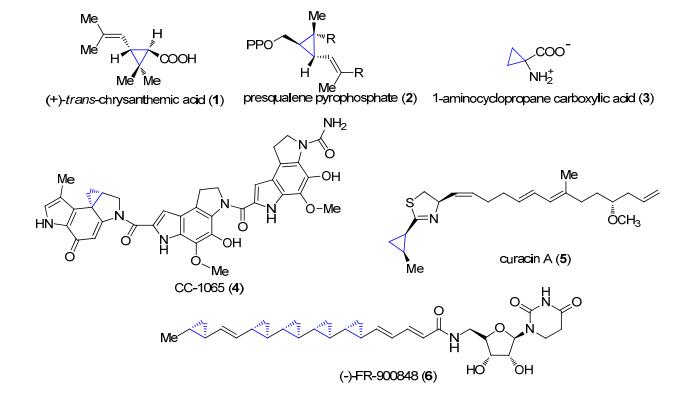
图 1.
含有环丙烷官能团的代表性天然产物
Figure 1.
Structures of representative cyclopropane-containing natural products

环丙烷是最简单的碳环结构, 是一种无色易燃气体, 可作为全身麻醉剂, 于1882年实现全合成[1].从除虫菊花中分离到的具有杀虫效果的除虫菊素(+)-trans- chrysanthemic acid (1)是第一个从自然界中分到的含有环丙烷官能团的天然产物(图 1)[2].后来陆续从其他植物、真菌和微生物中分离和鉴定了很多含有环丙烷的化合物[3].这类化合物中有些发挥着重要的生理功能, 例如presqualene pyrophosphate (PSQPP, 2)[4]和1-amino- cyclopropane carboxylic acid (ACC, 3)[5]分别是胆固醇和植物激素乙烯在生物合成过程中一个关键的中间体.环丙烷中特殊的环张力(115 kJ/mol)使其具有独特的性质和反应活性, 因此具有开发成先导药物的潜力.例如, CC-1065 (4)和多卡霉素具有极高的DNA烷基化活性, 其化学合成类似物已用来开发抗体药物偶联分子, 其中SYD985和MDX-1203已进入临床I期研究[6~8]; (+)-curacin A (5)具有结合微管蛋白和抑制有丝分裂活性, 因此具有抗肿瘤活性[9]; (-)-FR-900848 (6)具有抗植物致病真菌活性[10].科学家在20世纪60年代就已将环丙烷基团引入到药物分子中, 例如, 单胺氧化酶的抑制剂苯基环丙胺[11]和阿片受体拮抗剂环丙甲羟二羟吗啡酮[12].现在越来越多的药物分子引入了环丙烷基团[13].尽管环张力的存在使其化学合成富有极大的挑战性, 合成化学家们做了大量的研究工作, 并开发了许多策略来将环丙烷引入到分子中.本综述第一部分将对已开发的通过化学合成构筑环丙烷的策略进行简单的归纳和总结.从化学角度考虑, 自然界合成这种有张力的环丙烷化合物确实很有趣, 因此, 在综述的第二部分, 我们将简要介绍自然界的生物合成途径中所用到的合成策略.最后对这两部分进行了总结和展望, 希望能为相关领域的研究工作者们提供一些新的思路.
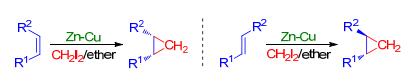
1958年, 杜邦公司的Simmons和Smith[14]报道了一种烯烃与二碘甲烷经过Zn-Cu处理之后产生的亚甲基物种发生环加成形成环丙烷的方法(Scheme 1).该方法由于具有较强的立体选择性以及宽泛的底物适应性而在化学合成中得到了广泛应用.
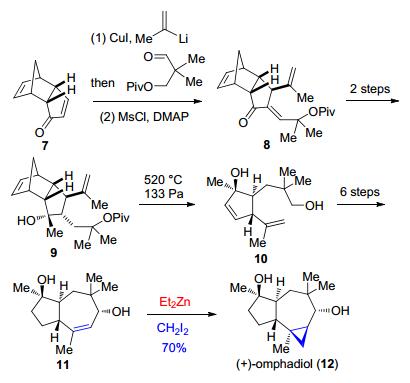
在含有5-7-3并环结构的倍半萜(+)-omphadiol (12)的全合成过程中, Kalesse等[15]在最后一步形成环丙烷的时候利用分子间的Simmons-Smith环丙烷化反应在5-7并环底物11的基础上构筑了剩下的一个三元环, 最终以双环烯酮化合物7为原料, 经过12步反应实现了(+)-omphadiol的全合成(Scheme 2).
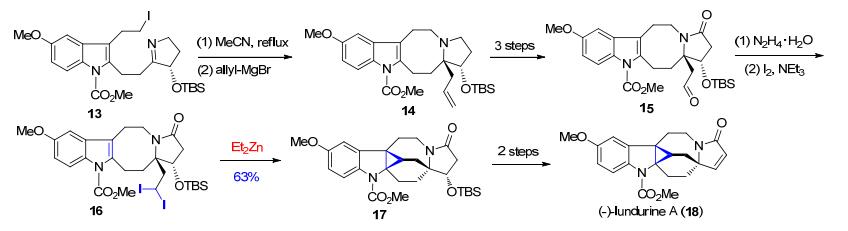
在Qin等[16]报道的吲哚类生物碱(-)-lundurine A (18)的全合成中, 在合成路径的后期采用了一个分子内的Simmons-Smith环丙烷化反应(Scheme 3).在该反应中, 将化合物16与过量的二乙基锌反应, 实现了17中环丙烷的形成.从16到17的环丙烷化反应同时解决了合成(-)-lundurine A中所面临的具有挑战性的环丙烷构筑以及复杂多环骨架合成的难题.
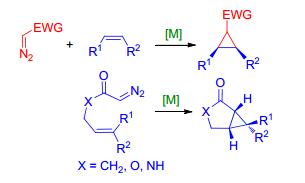
通过重氮化合物衍生的卡宾体来实现碳碳双键的环丙烷化, 需要先让重氮化合物在金属催化剂的作用下发生裂解产生卡宾体. 1906年, Silberrad和Roy[17]发现铜粉可以在加热条件下催化重氮醋酸酯的裂解.随后, 在20世纪60年代, Noyori和Moser等[18, 19]合成了可溶性的铜络合物, 将其应用到重氮化合物的均裂以及对烯烃进行环加成形成环丙烷的产物.十几年后, Teyssie等[20~22]发现Pd(OAc)2和Rh2(OAc)4可以作为比铜络合物更好的催化剂(Scheme 4).随后, 通过分子间或者分子内的重氮化合物衍生的卡宾体实现烯烃的环丙烷化反应就被广泛地应用到有机合成中的环丙烷官能团的构建.
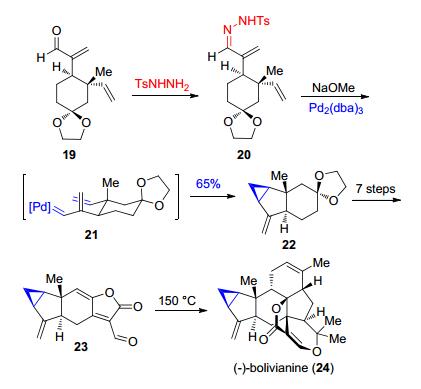
在Liu等[23]对二倍半萜类天然产物(-)-bolivianine (24)的全合成过程中, 通过底物20在Pd2(dba)3的催化下衍生形成的烯丙基卡宾体21发生分子内的环丙烷化形成22中的3-5并环结构, 再经由一系列后续的合成步骤构筑了含有环丙烷的高度拥挤的七环体系(Scheme 5).
20世纪60年代, Doering和Hoffmann[24]报道了氯仿在t-BuOH中在碱KOt-Bu的作用下发生α-消除产生的二氯卡宾对环己烯发生环加成反应, 从而实现了对环己烯上碳碳双键的环丙烷化(Scheme 6).该方法后来在天然产物全合成中也得到了较为广泛的应用.
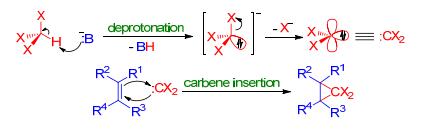
Ding等[25]在对二萜类天然产物steenkrotin A (30)的全合成策略中, 采取了该方法来构建其中的环丙烷.烯酮底物25转化成三甲基硅基烯醇醚26之后, 与2, 2-二溴丙烷在n-BuLi的作用下发生α-消除原位生成的高活性二甲基卡宾反应, 从而以70%的产率立体选择性地引入二甲基环丙烷得到单一构型的3-7并环产物27(Scheme 7).
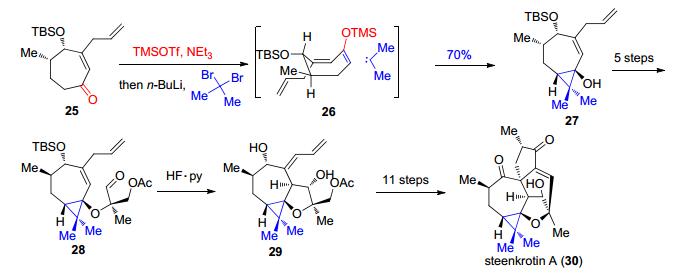
Boger等[26, 27]在抗肿瘤抗生素谷田霉素(yatakemycin, 32)的全合成中的最后一步都采用了分子内SN2反应使两种不同的环化底物在碱性条件下形成环丙烷. 2004年, 报道的是从31发生分子内SN2反应来构建最终产物中的环丙烷, 而在2006年, 报道的则是以离去基团连在六元环上的底物33发生分子内SN2反应, 从而形成产物中的3-5并环(Scheme 8, a).在2006年, Fukuyama等[28]也发表了32的全合成报道, 其合成策略的前面部分不同于Boger等的策略, 但在最后一步形成环丙烷的反应中, 同样采用了与前述33类似的底物34发生分子内SN2反应的方法, 最终实现32中环丙烷的构筑(Scheme 8, b).
早在1976年, Ohloff等[29]报道了含有烯炔结构的底物35在金属试剂ZnCl2的催化作用下加热生成六元环产物36的反应体系中, 检测到很少量的(5%)形成3-6并环的副产物37, 发现具有烯炔结构这种结构特征的底物可以在金属试剂的催化作用下发生环异构化形成环丙烷(Eq.1)[30].
|
|
(1) |
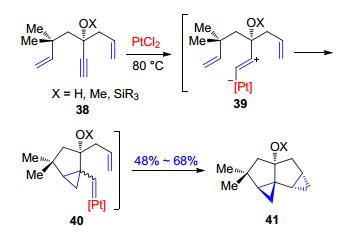
随后, Fensterbank和Malacria等[31, 32]对这种金属试剂催化含有烯炔结构底物的环异构化形成环丙烷的方法做了重大的改进(Scheme 9).他们发现, 具有两套烯炔结构的底物38在金属试剂PtCl2的催化作用下可以发生环丙烷化最终生成含有两套3-5并环的产物41, 并且推测了反应可能经历的过程:首先是底物的炔基和金属试剂PtCl2作用形成两性离子39, 39发生环丙烷化生成第一个3-5并环产物40并且同时生成40中的Pt-卡宾结构, 随后该金属卡宾对第二个烯基发生加成形成最终41中的第二个3-5并环.
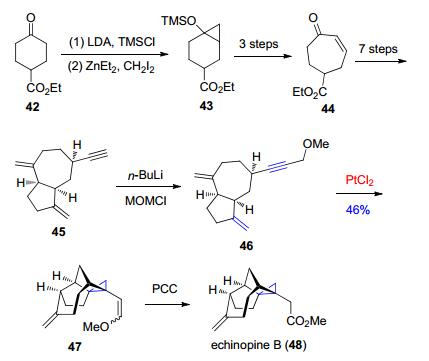
这种环异构化的方法在含有环丙烷的天然产物的全合成策略中也得以被巧妙地运用.在倍半萜类天然产物echinopine B (48)的全合成过程中, 关于其环丙烷的形成, 此前有报道是采用了Simmons-Smith环丙烷化反应和分子内经由卡宾体的环丙烷化方式来构建的. Vanderwal等[33]在设计合成路线的时候就采用了上述的环异构化的方法, 通过PtCl2催化烯炔底物46发生环异构化生成了47中的环丙烷结构, 同时构建了天然产物echinopine B中独特的拥挤的四环骨架(Scheme 10).
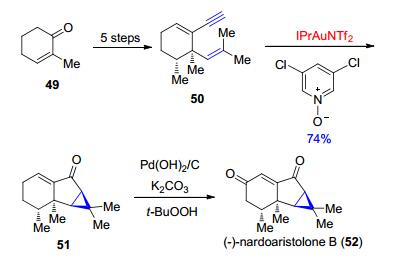
另外, Echavarren等[34]在天然产物(-)-nardoaris- tolone B (52)的全合成中也采用了类似的方法来构筑其中的环丙烷(Scheme 11).通过简单底物得到的1, 5-烯炔底物50在经过筛选确定下来的金属试剂IPrAuNTf2的催化作用下发生环丙烷化, 从而很简便地就构建了51中的6-5-3并环体系, 再经过随后的一步反应直接得到了天然产物(-)-nardoaristolone B.
除了上述这些环丙烷化的方法之外, 在有机合成中使用的其他的形成环丙烷的方法还有很多.例如Hodgson等[35, 36]在卡宾对烯烃的环加成反应的基础上发展的环丙烷化方法, 环氧化合物在LiTMP的作用下以较高的产率立体选择性地得到环丙烷化的产物.有趣的是, 该环氧化合物底物若采用前述Simmons-Smith环丙烷化的方法, 会因为氧的诱导效应而生成与Hodgson环丙烷化方法构型相反的产物, 也就是说这两种环丙烷化的方法在立体选择性上是可以互补的, 该方法在天然产物(-)-cubebol和chloranthalactone A的全合成策略中得到了巧妙的运用[37, 38].另外, Büchner和Perkel[39]报道了1-吡唑啉类化合物在加热条件下释放一分子N2, 同时生成环丙烷化产物的过程, 随后Van Auken和Rinehart[40]发现该反应在光照的条件下也能够实现, 这种通过1-吡唑啉类实现环丙烷化的方法就被用到了前面所述的天然产物(-)-lundurine A (18)中的环丙烷的构建[41].除此之外, Kulinkovich反应也是一种很实用的在分子中引入环丙烷的方法, 脂肪酸酯和乙基格氏试剂在Ti(OiPr)4的催化作用下直接形成环丙烷, 这种方法在全合成中也有很多应用, 例如在天然产物(-)-β-araneosene[42]和cyathins B2[43]的全合成过程中, 该方法就被用来在合成过程中引入环丙烷, 然后再在引入的环丙烷的基础上做进一步的官能团化的衍生.另外, 经过巧妙设计的分子内的环加成反应也可以非常高效简洁地形成环丙烷, 例如在Gaich和Mulzer[44]对天然产物(-)-penifulvin A的全合成过程中就设计了一个分子内的环加成反应, 一步构建了包含有环丙烷的复杂环系.
虽然关于不同类型天然产物的酶学催化机制研究的已经很多, 但是酶催化形成环丙烷基团的报道却很少.根据已报道的酶学催化机制研究, 可将酶催化形成环丙烷基团的生物合成过程分为三大类: (ⅰ)由碳正离子参与的环丙烷化反应、(ⅱ)由碳负离子参与的环丙烷化反应和(ⅲ)由碳自由基参与的环丙烷化反应.
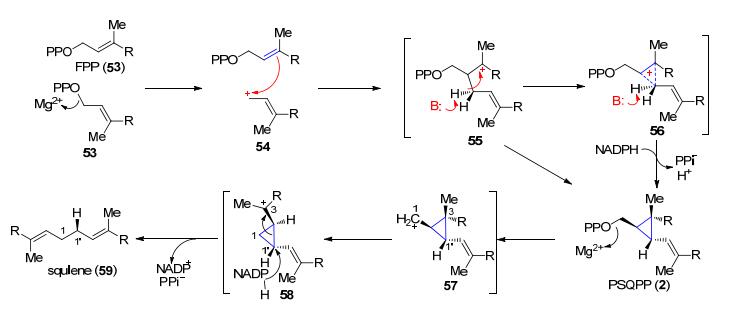
萜类天然产物是自然界中分布最多的化合物, 它们的核心骨架都是由异戊烯焦磷酸(IPP)和二甲基丙烯基焦磷酸(DMAPP)两种单体组成的, 并且是由异戊烯转移酶和萜类合酶参与催化的[45].这两类酶的催化机制共同点是产生一个高活性的碳正离子中间体, 因此, 萜类化合物中环丙烷的合成一般是由碳正离子参与的反应.例如, 胆固醇的前体鲨烯的合成需要经过一个环丙烷中间体2 (Scheme 12), 这个中间体是由鲨烯合酶(squalene synthase, SQS)催化完成的[4]. SQS属于异戊烯转移酶, 催化两分子焦磷酸法尼酯(FPP, 53)以1'-1连键形成鲨烯.第一步, 在SQS的酪氨酸残基辅助下, 一分子FPP的焦磷酸离去产生烯丙基碳正离子54, 此碳正离子发生对双键的亲电加成从而烷基化另一分子的FPP, 从而产生碳正离子中间体55, 随后55可异构化为具有环丙烷过渡态的中间体56, 56脱质子便产生了2.第二步, 2中焦磷酸的离去发生重排产生更加稳定的三级碳正离子58, 接着经过还原转化成鲨烯(59)[45, 46].
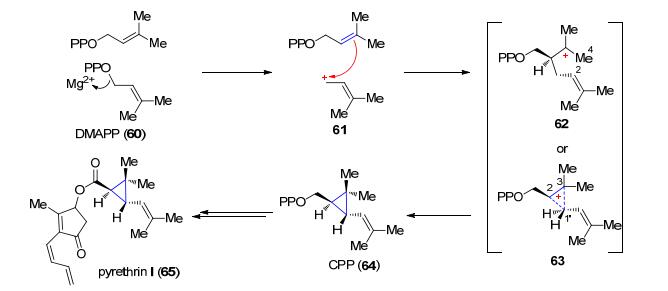
另一个异戊烯转移酶是菊酰基焦磷酸(chrysan- themyl pyrophosphate, CPP)合酶, 它催化两分子DMAPP (60)以1'-2-3连键形式合成除虫菊素类化合物的单萜前体CPP (Scheme 13)[45].同样的, CPP合酶催化反应的第一步也是产生烯丙基碳正离子61, 此碳正离子接着烷基化另一分子的DMAPP, 产生三级碳正离子62或者质子化的环丙烷中间体63, 随后失去C(1')位一个质子便产生了CPP (64), 64经过后续的一系列生物合成过程最终形成天然产物除虫菊素Ⅰ (pyrethrin Ⅰ) (65)[45, 46].
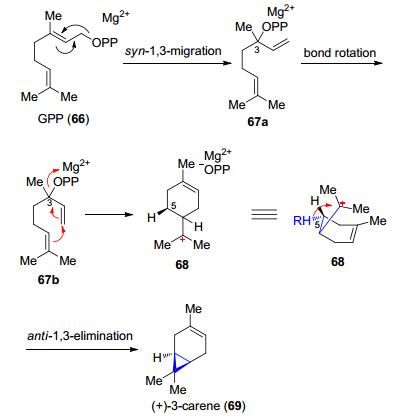
单萜类化合物(+)-3-蒈烯(carene)和三萜类化合物环阿屯醇(cycloartenol)的合成涉及的也是由碳正离子参与的环丙烷化反应. (+)-3-蒈烯(carene)是由Ⅰ类萜类合酶合成的, 而环阿屯醇(cycloartenol)是由Ⅱ类萜类合酶合成的.在(+)-3-carene合酶催化下牻牛儿基焦磷酸(GPP, 66)通过顺式-1, 3-迁移变成焦磷酸芳樟酯(linalyl pyrophosphate) (67a), 这种转化利于C(2)—C(3)的旋转, 将C(1)=C(2)双键变为顺式构象67b, 利于分子内环化变为68, 随后68发生C(5)位R构型的氢的消除, 最终转化为(+)-3-蒈烯(carene) (69) (Scheme 14)[46].
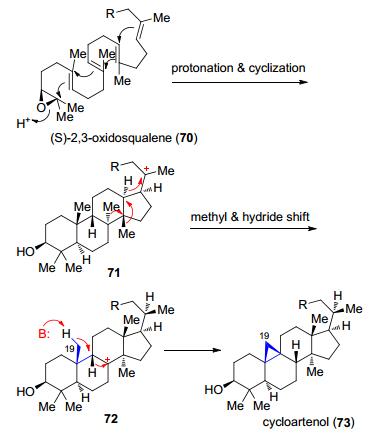
Cycloartenol的合成是以质子化(S)-2, 3-氧鲨烯(oxidosqualene) (70)作为起始的, 引起一系列的环化反应变为71, 经历1, 2-甲基和1, 3-氢迁移变为三级碳正离子72, 最后发生1, 2-氢迁移和C(19)甲基脱质子而产生了cycloartenol (73) (Scheme 15)[46].
在环丙烷脂肪酸和环丙烷分支菌酸中环丙烷的合成也是采取了碳正离子的形式, 但它们的一碳单位供体来源于S-腺苷甲硫氨酸(SAM).这类化合物是由环丙烷脂肪酸/环丙烷分支菌酸合酶负责催化完成的, 首先是底物中的双键亲核进攻SAM中的甲基产生一个碳正离子中间体, 接着获得的甲基被拔一个质子, 产生的亚甲基负离子捕获之前形成的碳正离子, 从而关环形成了环丙烷基团(Scheme 16)[47].
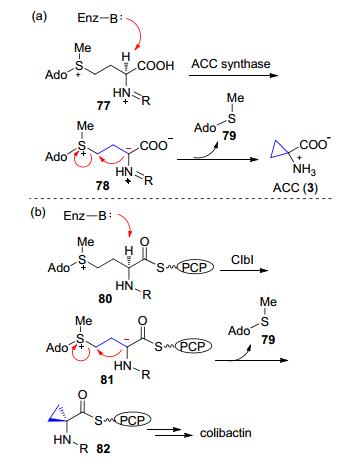
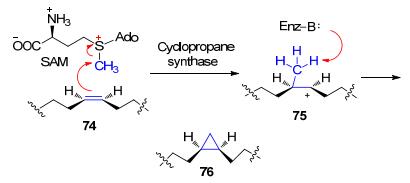
这类反应首先在C(α)位可以形成碳负离子, 并且在C(γ)位有一个好的离去基团, 这样产生的C(α)位的碳负离子对C(γ)位的离去基团发生分子内的SN2反应, 从而形成三元环.前述Boger和Fukuyama等[5]分别报道的对谷田霉素的全合成所采取的分子内的SN2反应就与这种生物合成策略类似.这种类型的反应最初是在植物激素乙烯前体ACC (3)的生物合成过程中发现的, ACC的合成以SAM作为直接底物, 因此这里的离去基团是SAM中的甲硫基腺苷(methylthioadenosine, 79) (Scheme 17, a).最近发现, 在colibactin的生物合成可能也采取了类似的机制(Scheme 17, b)[48].
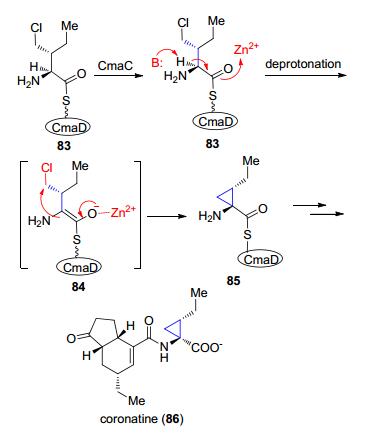
最近在一些聚酮、聚肽等天然产物生物合成过程中也发现了一些类似反应机理的酶, 这里的离去基团为氯离子.例如, 植物毒素coronatine (86)的一个模块单元为coronamic acid (1-amino-1-carboxy-2-ethylcyclopropane, CMA, 85), CMA是由异亮氨酸经过γ-氯-异亮氨酸中间体形成的(Scheme 18).首先Zn2+-依赖的CmaC蛋白拔取83上C(α)位的质子形成负离子中间体84, Zn2+的作用可能是用于稳定烯醇负离子中间体84, 接着通过SN2反应离去氯离子从而形成带环丙烷的85, 随后经过一系列后续的转化得到含有环丙烷的天然产物86[49, 50].后来发现在kutzneride 2[51]和curacin A(5)[52]也采用了类似的策略.
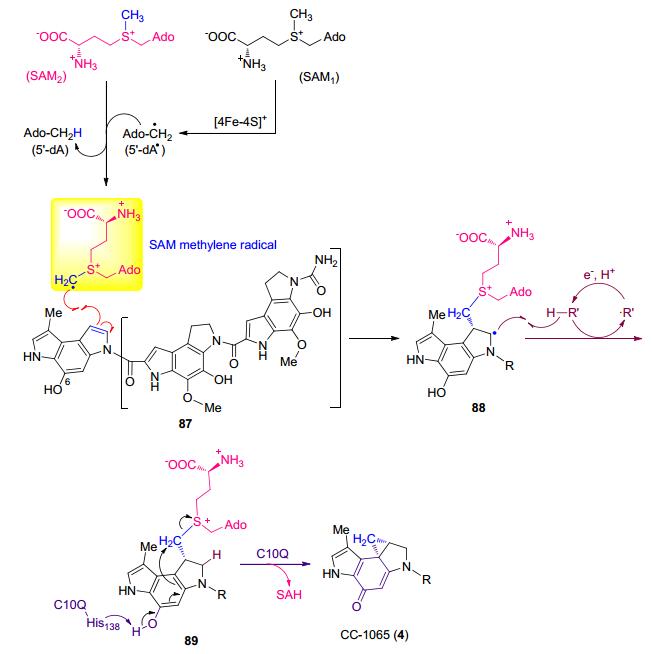
最近, 我们课题组[53, 54]在苯并二吡咯类天然产物生物合成研究中发现了一种新型的由自由基参与的环丙烷形成过程:即由一个属于HemN家族的自由基SAM蛋白(C10P)和一个甲基转移酶蛋白(C10Q)以SAM作为一碳单位供体首先催化C=C双键的SAM加成, 紧接着形成的SAM-底物共价中间体经分子内环化形成抗肿瘤抗生素CC-1065 (4)中的环丙烷(Scheme 19).通过质谱技术以及CD3-SAM和D2O标记实验证实了SAM-底物共价中间体89的存在, 这也是首次检测到HemN家族蛋白催化的关键中间体.基于同位素标记实验的结果以及这个关键中间体的检测, 我们提出了HemN蛋白的催化机制: HemN蛋白首先在电子供体作用下利用自身的[4Fe-4S]还原切割第一分子SAM产生5'-dA自由基; 这个自由基被用来拔取第二分子SAM甲基上的一个氢原子, 从而产生SAM亚甲基自由基; 紧接着, 这个SAM亚甲基自由基加成到底物87中一个吡咯环的C=C双键上形成自由基中间体88, 随后生成关键的中间体89.因此, 这些结果不仅说明SAM的甲基可以形成SAM亚甲基自由基, 而且由于硫鎓离子(sulfonium)的吸电子效应使得SAM亚甲基自由基属于亲电自由基.点突变实验表明甲基转移酶C10Q参与了SAM-底物共价中间体89的分子内关环反应. C10P和C10Q同源蛋白广泛存在于苯并二吡咯类天然产物生物合成基因簇中, 这说明这种环丙烷策略是保守的[54, 55].另外, 在生物碱cyclo- clavine中环丙烷的生物合成方式也可能采取了自由基反应的方式, 即EasH催化产生的铁氧四价物种攫取底物上的氢之后形成的自由基物种发生重排形成cyclo- clavine中的环丙烷结构[56].
随着天然产物化学研究的深入, 越来越多的环丙烷化合物被发现、研究以及开发成先导药物分子.早期的天然产物除虫菊素(+)-trans-chrysanthemic acid (1)就是一个经典的例子, 1是从除虫菊花中分离到的, 起到防止昆虫侵扰的作用, 也正以此化合物为灵感, 合成化学家们合成了一系列除虫菊素类杀虫剂[2].虽然合成化学家开发了若干成熟的环丙烷合成策略, 但对于化学合成来说, 化合物分子中的微环境起着决定性作用, 所以每一个化合物适用的策略都不一样, 因此环丙烷化方法的开发永远在路上.同样的, 随着生物合成研究的不断扩展和深入, 越来越多的用于环丙烷化的生化反应被揭示.生化反应本质上是生物分子的有机化学反应, 因此也是符合有机化学原理的.例如, 本文中介绍的Boger和Fukuyama等分别报道的对谷田霉素中环丙烷结构的构筑所采用的分子内的SN2反应与ACC (3)和colibactin生物合成过程中经过碳负离子中间体发生的环丙烷化反应就有异曲同工之妙.另外, Arnold等[57]利用蛋白质定向进化技术获得一个细胞色素P450蛋白通过卡宾转移实现了烯烃的环丙烷化.这一个全新的生化反应与通过重氮化合物衍生的卡宾体实现环丙烷化的策略实现了完美的统一.因此, 全合成的策略可以引导生物化学家发现或设计新的生化反应; 同样的, 新的生化反应的发现也可以指导合成化学家设计新的合成路线(仿生合成).
Freund, A. J. Prakt. Chem. 1882, 26, 367. doi: 10.1002/(ISSN)1521-3897
Staudinger, H.; Ruzicka, L. Helv. Chim. Acta 1924, 7, 177. doi: 10.1002/hlca.19240070124
Wessjohann, L. A.; Brandt, W.; Thiemann, T. Chem. Rev. 2003, 103, 1625. doi: 10.1021/cr0100188
Pandit, J.; Danley, D. E.; Schulte, G. K.; Mazzalupo, S.; Pauly, T. A.; Hayward, C. M.; Hamanaka, E. S.; Thompson, J. F.; Harwood, H. J. J. Biol. Chem. 2000, 275, 30610.
Capitani, G.; Hohenester, E.; Feng, L.; Storici, P.; Kirsch, J. F.; Jansonius, J. N. J. Mol. Biol. 1999, 294, 745. http://www.sciencedirect.com/science/article/pii/S0022283699932552
Menderes, G.; Bonazzoli, E.; Bellone, S.; Black, J.; Predolini, F.; Pettinella, F.; Masserdotti, A.; Zammataro, L.; Altwerger, G.; Buza, N.; Hui, P.; Wong, S.; Litkouhi, B.; Ratner, E.; Silasi, D. A.; Azodi, M.; Schwartz, P. E.; Santin, A. D. Clin. Cancer Res. 2017, 23, 5836. doi: 10.1158/1078-0432.CCR-16-2862
Tietze, L. F.; Krewer, B. Anticancer Agents Med. Chem. 2009, 9, 304.
Sievers, E. L.; Senter, P. D. Annu. Rev. Med. 2013, 64, 15.
Wipf, P.; Reeves, J. T.; Day, B. W. Curr. Pharm. Des. 2004, 10, 1417.
Hiratsuka, T.; Suzuki, H.; Kariya, R.; Seo, T.; Minami, A.; Oikawa, H. Angew. Chem., Int. Ed. 2014, 53, 5423.
Silverman, R. B.; Zieske, P. A. Biochemistry 1985, 24, 2128. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM3995007
Wentland, M. P.; Lu, Q.; Lou, R.; Bu, Y.; Knapp, B. I.; Bidlack, J. M. Bioorg. Med. Chem. Lett. 2005, 15, 2107. doi: 10.1016/j.bmcl.2005.02.032
Talele, T. T. J. Med. Chem. 2016, 59, 8712.
Lászlò, K.; Barbara, C. Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, Burlington, 2005, p. 412.
Parthasarathy, G.; Eggert, U.; Kalesse, M. Org. Lett. 2016, 18, 2320. http://www.ncbi.nlm.nih.gov/pubmed/27105390
Jin, S.; Gong, J.; Qin, Y. Angew. Chem., Int. Ed. 2015, 54, 2228.
Silberrad, O. R.; Roy, C. S. J. Chem. Soc. 1906, 89, 179.
Nozaki, H.; Moriuti, S.; Yamabe, M.; Noyori, R. Tetrahedron Lett. 1966, 7, 59.
Moser, W. R. J. Am. Chem. Soc. 1969, 91, 1141. doi: 10.1021/ja01033a018
Paulissen, R.; Hubert, A. J.; Teyssie, P. Tetrahedron Lett. 1972, 13, 1465.
Paulissen, R.; Reimlinger, H.; Hayez, E.; Hubert, A. J.; Teyssie, P. Tetrahedron Lett. 1973, 14, 2233. doi: 10.1016/S0040-4039(01)87603-6
Hubert, A. J.; Noels, A. F.; Anciaux, A. J.; Teyssie, P. Synthesis 1976, 600.
Yuan, C. C.; Du, B. A.; Yang, L.; Liu, B. J. Am. Chem. Soc. 2013, 135, 9291. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM23713856
Doering, W.; Hoffmann, A. K. J. Am. Chem. Soc. 1954, 76, 6162. doi: 10.1021/ja01652a087
Pan, S. Y.; Xuan, J.; Gao, B. L.; Zhu, A.; Ding, H. F. Angew. Chem., Int. Ed. 2015, 54, 6905.
Tichenor, M. S.; Kastrinsky, D. B.; Boger, D. L. J. Am. Chem. Soc. 2004, 126, 8396. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM15237994
Tichenor, M. S.; Trzupek, J. D.; Kastrinsky, D. B.; Shiga, F.; Hwang, I.; Boger, D. L. J. Am. Chem. Soc. 2006, 128, 15683.
Okano, K.; Tokuyama, H.; Fukuyama, T. J. Am. Chem. Soc. 2006, 128, 7136. doi: 10.1021/ja0619455
Strickler, H.; Davis, J. B.; Ohloff, G. Helv. Chim. Acta 1976, 59, 1328. doi: 10.1002/(ISSN)1522-2675
Bruneau, C. Angew. Chem., Int. Ed. 2005, 44, 2328.
Mainetti, E.; Mouries, V.; Fensterbank, L.; Malacria, M.; Marco-Contelles, J. Angew. Chem., Int. Ed. 2002, 41, 2132. doi: 10.1002/1521-3773(20020617)41:12<2132::AID-ANIE2132>3.0.CO;2-S
Lemiere, G.; Gandon, V.; Cariou, K.; Hours, A.; Fukuyama, T.; Dhimane, A. L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2009, 131, 2993. doi: 10.1021/ja808872u
Michels, T. D.; Dowling, M. S.; Vanderwal, C. D. Angew. Chem., Int. Ed. 2012, 51, 7572. doi: 10.1002/anie.201203147
Homs, A.; Muratore, M. E.; Echavarren, A. M. Org. Lett. 2015, 17, 461. doi: 10.1021/ol503531n
Hodgson, D. M.; Chung, Y. K.; Paris, J. M. J. Am. Chem. Soc. 2004, 126, 8664. doi: 10.1021/ja047346k
Hodgson, D. M.; Chung, Y. K.; Nuzzo, I.; Freixas, G.; Kulikiewicz, K. K.; Cleator, E.; Paris, J. M. J. Am. Chem. Soc. 2007, 129, 4456.
Hodgson, D. M.; Salik, S.; Fox, D. J. J. Org. Chem. 2010, 75, 2157. doi: 10.1021/jo9022974
Büchner, E.; Perkel, L. Ber. Dtsch. Chem. Ges. 1903, 36, 3774. doi: 10.1002/(ISSN)1099-0682
Buchner, E.; Curtius, T. Ber. Dtsch. Chem. Ges. 1885, 18, 2377.
Rinehart, K. L.; Van Auken, T. V. J. Am. Chem. Soc. 1960, 82, 5251.
Kirillova, M. S.; Muratore, M. E.; Dorel, R.; Echavarren, A. M. J. Am. Chem. Soc. 2016, 138, 3671. doi: 10.1021/jacs.6b01428
Kingsbury, J. S.; Corey, E. J. J. Am. Chem. Soc. 2005, 127, 13813.
Kim, K.; Cha, J. K. Angew. Chem., Int. Ed. 2009, 48, 5334. doi: 10.1002/anie.200901669
Gaich, T.; Mulzer, J. J. Am. Chem. Soc. 2009, 131, 452. doi: 10.1021/ja8083048
Walsh, C. T. ACS Chem. Biol. 2007, 2, 296.
Thibodeaux, C. J.; Chang, W. C.; Liu, H. W. Chem. Rev. 2012, 112, 1681. doi: 10.1021/cr200073d
Liao, R. Z.; Georgieva, P.; Yu, J. G.; Himo, F. Biochemistry 2011, 50, 1505.
Zha, L.; Jiang, Y.; Henke, M. T.; Wilson, M. R.; Wang, J. X.; Kelleher, N. L.; Balskus, E. P. Nat. Chem. Biol. 2017, 13, 1063. doi: 10.1038/nchembio.2448
Frederic, H.; Vaillancourt, E. Y.; Vosburg, D. A.; O'Connor, S. E.; Walsh, C. T. Nature 2005, 436, 1191.
Kelly, W. L.; Boyne, M. T.; Yeh, E.; Vosburg, D. A.; Galoni, D. P.; Kelleher, N. L.; Walsh, C. T. Biochemistry 2007, 46, 359.
Jiang, W.; Heemstra, J. R.; Forseth, R. R.; Neumann, C. S.; Manaviazar, S.; Schroeder, F. C.; Hale, K. J.; Walsh, C. T. Biochemistry 2011, 50, 6063. doi: 10.1021/bi200656k
Gu, L.; Wang, B.; Kulkarni, A.; Geders, T. W.; Grindberg, R. V.; Gerwick, L.; Hakansson, K.; Wipf, P.; Smith, J. L.; Gerwick, W. H.; Sherman, D. H. Nature 2009, 459, 731.
Wu, S.; Jian, X. H.; Yuan, H.; Jin, W. B.; Yin, Y.; Wang, L. Y.; Zhao, J.; Tang, G. L. ACS Chem. Biol. 2017, 12, 1603. doi: 10.1021/acschembio.7b00302
Jin, W. B.; Wu, S.; Jian, X. H.; Yuan, H.; Tang, G. L. Nat. Commun. 2018, 9, 2771. doi: 10.1038/s41467-018-05217-1
Wang, X.; Wu, S.; Jin, W.; Xu, B.; Tang, G.; Yuan, H. Acta Biochim. Biophys. Sin. 2018, 50, 516.
Jakubczyk, D.; Caputi, L.; Hatsch, A.; Nielsen, C. A.; Diefenbacher, M.; Klein, J.; Molt, A.; Schröder, H.; Cheng, J. Z.; Naesby, M.; O'Connor, S. E. Angew. Chem., Int. Ed. 2015, 54, 5117. doi: 10.1002/anie.v54.17
Coelho, P. S.; Brustad, E. M.; Kannan, A.; Arnold, F. H. Science 2013, 339, 307. doi: 10.1126/science.1231434

图 1 含有环丙烷官能团的代表性天然产物
Figure 1 Structures of representative cyclopropane-containing natural products

图式 2 Kalesse等报道的(+)-omphadiol的全合成
Scheme 2 Total synthesis of (+)-omphadiol reported by Kalesse et al.

图式 3 Qin等报道的对(-)-lundurine A的全合成
Scheme 3 Total synthesis of (-)-lundurine A reported by Qin et al.

图式 5 Liu等报道的对(-)-bolivianine的全合成
Scheme 5 Total synthesis of (-)-bolivianine reported by Liu et al.

图式 6 卡宾对烯烃的环加成反应
Scheme 6 Cyclopropanation through addition of free carbenes to olefins

图式 7 Ding等报道的对steenkrotin A的全合成
Scheme 7 Total synthesis of steenkrotin A reported by Ding et al.

图式 8 Boger和Fukuyama课题组报道的对谷田霉素的全合成
Scheme 8 Total synthesis of yatakemycin reported by Boger and Fukuyama, respectively

图式 9 Fensterbank和Malacria等报道的PtCl2催化的环异构化反应
Scheme 9 Cycloisomerization catalyzed by PtCl2 reported by Fensterbank and Malacria et al.

图式 10 Vanderwal等报道的对echinopine B的全合成
Scheme 10 Total synthesis of echinopine B reported by Vanderwal et al.

图式 11 Echavarren等报道的对(-)-nardoaristolone B的全合成
Scheme 11 Total synthesis of (-)-nardoaristolone B reported by Echavarren et al.

图式 12 经过含有环丙烷结构的PSQPP中间体的鲨烯的生物合成过程
Scheme 12 Biosynthesis of squalene preceeds through the stable cyclopropane intermediate PSQPP

图式 13 经过含有环丙烷结构的CPP中间体的除虫菊素Ⅰ的生物合成过程
Scheme 13 Biosynthesis of pyrethrin Ⅰ preceeds through the cyclopropane intermediate CPP

图式 17 ACC和colibactin中环丙烷的生物合成过程
Scheme 17 Biosynthesis of cyclopropane in ACC and colibactin

图式 18 Coronatine中环丙烷的生物合成过程
Scheme 18 Biosynthesis of coronatine preceeds through the cyclopropane intermediate CMA

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:






 下载:
下载: