表 1
羰基还原酶在重大药物品种的工业化应用
Table 1.
Pharmaceutical drugs applied with carbonyl reductases in industry

不对称催化包括三种类型:金属配体催化、有机小分子催化和生物催化.生物催化又称为生物转化, 是指利用酶或有机体(细胞、细胞器等)作为催化剂实现化学转化的过程[1].在多样的生物催化剂中, 水解酶、氧化还原酶和转移酶应用较为广泛.据统计, 水解酶使用占比约60%, 氧化还原酶类使用占比约20%[2].羰基还原酶(Carbonyl Reductase CRED), 又称酮还(Ketoredutase KRED)或者醇脱氢酶(Alcohol Dehydrogenase, ADH), 属于氧化还原酶类, 广泛存在于细菌、真菌、酵母和动植物体内.在辅酶NADH或NADPH条件下, 羰基还原酶能将酮或醛转化成醇类化合物[3].由于辅酶价格昂贵, 研究人员成功开发了高效且低成本的辅酶再生方法, 以减少辅酶用量.工业上常利用醇脱氢酶/异丙醇[4]、甲酸脱氢酶/甲酸铵[5]或葡萄糖脱氢酶/葡萄糖[6]三种组合方法实现辅酶NAD(P)H的循环.特别地, 对于醇脱氢酶/异丙醇体系, 部分醇脱氢酶具有双向催化特征:一是催化还原羰基; 二是催化氧化异丙醇脱氢, 实现辅酶再生[7], 单酶即可实现双酶的催化功能.近些年, 利用整细胞或分离酶催化该类还原反应已成功应用于一系列工业转化过程中[8~19](表 1和图 1).
 下载:
导出CSV
下载:
导出CSV
羰基还原酶是一种实用的能够实现不对称还原的生物催化剂, 因其具有化学、区域与高立体选择性的特点, 正逐渐成为合成手性醇的主要方法之一[20].羰基还原酶应用于工业化生产的生物转化案例, 其特征主要表现为底物浓度、转化率、手性纯度、酶的用量和反应时间等参数应达到表 2所列的数值, 如底物浓度≥100 g/L, 手性纯度≥99%[21].从生物催化剂成本考虑, 由于DNA测序与合成技术的突破, 生产酶的成本已经有大幅度的降低, 且随着生产规模的扩大, 酶的成本会进一步降低[22], 目前在酶法还原步骤中, 酶的成本在50~100元/千克产品(如表 3).随着新的羰基还原酶持续不断的涌现, 为有机合成专家提供了丰富的手性催化剂工具箱, 同时越来越多的合成药物学家在早期新药合成工艺开发的阶段, 已经尝试将生物催化反应应用于路线的设计中[23, 24].
下载:
导出CSV
| 工艺参数 | 数值 |
| 底物浓度 | ≥100 g/L |
| 转化率 | ≥98% |
| 手性纯度 | ≥99% |
| 反应时间 | ≤24 h |
| 酶用量 | ≤5 g/L |
下载:
导出CSV
| 工业规模 | 产品成本/(元•kg-1) | 酶成本/(元•kg-1) |
| 药物 | >760 | 76 |
| 精细化工 | >114 | 11.4 |
| 特殊化学品 | 38 | 1.9 |
| 大宗产品 | 7.6 | 0.38 |
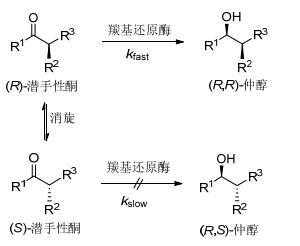
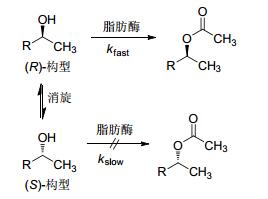
羰基还原酶在动态动力学拆分中应用的案例多散落在不同的文献, 很少有研究者对其进行梳理与归纳, 本文以此为切入点对其进行综述.为便于理解, 首先介绍动力学拆分(KR)与动态动力学拆分(DKR)的基本概念.以脂肪酶拆分仲醇制备单一异构体为例加以解释动力学拆分[25].动力学拆分是指在脂肪酶的催化下, 仲醇中的一个对映体发生成酯反应, 而另一个对映异构体不参与或少量参与反应, 其理论收率为50%, 而不需要的异构体只能丢弃或通过消旋化方法进行回收(Eq. 1).动态动力学拆分能够克服上述不足[26~28], 其可以对仲醇进行成酯拆分的同时, 实现对不需要的异构体进行原位消旋, 拆分与消旋的过程在同一条件下进行, 该拆分理论收率为100% (Scheme 1).消旋化方法根据底物结构的不同而有所差异, 主要有化学法[29]和生物法两种[30].动态动力学拆分在传统的化学拆分中, 已有大量案例报道, 如将其用于抗凝药氯吡格雷关键中间体(R)-邻氯苯甘氨酸的合成案例中[31, 32].
|
|
(1) |
上述介绍的动态动力学拆分, 不论是使用有机拆分剂还是生物催化剂, 均是制备具有单一手性中心化合物的方法, 是否可以通过一步反应同时制备含两个或两个以上手性中心的化合物?在金属配体参与催化的动态动力学拆分中, 已有大量相关案例报道[33~35].而通过整细胞或分离酶参与的动态动力学拆分案例也有少量报道, 最早可追溯至1976年, 研究者使用面包酵母还原氨基酮类底物, 依底物结构不同, 整细胞表现出不同的立体选择性.随后研究者陆续报道, 通过使用含有羰基还原酶的野生菌对类似反应进行研究, 对于部分底物羰基还原酶显示出良好的立体选择性[36].随后的二十年间, 类似的研究案例报道较少, 推测主要有三方面的原因:一是该方法对酶催化的立体选择性的特性要求高, 符合条件的酶难以直接从自然界获得; 二是受限于基因测序与合成技术的发展滞后, 酶的筛选与分离表达研究工作进展缓慢, 导致供研究的酶的类别较少; 三是有机合成专家与生物催化专家缺乏有效的合作.近十年来, 由于DNA测序与合成技术的突破与快速发展, 羰基还原酶研究领域取得了长足的进步, 越来越多的野生型羰基还原酶被鉴定、分离与表达[37, 38];同时, 通过分子生物学、计算机辅助的蛋白质定向进化技术对野生型酶进行改造以适应工业化化学反应的要求已成为可能[39~41], 因此将羰基还原酶与动态动力学拆分相结合展现出了潜在的基础与应用前景.
当羰基还原酶与动态动力学拆分结合, 我们便可经一步还原反应制备含两个手性中心的醇类化合物, 其原理为:通过羰基还原酶立体选择性地还原单一构型的潜手性酮(如R-构型), 同时另一构型的潜手性酮(如S-构型)通过羰基烯醇互变或其他条件, 实现α-位手性构型的消旋, 还原与消旋在同一反应条件下进行, 实现高效制备含两个手性中心仲醇的目的(Scheme 2).此方法有两个关键点:一是羰基还原酶对单一构型的潜手性酮的立体选择性还原; 二是消旋条件与还原条件的兼容性, 后面会加以详细论述.
本文重点对羰基还原酶参与的动态动力学拆分的研究案例进行梳理, 并结合实际工作, 尝试对其研究方法进行整理与分析, 为后续研究提供借鉴.本综述主要分为两部分:应用案例和研究方法.
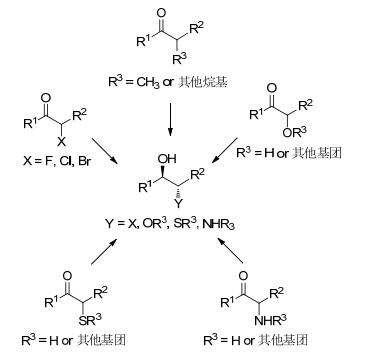
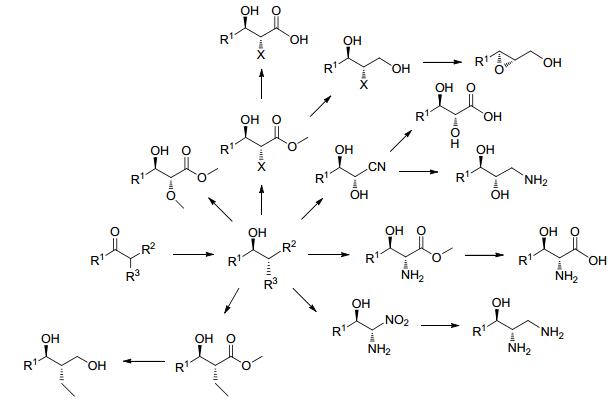
基于上述原理, 从不同底物结构出发, 应用该方法可以制备结构多样的含有多手性中心的仲醇, 包括烷烃醇、卤代醇、硫醚醇、双醇和氨基醇(Scheme 3).
当R1为非氢原子或基团, R2为酯基、氰基、硝基或其他吸电子基团, R3为烷烃基团时, 羰基还原酶催化还原可制备含两个手性中心碳的烷烃醇(Scheme 3).羰基还原酶动态不对称还原该类潜手性酮的研究案例较为丰富.
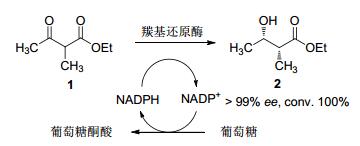
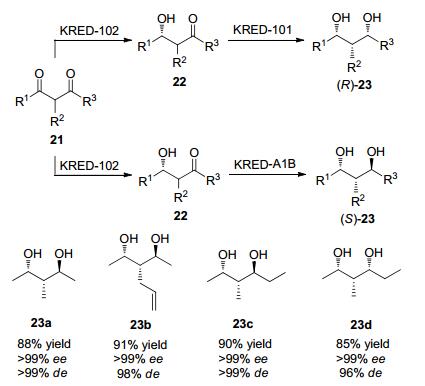
2005年Smonou及其团队[42]在对底物酮酯与双酮等底物进行立体选择性还原时, 筛选了20种分离的羰基还原酶, 当底物结构为α-甲基乙酰乙酸酯时, KRED-102表现出显著的立体选择性, ee>99%, de>99%且转化率为100%.当α位的基团变化时, 不同的羰基还原酶表现出不同的立体选择性(Scheme 4).
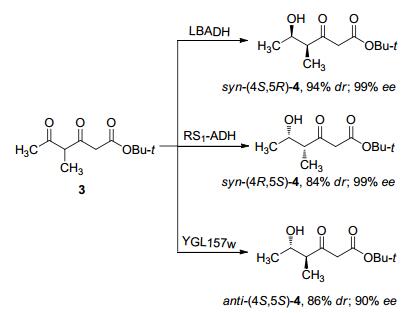
金属配体参与的不对称催化合成中, 对双酮进行区域与立体选择性还原是具有挑战性的研究领域. 2009年Lüdeke等[43]尝试开展对双酮进行羰基还原酶参与的动态动力学拆分的应用研究, 期望实现区域与立体选择性地制备含两手性中心碳的仲醇.研究者通过筛选商业化和自主构建的羰基还原酶, 对底物进行还原, 结果发现不同的酶对相同的底物可以实现定向合成四个异构体中的三个, 实现了双酮的区域选择性还原(Scheme 5).
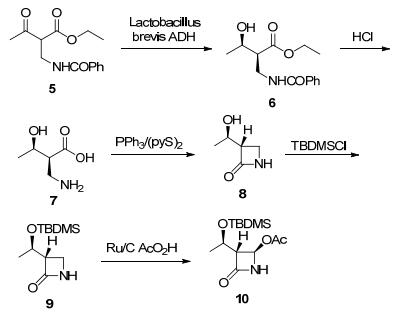
2005年Yasohara等[44]研究者在专利中报道了碳青霉烯类抗生素通用中间体4-AA的化学-酶法路线. 4-AA化学结构中含三个手性碳, 其中两个是通过动态还原动力学拆分的方法实现的.研究者经筛选发现短乳酸杆菌(Lactobacillus brevis)醇脱氢酶显示高的立体选择性, ee>99%, de=92%.随后2007年, Codexis公司报道对短乳酸杆菌(Lactobacillus brevis)醇脱氢酶进行蛋白质改造, 以提高酶的催化效率、底物浓度、溶剂、pH与温度耐受性为多目标, 经多轮进化, 最终筛选到满足工业化要求的多个序列酶.在最优实施例中使用KRED10酶还原时, 反应底物浓度高达300 g/L, 反应转化率>96%, 异丙醇浓度达50%, 生物转化一步还原形成两个手性中心, ee>99%, de>99%[45].酶法的突破为实现该路线的产业化开发提供了良好的基础(Scheme 6).
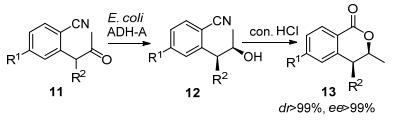
2013年Gotor及其团队[46]通过使用还原酶ADH-A, 经动态还原动力学拆分制得手性醇, 后经氰基水解, 关环成酯制备苯并内酯环, 其中还原反应具有显著的立体选择性, ee>99%, dr>99%.该研究对羰基还原酶参与的动态动力学拆分技术提供了较完整的研究思路, 研究人员初始时发现, 该还原反应仅表现为动力学拆分的特点, 即收率<50%, 随后研究者调整思路, 尝试对不需要异构体潜手性酮进行消旋化研究, 由于底物结构中羰基α-H表现一定酸性, 经筛选发现向反应体系中加入1%~3%三乙胺, pH约为8时, 可实现其消旋, 最后将消旋化与还原在同一反应体系下进行, 实现该步的动态动力学拆分(Scheme 7).
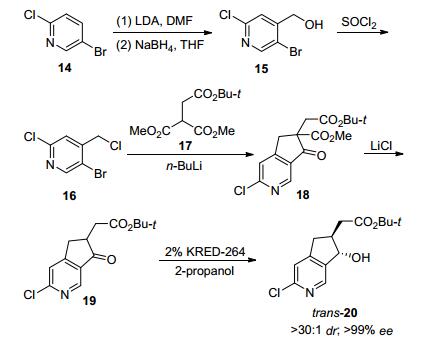
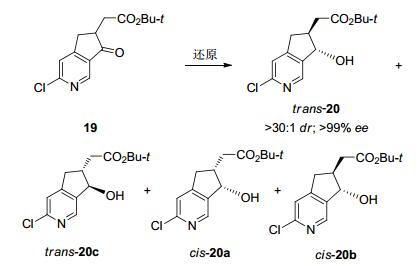
同样地, 2016年由Merck公司、Codexis公司与上海合全公司组成的研究团队, 为大量供应在研药物MK-8666三期临床实验的使用, 成功开发了一条羰基还原酶参与的动态动力学拆分路线.该路线主要集中对中间体19进行还原研究, 研究者希望通过动态动力学拆分制备两个手性碳的仲醇trans-20 (Schemes 8、9).底物19仅具有一个酮羰基, 其α-H的酸性预测值为pKa18.5, 需要在较强的碱性条件才能实现消旋, 所以要求酶对碱性条件的耐受性更强.研究者对金属配体与羰基还原酶进行了筛选, 金属配体催化条件下主要获得顺式产物, 而不同的酶可制备不同构型的顺或反式产物.初筛中发现KRED-208表现一定选择性trans-20/cis- 20a=7/3, 后经六轮蛋白质定向进化, 获得具有显著立体选择性与高催化活性的羰基还原KRED264, 转化的底物浓度提至50 g/L、立体选择性提高至ee>99%、dr>30:1, 反应在pH=9, 50 ℃条件下便可实现底物的消旋[47].
Dimitris团队[48]报道了羰基还原酶对双酮羰基化合物进行区域与立体选择性还原制备含3个手性中心碳的双醇的方法.第一阶段筛选到KRED-102酶经动态动力学拆分, 区域选择性地还原一个酮羰基, 先制备两个手性中心的羟酮, 第二阶段筛选到KRED-101与KRED- A1B酶对第二个羰基进行还原形成第三个手性中心, 进而制备双醇(Scheme 10).研究者发现第一阶段的反应转化完全且无副反应发生, 所以将两个阶段在同一反应缓冲液内进行, 实现一锅法制备含三手性中心的双醇, 该法充分表现出酶催化的绿色与高效.最后研究者对反应的底物谱进行扩展, 该法表现一定的通用性.
上述案例主要围绕脂肪和芳香酮酯的还原, 对于环状的酮酯进行动态还原动力学拆分同样有相关的案例报道. Danchet等[49]开发了整细胞催化还原7、8元环酮酯的动态动力学拆分方法.作者筛选了11种野生菌, 结果表明对于7、8元环表现出不同的立体选择性.克勒克酵母菌(Kloekera magna)对于7元环表现显著的立体选择性, ee=94%, de=100% (Eq. 2);然而对于8元环, 却难以催化反应的进行.令人惊喜的是球孢白僵菌(Beauveria bassiana)与灰蓝毛霉菌(Mucorgriseocyanus)可以高立体选择性的催化底物(Eq. 3).
|
|
(2) |
|
|
(3) |
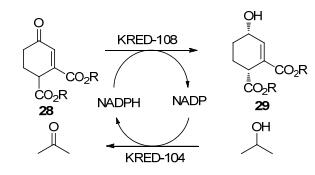
Merck公司研发人员[50]报道了α, β-不饱和酮酯的还原方法, 包括金属配体催化氢化与羰基还原酶催化.对底物28进行动态动力学拆分时, 金属配体催化氢化可以高对映选择性地制备29, 但缺乏非对映选择性.随后Kosjek团队使用羰基还原酶进行还原, 发现部分酶表现出良好的对映选择性ee>99%, 具有一定的非对映选择性de=60%.接着研究人员改变底物结构, 比较不同酶催化的非对映选择性, 发现将乙酯改为甲酯即能将非对映选择性de值由60%提高至95% (Scheme 11).该案例有两点启示:一是可以通过对α, β-不饱和酮酯的动态不对称还原制备含两个手性中心碳的烯丙醇; 二是酶与底物之间立体选择性关系既可以通过对酶的改造来实现, 也可以通过对底物结构的变化来影响.
当R1为非氢原子或基团, R2为酯基、氰基、硝基或其他吸电子基, R3为卤素时, 羰基还原酶可还原底物制备含两个手性中心碳的卤代仲醇(Scheme 3). Musa等[51]的研究工作说明可以通过理性设计与定点突变技术实现对酶的改造, 进一步扩大酶的底物谱与提高催化活性.作者对来自于嗜热厌氧乙醇杆菌(Thermoanaero- bacter ethanolicus ADH, TeSADH)进行蛋白质改造, 改变其催化口袋大小, 改变其催化的立体选择性, 筛选获得突变株W110A, 实现了对α-氯代酮衍生物不对称的动态还原, 获得ee=99%, de=84% (Eq. 4).
|
|
(4) |
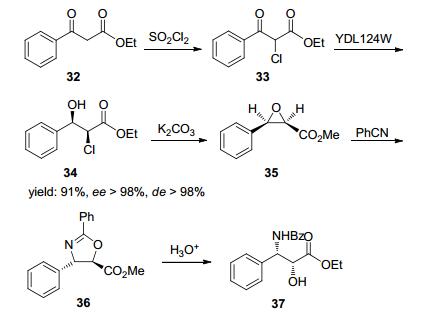
Feske等[52]报道了一条酶法制备紫杉醇侧链37的路线.首先对起始物料4进行氯代制得氯代酮酯化合物33, 然后筛选了分离的羰基还原酶, 其中YDL124W酶可高立体选择性地制得34, 收率91%, ee>98%, de>98%.由于底物与产物对酶活性有明显的抑制作用, 底物浓度仅能达到约1.3 g/L.另外文献报道反应在pH 5.6条件下进行, 说明底物易于消旋.然后化合物34经环氧化, 苯异氰酸酯关环、水解制得侧链37 (Scheme 12).该路线设计简洁, 原子经济性高.由于被还原底物的分子结构易于消旋, 如果尝试更多筛选工作或对现有酶进行改造, 有望克服现有路线中底物浓度较低的不足.
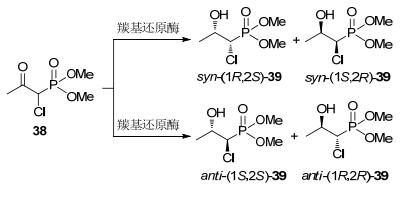
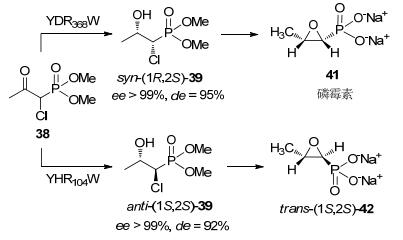
2011年Marocco等[53]报道了一条磷霉素的化学-酶法路线, 该路线关键步骤为应用羰基还原酶经动态动力学拆分制备化合物syn-(1R, 2S)-39 (Scheme 13).研究人员筛选了20种羰基还原酶, 其中三种酶YBR149W、YHR104W、YDR368W表现出高的立体选择性, ee>99%, de>90%, 转化率>98%, 底物浓度约1 g/L.反应条件pH 7.0与温度28 ℃, 表明化合物38易于消旋.使用YDR368W制备syn-(1R, 2S)-39产物后, 再经两步反应制备磷霉素.较现有传统拆分工艺, 该路线简洁, 有一定应用开发前景(Scheme 14).
下载:
导出CSV
| No. | 催化剂 | syn-(1R, 2S)-39 | anti-(1S, 2S)-39 |
| 1 | YOL151w | ND | 64% de |
| 2 | YGL157w | 12% de | ND |
| 3 | YGL039w | ND | 56% de |
| 4 | YNL331c | 79% de | ND |
| 5 | YOR120w | 77% de | ND |
| 6 | YBR149w | 95% de | ND |
| 7 | YHR104w | ND | 92% de |
| 8 | YDR368w | 95% de | ND |
| 9 | 面包酵母 | 18% de | ND |
当R1为非氢原子或基团, R2为酯基、氰基、硝基或其他吸电子基, R3为羟基、烷氧基或烷巯基时, 羰基还原酶可以通过立体选择性的还原该类潜手性酮制备含两个手性中心碳的双醇或醇醚衍生物(Scheme 3).由于羟基酮底物较难制备, 因此现有文献报道案例较少.
Perrone等[54]研究者对底物芳醚酮酯类化合物43进行动态动力学拆分研究.研究人员筛选了5种野生菌, 进行整细胞催化, 发现静息细胞比生长细胞的催化效率高.作者幸运的筛选到野生菌马克思克鲁维酵母(Kluy- veromyces marxianus)可以高立体选择性(ee=97%, de>99%)制备syn-(2R, 3S)-44 (Eq. 5).
|
|
(5) |
当R1为非氢原子或基团, R2为酯基、氰基、硝基或其他吸电子基, R3为氨基或其衍生基团时, 羰基还原酶可以通过立体选择性的还原该类潜手性酮制备含两个手性中心碳的氨基醇及其衍生物(Scheme 3).
1993年Ramesh团队[55]在合成紫杉醇侧链时, 通过对含有羰基还原酶的微生物进行大量筛选, 最终获得一株具有良好立体选择性的多型汉逊酵母(Hansenula polymorpha) SC13865, ee>99%.然而对于顺反立体异构的区域选择性(de>50%)尚不理想(Eq. 6).分析原因可能是微生物体内存在多种羰基还原酶, 干扰了彼此的立体选择性, 如果进行单酶的离体表达, 还原效果可能会有改观.
|
|
(6) |
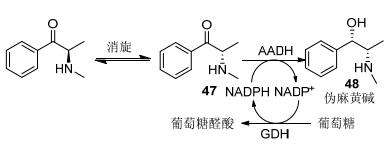
Kataoka等[56, 57]研究人员设计了一条伪麻黄碱的生物制备方法, 即以氨基酮盐酸盐为底物, 广泛筛选羰基还原酶, 经动态动力学拆分高立体选择性的制备伪麻黄碱.该团队2006年从圆平球菌(Rhodococcus erythropolis) MAK154筛选到氨基丙醇脱氢酶, 该羰基还原酶表现良好的立体选择性, ee>99%, de>99% (Scheme 15).然而该酶表现明显的底物与产物抑制现象, 底物浓度仅为40 g/L.随后2011年, Kataoka等[58]针对野生酶存在的缺陷, 对其开展了蛋白质改造工作, 最终筛选到催化活性更优的突变体:对底物与产物的耐受性显著提高; 底物浓度可提高至100 g/L.该制备方法表现出潜在的工业化应用前景.
2013年为临床提供候选药物49时, Merck公司研发人员[59]成功开发一条对关键底物氨基酮酯进行动态动力学还原的合成路线.研究人员通过对自有酶库进行筛选获得具有潜在开发前景的羰基还原酶CDX-901.在放大过程中由于底物溶解性低的原因, 导致反应转化率降低, 随后研究者对该酶进行改造, 提高其对有机溶剂的耐受性.经过筛选获得突变体CDX-108, 转化反应底物浓度提高至100 g/L, 转化率>95%, 产物ee>99%, de>99% (Eq. 7).
|
|
(7) |
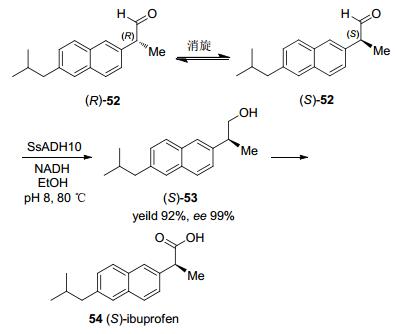
羰基还原酶能够还原醛基为羟基的研究案例较少.因为该方法不涉及手性中心的变化, 而现有的化学还原方法选择较多, 如硼系列的还原体系与氢气还原体系. Friest等[60]报道了可以高立体选择性的还原醛基的醇脱氢酶SsADH10, 该酶最大耐受温度高达80 ℃.将该脱氢酶与动态动力学拆分结合, 为制备非甾体抗炎药(S)-布洛芬提供了新颖的研究思路(Scheme 16).
由前述案例分析知, 羰基还原酶参与的动态动力学拆分包括三部分:一是羰基还原酶对需要的单一手性异构体潜手性酮的立体选择性还原; 二是对不需要的单一手性异构体潜手性酮的消旋化; 三是还原与消旋两部分的兼容与平衡.
首先, 在确定并完成底物的合成后, 需要去寻找满足研究要求的羰基还原酶.研发早期主要有三条途径:一是阅读分析文献, 根据底物相似性, 通过克隆表达, 自制羰基还原酶, 供筛选使用; 二是筛选已建立的羰基还原酶库, 该库可以为内部积累亦可为外部资源, 包括拥有商业化酶库的企业或高校团队, 三是从自然界中以目标底物为模板, 建立筛选模型, 筛选含羰基还原酶的野生菌.羰基还原酶已有大量的工业化案例报道, 企业酶库中多为已实现产业化品种的酶, 工业化前景较好, 可优先尝试.若将羰基还原酶应用于动态动力学拆分, 须对酶的特性有两点要求:一是高的对映选择性(ee>99%); 二是高的非对映选择性(de>95%).对映选择性易于实现, 非对映选择性要求酶在还原羰基的同时对潜手性酮α-位的手性异构体R或S构型具有相对映的立体选择性.因此, 在前三条途径无法筛选到同时满足上述两种立体选择性的情况下, 研究者可以优先选择具有高对映选择性(ee>99%), 但具有部分非对映选择性(如de>80%)的羰基还原酶作为初始模板, 通过蛋白质改造的手段, 对酶进行再设计, 建立突变体库, 进行有效筛选, 以期获得满足立体选择性要求的酶.目前, 该类技术已经有大量的研究案例报道, 如上述案例卤代醇[51]和氨基醇[59]均涉及到对酶进行改造.
其次, 对不需要异构体潜手性酮的消旋化研究. 2013年Gotor团队[46]的研究思路就涉及该部分内容(Scheme 7).研究者通过筛选不同碱性条件对不需要的潜手性酮进行消旋, 发现在1%~3%三乙胺条件下, 可将底物进行消旋.上述案例消旋化的原理均在碱性条件下, 通过羰基烯醇互变对α-位手性中心进行消旋化, 所以α-H的酸性强弱就直接决定了消旋化的难易. Applegate等[62]总结动态还原动力学拆分案例, 发现通常α-H的pKa值在7.5~12.5 (H2O或DMSO)易于消旋化.但是, 这并非教条, 2016年Merck团队[47]开发的路线中, 底物预测的pKa值为18, 仍可在pH 9的条件下实现动态动力学拆分(Scheme 9).碱性消旋在前述案例中占绝大多数.但其他条件如酸性条件、加热、金属离子催化或通过氨基与醛形成席夫碱进行消旋, 上述各法均值得探究.
最后将羰基还原酶催化条件与消旋化条件相结合, 实现两者反应速率的平衡.两者结合过程中, 可能会出现一种不利情况:即消旋速度显著慢于还原的速度, 此时容易导致羰基还原酶识别不需要的潜手性酮进行还原, 从而降低非对映选择性.因此当非对映选择性在多批次反应中存在差异时, 可以通过调整消旋的速度, 以与酶还原速率相匹配.这样便可在同一反应容器内进行, 同时高效构建两个手性中心碳.
本文主要对羰基还原酶参与的动态动力学拆分的应用案例进行了梳理与分析, 介绍该技术的应用进展, 并尝试提供一种可行的研究思路:首先是酶的筛选与改造; 其次是消旋化研究; 最后实现两者反应速率的平衡.将该方法应用于不同的底物可制备不同类型的手性化合物, 如具有多手性中心的氨基醇、双醇、三醇、手性烷烃醇及羟基氨基酸等(Scheme 17), 该法在有机合成官能团转化中表现良好的应用前景.
羰基还原酶催化的动态动力学拆分与金属配体催化的动态动力学拆分形成优势互补, 但具有自身四个特点:羰基还原酶具有一定成本优势; 转化反应在水中进行; 反应条件温和; 催化剂的选择范围广, 商业化的羰基还原酶种类丰富以及酶的改造手段逐渐成熟.一经筛选到合适的羰基还原酶, 便可快速地实现酶转化工艺的产业化.将生物催化与传统有机化学反应相结合, 可以更丰富有机反应的类型, 并产生更为巧妙的化学结果.如对于本文动态动力学拆分方法, 若将酶的范围由羰基还原酶扩展至转氨酶, 并与动态动力学拆分结合, 便可经一步转氨反应, 制备含两个手性中心碳的仲胺, 已有相关案例报道[63], 该法是传统金属配体催化难以实现的, 同理我们可以依据不同化学反应特点, 与不同种类的生物催化剂进行有效结合, 因此有机合成专家可以更加广泛的视角去设计适宜工业化生产的合成路线.要实现化学合成与酶催化的有效结合, 首先需要有机合成专家建立基于生物催化基元反应的逆合成分析意识, 其次需要跨学科合作, 包括生物催化、有机合成以及蛋白改造等领域.随着新酶数量与种类的不断增长, 酶基因挖掘技术的进步与蛋白改造技术的逐渐成熟, 生物催化剂将会与金属配体催化剂互补协作在手性药物的制备中展现更为广泛的基础与应用价值.
林国强, 李明月, 陈耀全, 孙兴文, 陈兴滋, 手性合成-不对称反应及其应用(第四版), 科学出版社, 北京, 2010, p. 584.Lin, G.-Q.; Li, M.-Y.; Chen, Y.-Q.; Sun, X.-W.; Chen, X.-Z. Chiral Synthesis-Asymmetric Reactions and Its Application, Science Press, Beijing, 2010, p. 584 (in Chinese).
Straathof, A. J.; Panke, S.; Schmid, A. Curr. Opin. Biotechnol. 2002, 13, 548. doi: 10.1016/S0958-1669(02)00360-9
陶军华, 林国强, 安德烈亚斯•李斯, 生物催化在制药工业的应用——发现、开发与生产, 化学工业出版社, 北京, 2010, p. 3.Tao, J.-H.; Lin, G.-Q.; Liese, A. Biocatalysis for the Pharmaceutical Industry Discovery, Development and Manufaturing, Chemical Industry Press, Beijing, 2010, p. 3 (in Chinses).
Truppo, M. D.; Pollard, D.; Devine, P. Org. Lett. 2007, 9, 335. doi: 10.1021/ol0627909
Kavanagh, K. L.; Klimacek, M.; Nidetzky, B.; Wilson, D. K. Biochemistry 2002, 41, 8785. doi: 10.1021/bi025786n
Xu, Z.; Jing, K.; Liu, Y.; Cen, P. J. Ind. Microbiol. Biotechnol. 2007, 1, 83. doi: 10.1007/s10295-006-0168-2
Inoue, K.; Makino, Y.; Itoh, N. Appl. Environ. Microb. 2005, 7, 3633.
Ma, S. J.; Gruber, J.; Davis, C.; Newman, L.; Gray, D.; Wang, A.; Grate, J.; Huisman, G. W.; Sheldon, R. A. Green Chem. 2010, 12, 81. doi: 10.1039/B919115C
Liang, J.; Lalonde, J.; Borup, B.; Mitchell, V.; Mundorff, E.; Trinh, N.; Kochrekar, D. A.; Cherat, R. N.; Pai, G. G. Org. Process Res. Dev. 2010, 193.
Nurit, P.; Ayelet, M. WO 2008151324. 2008.
Bong, Y. K.; Vogel, M.; Collier, S. J.; Mitchell, V.; Mavinahalli, J. WO 2011005527, 2010.
Li, H. G.; Moncecchi, J.; Truppo, M. D. Org. Pro. Res. Dev. 2015, 19, 695. doi: 10.1021/op5003215
Savile, C.; Gruber, J. M.; Mundorff, E.; Huisman, G. W.; Collier, S. J. WO 20100252, 2009.
Isotani, K.; Kurokawa, J. J.; Itoh, N. Int. J. Mol. Sci. 2012, 13, 13542. doi: 10.3390/ijms131013542
Zheng, W.-X.; Xu, G.-C.; Huang, L.; Pan, J.; Yu, H.-L.; Xu, J.-H. Org. Lett. 2013, 19, 4917.
Zhang, F.-L.; Ni, G.-W. Chen, S.-X.; Ju, D.-W.; Tang, J.-W.; Tan, Z.-M.; Zou, J.; Guo, X.; Wang, Z.-W. CN 201611008108, 2016.
Zhang, F.-J.; Liu, Y.; Peng, X.-Q.; Guo, C.; Wu, Z.-L. Appl. Microbiol. Biotechnol. 2016, 5, 1.
Liang, J.; Jenne, S. J.; Mundorff, E.; Ching, C.; Gruber, J. M.; Krebber, A.; Huisman, G. W. WO 2009036404, 2008.
Xu, G.-P.; Wang, H.-B.; Wu, Z.-L. Process Biochem. 2016, 51, 881. doi: 10.1016/j.procbio.2016.04.008
陶军华, 林国强, 安德烈亚斯•李斯, 生物催化在制药工业的应用——发现、开发与生产, 化学工业出版社, 北京, 2010, p. 121.Tao, J.-H.; Li, G.-Q.; Liese, A. Biocatalysis for the Pharmaceutical Industry-Discovery, Development and Manufaturing, Chemical Industry Press, Beijing, 2010, p. 121 (in Chinses).
Luetz, S.; Giver, L.; Lalonde, J. Engineered Enzymes for Chemical Production. Biotechnology and Bioengineering, 2008, 4, 647.
Tufvesson, P.; Lima-Ramos, J.; Nordblad, M.; Woodley, J. M. Org. Process. Res. Dev. 2011, 15, 266. doi: 10.1021/op1002165
Xin, L. H.; Nicholas, W.; Hans, I.; Vera, J.; Zhang, H. M.; Koenig, S. G.; Gossselin, Francis. Org. Process Res. Dev. 2017, 3, 387.
Hou, X. P.; Zhang, H. P.; Chen, B. C. Guo, Z. W.; Singh, A.; Goswami, A.; Gilmore, J. L.; Sheppeck, J. E.; Dyckman, A. J. Carter, P. H.; Mathur, A. Org. Process Res. Dev. 2017, 2, 200.
Ginotra, S. K. J.; Friest, A.; Berkowitz, D. B. Org. Lett. 2012, 14, 968. doi: 10.1021/ol203088g
Pellissier, H. Adv. Synth. Catal. 2011, 353, 659. doi: 10.1002/adsc.201000751
程咏梅, 徐刚, 吴坚平, 杨丽蓉, 有机化学, 2010, 31, 1695. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract339376.shtmlCheng, Y.-M.; Xu, G.; Wu, J.-P.; Yang, L.-R. Chin. J. Org. Chem. 2010, 31, 1695 (in Chinses). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract339376.shtml
张占辉, 刘庆彬, 有机化学, 2005, 25, 780. doi: 10.3321/j.issn:0253-2786.2005.07.005Zhang, Z.-H.; Liu, Q.-B. Chin. J. Org. Chem. 2005, 25, 780 (in Chinses). doi: 10.3321/j.issn:0253-2786.2005.07.005
Larsson A. L. E.; Persson, B. A.; Bäckvall, J.-E. Angew. Chem., Int. Ed. 1997, 36, 1211. doi: 10.1002/(ISSN)1521-3773
Sakulsombat, M.; Vongvilai, P.; Ramstr m, O. Chem.-Eur. J. 2014, 20, 11322. doi: 10.1002/chem.v20.36
Lhum, M. D.; Toulouse, J. R. US 5204469, 1991.
Reddy, B. S. WO 2006003671, 2004.
Wang, X.-L.; Xu, L.-J.; Yan, L.-J.; Wang, H.-F.; Han, S.; Wu, Y.; Chen, F. Tetrahedron 2016, 72, 1787. doi: 10.1016/j.tet.2016.02.045
Sayo, N.; Satio, T.; Okeda, Y.; Nagashima, H.; Kumobayashi, H. US 4981992, 1991.
Li, X.-M.; Tao, X.-M.; Ma, X.; Li, W.-F.; Zhao, M.-M.; Xie, X.-M.; Ayad, T.; Ratovelomanana-Vidal, V.; Zhang, Z.-G. Tetrahedron 2013, 34, 7152.
Deol, B. S.; Ridley, D.; Simpson, G. Aust. J. Chem. 1976, 29, 2459. doi: 10.1071/CH9762459
Gamenara, D.; Sevane, G. A.; Mendez, P. S.; Maria, P. D. Redox Biocatalysis, Wiley, New Jewery 2013, Chapter 3.
Faber, K.; Fessner, W. D.; Turner, N. J. Biocatalysis in Organic Synthesis 2, Georg Thieme Verlag KG, New York, 2015, pp. 421~458.
Bornscheuer, U. T.; Huisman, G. W.; Kazlauskas, R. J.; Lutz, S.; Moore, J. C.; Robins, K. Nature 2012, 485, 185. doi: 10.1038/nature11117
Savile, C. K.; Janey, J. M.; Mundorff, E. C.; Moore, J. C.; Sarena, T.; Jarvis, W. R.; Colbeck, J. C.; Krebber, A.; Fleitz, F. J.; Brands, J.; Devine, P. N.; Huisman, G. W.; Hughes, G. J. Science 2010, 329, 305. doi: 10.1126/science.1188934
Zheng, M.-M.; Chen, K.-C.; Wang, R.-F.; Li, H.; Li, C.-X.; Xu, J.-H. J. Agric. Food Chem. 2017, 6, 1178.
Kalaitzakis, D.; Rozzell, J. D.; Kambourakis, S.; Smonou, L. Org. Lett. 2005, 7, 4799. doi: 10.1021/ol051166d
Lüdeke, S.; Richter, M.; Müller, M. Adv. Synth. Catal. 2009, 351, 253. doi: 10.1002/adsc.200800619
Yasohara, Y.; Yano, M.; Kawano, S.; Kizaki, N. JP 200521371, 2005.
Onorato, C.; Emily, M.; Birthe, B.; Rama, V. US 9719071, 2007.
Juan, M. S.; Busto, E.; Gotor, V.; Vicente, G. F. Org. Lett. 2013, 15, 3872. doi: 10.1021/ol401606x
Hyde, A. M.; Liu, Z. J.; Kosjek, B.; Tan, L. S.; Klapars, A.; Ashley, E. R.; Zhong, Y. L.; Alvizo, O.; Agard, N. J.; Liu, G. Q.; Gu, X. Y.; Yasuda, N.; Limanto, J.; Huffman, M. A.; Tschaen, D. M. Org. Lett. 2016, 18, 5888. doi: 10.1021/acs.orglett.6b02910
Dimitris, K.; Ioulia, S. J. Org. Chem. 2010, 4, 8659.
Danchet, S.; Bigot, C.; Buisson, D.; Azerad, R. Tetrahedron: Asymmetry 1997, 11, 1735.
Kosjek, B.; Tellers, D. M.; Biba, M.; Farr, R.; Moore, J. Tetrahedron: Asymmetry 2006, 17, 2798. doi: 10.1016/j.tetasy.2006.10.012
Musa, M. M.; Ziegelmann-Field, K. I.; Vieille, C.; Zeikus, J. G.; Phillips, R. S. J. Org. Chem. 2007, 72, 30. doi: 10.1021/jo0616097
Feske, B. D.; Kaluzna, I. A.; Stewart, J. D. J. Org. Chem. 2005, 70, 9654. doi: 10.1021/jo0516077
Marocco, C. P.; Davis, E. V.; Finnell, J. E.; Nguyen, P. H.; Mateer, S. C.; Ghiviriga, I.; Padgett, C. W.; Feske, B. D. Tetrahedron: Asymmetry. 2011, 22, 1784. doi: 10.1016/j.tetasy.2011.10.009
Perrone, M. G.; Santandrea, E.; Scilimati, A.; Syldatk, C.; Tortorella, V.; Capitellic, F.; Bertolasi, V. Tetrahedron: Asymmetry 2004, 15, 3511. doi: 10.1016/j.tetasy.2004.08.028
Patal, R.; Banerjso, A.; Howell, J. M.; Mcnameo, C. G.; Brozozowski, D.; Mirfakhras, D.; Naaduri, V.; Thottathll, J. K.; Starka, L. J. Tetrahedron: Asymmetry 1993, 9, 2069.
Kataoka, M.; Nakamura, Y.; Urano, N.; Ishige, T.; Shi, G.; Kita, S.; Sakamoto, K.; Shimizu, S. Lett. Appl. Microbiol. 2006, 43, 430. doi: 10.1111/lam.2006.43.issue-4
Kataoka, M.; Ishige, T.; Urano, N.; Nakamura, Y.; Sakuradani, E.; Fukui, S.; Kita, S.; Sakamoto, K.; Shimizu, S. Appl. Microbiol. Biotechnol. 2008, 80, 597. doi: 10.1007/s00253-008-1563-6
Urano, N.; Fukui, S.; Kumashiro, S.; Ishige, T.; Kita, S.; Sakamoto, K.; Kataoka, M.; Kita, S.; Shimizu, S. J. Biosci. Bioeng. 2011, 3, 266.
Xu, F.; Chung, J. Y. L.; Moore, J. C.; Liu, Z. Q.; Yoshikawa, N.; Hoerrner, R. S.; Lee, J.; Royzen, M.; Cleator, E.; Gibson, A. G.; Dunn, R.; Maloney, K. M.; Alam, M.; Goodyear, A.; Lynch, J.; Yasuda, N.; Devine, P. N. Org. Lett. 2013, 6, 1342.
Friest, J. A.; Maezato, Y.; Broussy, S.; Blum, P.; Berkowitz, D. B. J. Am. Chem. Soc. 2010, 132, 5930. doi: 10.1021/ja910778p
Applegate, G. A.; Berkowitz, D. B. Adv. Synth. Catal. 2015, 357, 1619. doi: 10.1002/adsc.201500316
Limanto, J.; Ashley, E. R.; Yin, J. J.; Beutner, G..; Grau, B. T.; Klapars, A. M.; Kim, M. M.; Klapars, A. P.; Liu, Z. J.; Strotman, H. R.; Truppo, M. D. Org. Lett. 2014, 16, 2716. doi: 10.1021/ol501002a
Peng, Z. H.; Wong, J. H.; Hansen, E. C.; Puchlopek-Dermenci, A. L. A.; Clarke, H. J. Org. Lett. 2014, 16, 860. doi: 10.1021/ol403630g

图式 2 羰基还原酶参与的动态动力学拆分
Scheme 2 Dynamic kinetic resolution catalyzed by carbonyl reductases
表 1 羰基还原酶在重大药物品种的工业化应用
Table 1. Pharmaceutical drugs applied with carbonyl reductases in industry
 下载: 导出CSV
下载: 导出CSV
表 2 羰基还原酶的工业化标准
Table 2. Industrial standard for carbonyl reductase
| 工艺参数 | 数值 |
| 底物浓度 | ≥100 g/L |
| 转化率 | ≥98% |
| 手性纯度 | ≥99% |
| 反应时间 | ≤24 h |
| 酶用量 | ≤5 g/L |
下载: 导出CSV
表 3 不同生产规模下酶的成本比较
Table 3. Comparison of cost in different scales
| 工业规模 | 产品成本/(元•kg-1) | 酶成本/(元•kg-1) |
| 药物 | >760 | 76 |
| 精细化工 | >114 | 11.4 |
| 特殊化学品 | 38 | 1.9 |
| 大宗产品 | 7.6 | 0.38 |
下载: 导出CSV
表 4 筛选结果
Table 4. Results of screening
| No. | 催化剂 | syn-(1R, 2S)-39 | anti-(1S, 2S)-39 |
| 1 | YOL151w | ND | 64% de |
| 2 | YGL157w | 12% de | ND |
| 3 | YGL039w | ND | 56% de |
| 4 | YNL331c | 79% de | ND |
| 5 | YOR120w | 77% de | ND |
| 6 | YBR149w | 95% de | ND |
| 7 | YHR104w | ND | 92% de |
| 8 | YDR368w | 95% de | ND |
| 9 | 面包酵母 | 18% de | ND |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


































 下载:
下载: