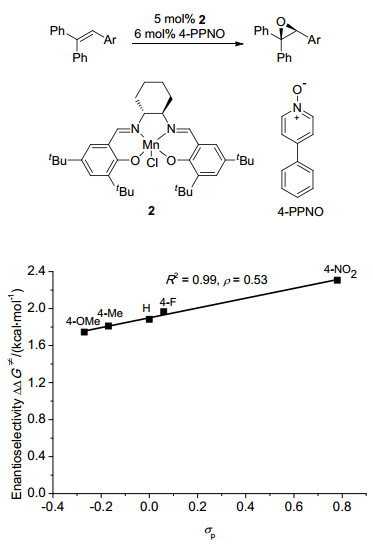
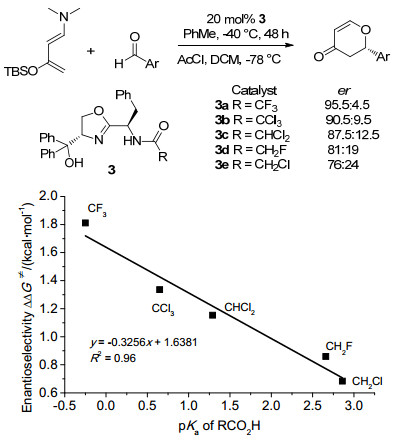
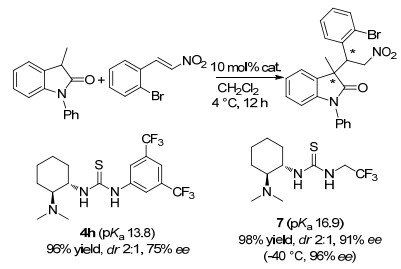
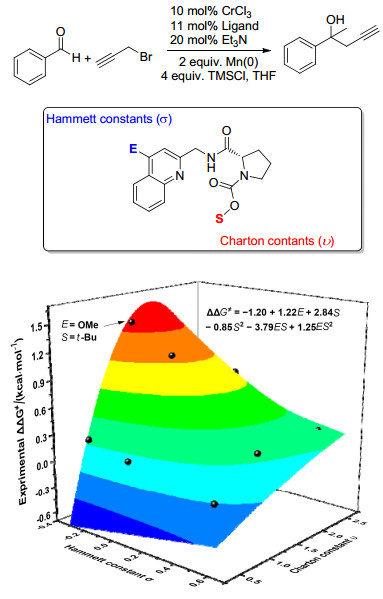
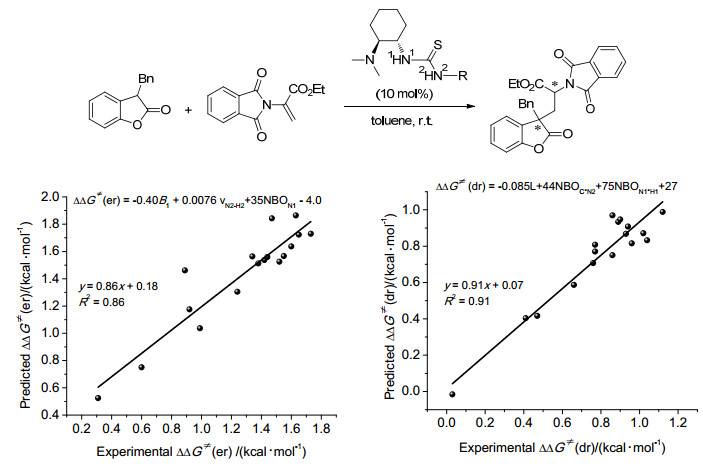
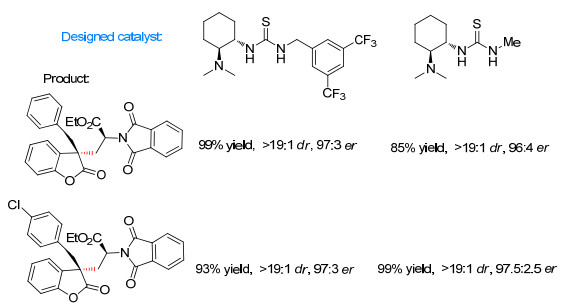
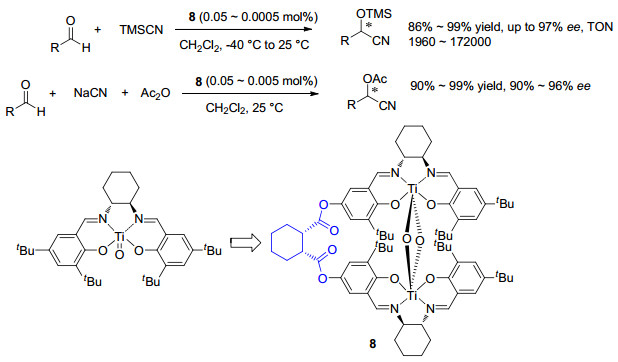
图 1
环氧化对映选择性与催化剂Hammett常数相关
Figure 1
Correlations of Enantioselectivity with Hammett constant of catalyst in asymmetric epoxidation≠

 下载:
下载:


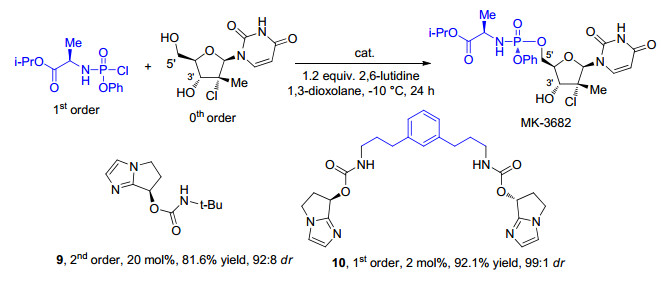
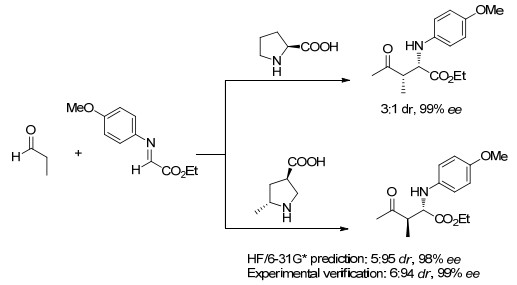
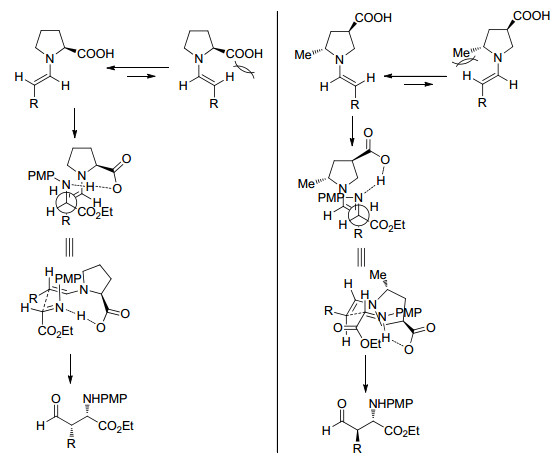
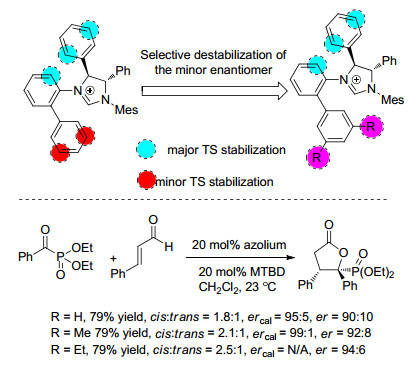

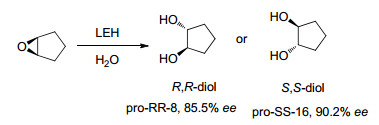
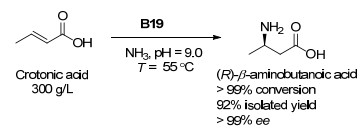
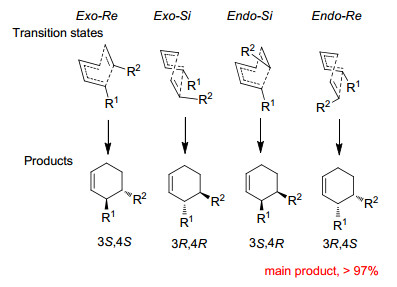

 下载:
下载:



























 下载:
下载:
