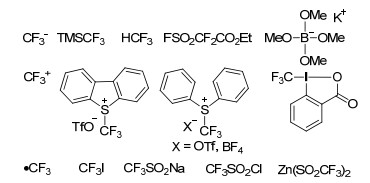
图 1.
常见的三氟甲基化试剂
Figure 1.
Common trifluoromethyl reagents

由于三氟甲基具有强吸电子诱导效应、亲脂性以及含有稳定的C—F键, 将其引入到有机分子中能够显著改变目标分子的各种性质.比如, 往药物分子中引入三氟甲基可以有效延长其在生物体内的作用时间, 增强代谢稳定性; 同时三氟甲基的引入通常会增加药物分子的脂溶性, 从而有助于药物分子在生物体内的吸收、传递和扩散[1].抗癌药索拉非尼(Sorafenib tosylate)、抗抑郁药氟西汀(Fluoxetine)[2]、新型植物广谱杀菌剂肟菌酯(Trifloxystrobin)[3]和液晶显示屏典型材料ZLI-2857[4]等都含有三氟甲基.因此, 含有三氟甲基的有机物在医药、农药及液晶材料的合成中具有广泛的应用价值.在温和的反应条件下, 将三氟甲基引入到有机化合物中一直是科研人员研究的热点.然而, 三氟甲基化合物的传统合成法反应条件一般比较苛刻, 且官能团兼容性也较差[5].近几年, 随着多种商业化三氟甲基试剂的出现, 过渡金属催化三氟甲基化反应得到迅速发展, 底物适用范围不断扩大, 新型三氟甲基试剂以及新的合成方法也不断涌现[6~9].其中, 过渡金属铜较早地应用于三氟甲基化反应的研究. 2013年, 戴建军课题组[10]就从发生三氟甲基化反应的底物出发, 综述了近年来铜促进卤代烃、硼酸类化合物、芳香化合物等三氟甲基化反应研究进展; 2011年至2012年, 刘丹课题组[11]和段春迎课题组[12]都以芳香化合物为底物, 撰写了过渡金属催化三氟甲基化反应的研究进展; 同时, 施章杰等[13]也综述了过渡金属催化底物C—H键的三氟甲基化反应, 但该领域的反应主要集中于端炔的sp型C—H键活化、杂环化合物和芳烃的sp2型C—H键活化以及烯丙基sp3型C—H键活化的三氟甲基化反应.本文将重点对近几年过渡金属促进三氟甲基化反应的研究进展进行综述, 包括银、钯、铁、镍、铑和钴等金属促进三氟甲基化反应, 所涉及到的底物范围比较广泛, 包括烯烃、炔烃、芳烃、胺类、醇类和羧酸类等; 所应用到的三氟甲基试剂包括Ruppert-Prakash试剂、Togni试剂、Umemoto试剂等以及一些新型的三氟甲基试剂(图 1).此外, 本文对部分过渡金属参与的三氟甲基化反应机理也进行了一些探讨.
2011年, Sanford课题组[14]以AgOTf为介导, 通过一步反应实现了芳烃1与TMSCF3 (2)的三氟甲基化(Eq. 1).该反应在无配体的情况下, 可以高效地将芳烃直接三氟甲基化, 得到目标产物3.其中, 当使用对苯二甲醚作为底物时, 目标产物3的产率能达到88%.此外, N-甲基吡咯、噻吩、咖啡因等含杂环的芳烃也能很好地兼容该反应, 产率在72%左右.作者发现在无碱条件下, 反应不能发生.但是该反应对芳环上三氟甲基化的位置并没有选择性.
|
|
(1) |
2011年, Xu等[15]以AgF为催化剂, 使用较易获得的仲氨基炔烃4作为原料, 通过“一锅法”实现了其与三氟甲基试剂2的串联胺化-三氟甲基化反应(Scheme 1).研究表明:该反应所有产物的区域选择性均遵循鲍德温(Baldwin)规则, 且产率都比较高(87%~95%).当使用手性氨基炔9作为底物时, 相应的三氟甲基化产物10产率高达90%, 同时产物具有良好的非对映选择性(Eq. 2).作者还使用其他催化剂和亲核试剂(如三甲基氰硅烷6)来进行反应, 也成功得到了胺化-氰化产物8, 产率为66%~99%.

作者推测:仲氨基炔烃4在催化剂作用下首先会形成关键的烯胺中间体5, 接着才与亲核三氟甲基试剂反应.他们使用氘代标记的底物进一步探究了该反应的机制, 结果发现在环上及环外的碳都能检测到氘.这表明烯胺中间体5可能在亲核试剂进攻前就已经发生了其他转化, 如逆氢胺化或互变异构化等.作者将进一步探究可能的反应机理.
|
|
(2) |
2012年, Br se等[16]在Sanford报道的基础上(Eq. 1), 将三氮烯结构单元引入到芳烃1中, 报道了芳基三氮烯11在AgF参与下的三氟甲基化反应(Eq. 3).反应可以选择性地在三氮烯邻位进行三氟甲基化, 得到目标产物13, 产率为39%~74%.该反应操作简单, 使用二异丙基取代三氮烯11作为底物可显著降低三氮烯的毒性, 同时该反应对许多官能团也具有良好的兼容性, 特别是碘化物和溴化物.此外, 三氮烯结构单元还很容易转化为其他基团, 例如叠氮基、氨基、羟基等.因此进一步扩大了该反应的应用范围.
|
|
(3) |
2014年, Wang等[17]以AgF为介导, 通过分步法实现了芳香胺的三氟甲基化反应.他们先将芳香胺15重氮化得到其重氮盐16, 然后在-78 ℃低温下, 将AgF与TMSCF3混合, 然后缓慢升至室温制备出关键的AgCF3 (14) (Eq. 4), 最后再与重氮盐16发生反应, 成功得到了三氟甲基化产物3 (Scheme 2).当芳胺15上R=CH3时, 相应的三氟甲苯化合物3的产率高达97%.值得注意的是:当使用4-吡啶胺作为底物时, 相应的三氟甲基吡啶产率低至10%.作者认为造成该影响的原因可能有两种: (1)吡啶环上的N与AgCF3具有强配位作用; (2)相应的吡啶基重氮盐在酸性条件下溶解度较低.该反应条件较温和, 底物15具有较好的官能团兼容性, 同时氨基容易被保护及脱除, 因此该反应具有良好的应用前景.
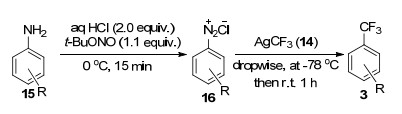
2013年, Qing等[18]以AgNO3为催化剂, 催化末端烯烃17与2的三氟甲基化反应(Eq. 5), 形成目标产物19, 同时该反应还伴随着少量副产物20和21的生成.
|
|
(4) |
与之前Brse等其他课题组报道过的反应不同, 该反应没有使用AgF参与来形成关键的活性中间体AgCF3.作者推测:目标产物可能是由反应过程中产生的自由基中间体23发生歧化反应和攫氢反应来得到.该中间体23还可能会进一步被氧化为阳离子活性种24, 随后发生脱质子化得到副产物20和21 (Scheme 3).据此推测, 作者认为应该往该体系中加入足够的H供体来抑制副反应的发生.经过系列筛选后, 作者得出添加1, 4-环己二烯(18, 1, 4-cyclohexadiene, 1, 4-CHD)能够最有效抑制副反应的发生, 提高主产物19的产率.当R=Ph时, 目标产物19的产率高达92%.
2013年, Lee课题组[19]以各种含银、氟的化学计量试剂为介导, 在温和条件下高效实现了炔烃26的自身聚合与氟化反应(Eq. 6), 目标产物为芳烃27, 主要包括二氢吲哚和异吲哚啉衍生物.其中, 当使用AgF和TMSCF3试剂进行反应时, 能够得到相应的三氟甲基化产物27, 产率最高可达到78%.该反应区域选择性较好, 对药物的合成具有重要的意义.
|
|
(6) |
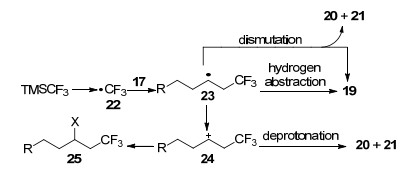
2013年, Hu等[20]以2-(三甲基硅烷基)三氟甲烷磺酸苯酯28作为苯炔前体, 首次报道了苯炔化合物47在银参与下与2-碘苯乙炔(29)发生的邻位三氟甲基化-碘化反应(Eq. 7).该反应能快速地将CF3和I基团引入到苯环上, 得到目标产物三氟甲基碘苯化合物31.反应中使用的AgCF3需要新鲜制备(Eq. 4).此外, 作者还研究了配体对目标产物产率的影响:当R=H时, 在无配体下反应生成目标产物31的产率只有31%;而选用正丙胺作为配体时, 产率可提高至59%;当加入2, 2, 6, 6-四甲基哌啶(30, 2, 2, 6, 6-tetramethylpiperidine, TMP)作为配体时, 产率能达到70%.
|
|
(7) |
作者认为TMP会立即与AgCF3生成新的银胺络合物AgCF3•TMP (34), 并提出了可能的反应机理:原位生成的苯炔化合物32首先与AgCF3•TMP发生金属银碳化反应生成中间体35, 35通过氢键作用再与TMP结合得到中间体36, 随后进一步发生脱质子化得到阴离子中间体37, 接着37通过卤素键与2-碘苯乙炔29配位得到中间体38, 最后发生分子内亲核进攻生成39, 在银促进下, 39很容易转化为邻位三氟甲基化的碘苯化合物31和副产物苯乙炔银33 (Scheme 4).
2013年, Liu等[21]设计了AgNO3与2-噁唑基吡啶41络合形成的催化体系, 通过“一锅法”实现2-炔基苯甲醛亚胺40与N-氟代双苯磺酰胺(42, N-fluorobenzenesul- fonimide, NFSI)的分子环内胺氟化反应, 并进一步与2及四甲基氟化铵(44, tetramethylammonium fluoride, TMAF)反应得到目标产物1-(三氟甲基)-4-氟-1, 2-二氢异喹啉45 (Scheme 5).实验结果表明: R1官能团如F、Cl、OCH3等都与反应条件相容(45a~45d), 且亚胺上的取代基R3对反应并没有影响(45e~45g).为了实现不对称的三氟甲基化, 作者将手性胺引入到底物中(45h~45i), 目标产物的产率虽然可达到70%, 但其非对映选择性很低.该反应为合成氟代杂环化合物提供了新的途径, 有望进一步应用于药物合成中.
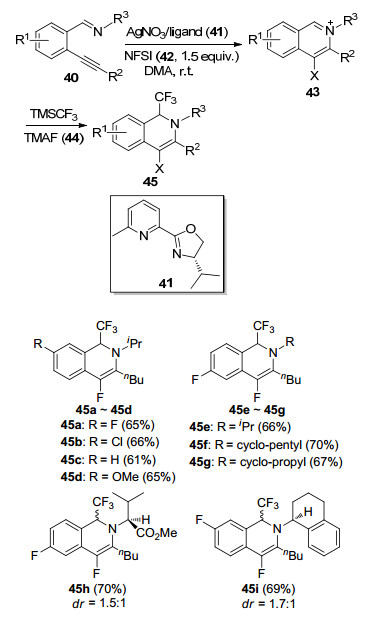
2013年, Zhang等[22]报道了以路易斯酸AgSbF6作为催化剂, 催化2-炔基苯甲醛亚胺40与三氟甲基试剂2的环化-三氟甲基化反应(Eq. 8), 目标产物为1-(三氟甲基)-1, 2-二氢异喹啉(45).作者发现:往体系中加入乙酸能够有效提高目标产物45的产率.他们推测乙酸的存在可能是作为Brnsted酸来活化亚胺基团.该反应条件比较温和, 在室温下就能得到较高的产率, 同时反应具有良好的底物适应性.例如当底物为40a时, 目标产物45的产率为88%;当底物为40b时, 产物45的产率达到91%;当底物为40c时, 产物45的产率高达95%.然而, 当苯环上连有吸电子基时, 产物45的产率会大幅下降.
|
|
(8) |
2015年, Qing课题组[23]以银为介导, 在氧化条件下通过使用TMSCF3和Selectfluor试剂来实现未活化烯烃的氟代三氟甲基化反应(Eq. 9).
|
|
(9) |
2013年, Greaney等[24]在Sanford的基础上(Eq. 1), 设计了不同取代基的芳烃1与2的三氟甲基化反应(Eq. 10).该反应具有良好的官能团耐受性:当底物1上R为NMe2时, 产物3的产率为75%;当使用N-甲基吡咯作为底物时, 相应产物的产率高达94%.作者在相同条件下尝试用三氟甲基三乙基硅烷来进行反应, 反应产率虽然略有所提高, 但考虑到其价格昂贵, 所以仍然选用TMSCF3作为三氟甲基化试剂.
|
|
(10) |
2015年, Cheng等[25]首次报道了以Ag2CO3和配体49组成的催化体系, 催化亚砜亚胺化合物48与2的N-三氟甲基化反应(Eq. 11).研究表明:温度的变化对反应结果有着显著影响.当底物48上R1=R2=Ph时, 在50 ℃下目标产物50的产率为71%;当温度上升到60 ℃时, 产率提高至85%;当温度继续上升至100 ℃时, 产率反而降低至67%.此外, 作者还发现底物在空气或氮气条件下反应的产率要远低于在氧气条件下反应的产率.由此推测氧气在反应过程中可能参与构成了催化剂的循环体系.
|
|
(11) |
C—H和N—H键的三氟甲基化已经被相继实现, 而有关O—H键三氟甲基化的报道却很少. 2015年, Qing等[26]以AgOTf为介导, 2-氟吡啶52为配体, 首次报道了烷基醇51与2的三氟甲基化反应(Eq. 12), 实现了烷基醇的O-三氟甲基化, 得到目标产物烷基三氟甲基醚53, 产率为31%~92%.通过使用该方法, 各种伯醇、仲醇和叔醇以及一系列高度复杂的药物分子都能够被有效地三氟甲基化.
|
|
(12) |
同年, 该课题组[27]在此基础上, 通过引入NFSI (42)作为氧化剂, 实现了苯酚化合物54与2的三氟甲基化反应, 得到芳基三氟甲基醚55 (Eq. 13), 产率在44%~77%之间.
|
|
(13) |
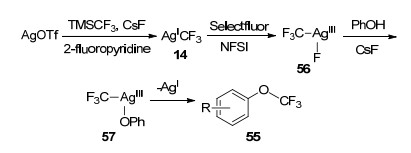
其中, 当使用Selectfluor或NFSI作为唯一的外源氧化剂时, 产率会明显降低.作者推测:该转化是由AgOTf与TMSCF3和CsF反应原位生成的AgCF3来引发. AgCF3首先与Selectfluor/或NFSI进行氧化加成形成[AgⅢ(CF3)(F)]复合物56, 随后在56中氟化物与酚氧化物交换得到关键的中间体[AgⅢ(CF3)(OPh)] (57), 最后中间体57经历还原消除得到目标产物55 (Scheme 7).
2016年, Cai等[28]以AgF为介导, TMSCF3为三氟甲基源, 同样实现了未活化烯烃58的氧化三氟甲基化反应(Eq. 14).目标产物α-三氟甲基酮59的产率为60%~90%.该反应条件较温和, 在室温下即可实现烯烃的转化.与Maiti的反应相比, 该反应虽选用价格更加昂贵的TMSCF3作为三氟甲基试剂, 但用O2代替K2S2O8作为氧化剂, 有效避免了环境污染, 且两者的反应产率相差不大.因此也是合成α-三氟甲基酮化合物的一个新思路.
|
|
(14) |
2013年, Maiti等[29]以K2S2O8为氧化剂, 报道了未活化烯烃58与三氟甲基亚磺酸钠(60)的氧化三氟甲基化反应(Eq. 15).作者使用AgNO3作为催化剂, 以廉价的CF3SO2Na作为三氟甲基源, 在室温下用敞口烧瓶进行反应, 成功得到目标产物α-三氟甲基酮59, 产率为50%~95%.该反应操作简单, 反应条件较温和, 并且具有很强的底物适用性及官能团耐受性.因此在合成、医药和农业化学中有着广泛的应用价值.
|
|
(15) |
2014年, Xiao等[30]以AgNO3为催化剂, 实现了N-芳基肉桂酰胺61与脂肪族羧酸62在水溶液中的串联脱羧自由基加成-环化反应(Eq. 16), 目标产物为3, 4-二取代二氢喹啉-2(1H)-酮衍生物63.为了在产物的3号位上引入三氟甲基, 作者对该反应进行了扩展.他们尝试使用三氟乙酸和3, 3, 3-三氟丙酸来进行反应, 结果都未能观察到预期产物的生成.值得一提的是:当选用CF3SO2Na (60)来进行反应时, 在相同的条件下作者成功实现了底物61的三氟甲基化-环化反应(Eq. 17), 但产率不高.当Ar1=Ar2=Ph, R=CH3时, 目标产物64的产率为43%.
|
|
(16) |
|
|
(17) |
2014年, Tan等[31]仍以AgNO3为催化剂, 报道了N-芳基丙烯酰胺65与CF3SO2Na的三氟甲基化-环化反应(Eq. 18), 目标产物为2-吲哚酮衍生物66.作者研究了温度变化对反应产率的影响.结果表明:反应对温度变化较为敏感, 其中最适宜的温度为40 ℃.当使用65a作为底物时, 产物66a的产率可达到81%.作者发现:底物的苯环对位上无论连的是给电子基还是吸电子基, 都能够有效地进行反应, 所得产物的产率为52%~79%;而苯环邻位连有取代基的底物虽然也能够实现转化, 但产率偏低.这表明该转化对空间位阻比较敏感.值得注意的是:当R2=Ac或H时, 反应得不到预期的目标产物66.
|
|
(18) |
炔烃通常是作为合成α-三氟甲基酮的前体.然而, 有关以炔烃为原料来制备α-三氟甲基酮的报道却很少. 2013年, Maiti等曾报道过用未活化烯烃来制备α-三氟甲基酮的反应(Eq. 15), 在此基础上, 他们[32]用O2代替K2S2O8作为氧化剂, 于2014年报道了(杂)芳基乙炔67与60的氧化三氟甲基化反应(Eq. 19).产物68的产率为45%~88%.该反应的底物适用性很广, 与乙炔相连的基团可以是烷基、芳基或杂芳基.当以苯乙炔67作为底物时, 相应的三氟甲基酮68产率最高, 达到88%;当乙炔连接的是杂芳基时, 产率有所降低.如以5-乙炔基嘧啶作为底物时, 产率降低至51%.机理研究表明该反应也是一种自由基反应.
|
|
(19) |
2015年, Hajra等[33]首次报道了以AgNO3为催化剂, 以叔丁基过氧化氢(70, tert-butyl hydroperoxide, TBHP)为氧化剂, 催化咪唑并吡啶69与60的三氟甲基化反应(Eq. 20).该反应可将CF3基团选择性地引入到咪唑环的3号位上, 得到3-(三氟甲基)咪唑并[1, 2-a]吡啶71.实验结果表明:反应必须在有氧条件下进行, 在氩气环境中反应只能得到痕量的产物.当R=H, Ar=Ph时, 71的产率达到最高的74%;作者发现:当咪唑环2号位上连接的是烷基时, 反应未能得到预期产物.
|
|
(20) |
近年来, 作为构建Cvinyl—R基团的有效方法, β-硝基苯乙烯的脱硝反应引起了广泛的关注. 2015年, Yi课题组[47]曾报道过β-硝基苯乙烯72与Togni试剂发生的脱硝三氟甲基化反应.由于Togni试剂价格昂贵, Li等[34]以60为三氟甲基源, 于2016年报道了β-硝基苯乙烯72在AgNO3催化下的脱硝三氟甲基化反应(Eq. 21).产物73的产率为30%~78%.研究表明:当苯环上的取代基R2为给电子基时, 能够获得良好的产率; 当取代基为吸电子基时, 产率较差; 而当取代基为卤素时, 对产率影响并不大.由于共轭效应, 对苯基取代的β-硝基苯乙烯72也仅以48%的产率得到相应产物73.
2015年, Zhang等[35]以AgNO3为催化剂, 叔丁基过氧化氢(70, TBHP)为氧化剂, 实现了芳基异腈74与60的串联三氟甲基化-环化反应(Eq. 22).目标产物为6-(三氟甲基)啡啶化合物75.实验结果表明:加入碱能有效提高反应的产率.当R1=R2=H时, 在无碱条件下产物75的产率只有58%;当加入Na2CO3作为碱时, 75的产率达到最高的85%.
|
|
(21) |
|
|
(22) |
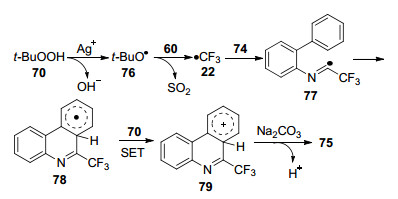
作者还提出了可能的反应机理:首先, 在痕量AgNO3存在下, 叔丁基过氧化氢70发生分解原位生成叔丁氧基自由基76, 76继而与CF3SO2Na 60反应生成关键的•CF3, •CF3随后加成到底物74上得到亚氨基自由基中间体77, 77通过分子内亲电环化转化为环己二烯基78, 78通过70的单电子转移氧化得到环己二烯基阳离子79, 最后在碱的作用下79脱质子化得到产物75 (Scheme 7).
2015年, Zhang等[36]以Ag2CO3为催化剂, 报道了芳烃1的另一种三氟甲基化方法(Eq. 23).与之前大部分反应不同, 作者选用廉价易得的三氟乙酸(80, trifluoroacetic acid, TFA)作为三氟甲基源, 除得到单取代三氟甲基的产物3外, 还得到少量双取代三氟甲基产物81.当以对苯二腈作为底物时, 作者发现:在反应烧瓶密封的情况下, 产率只有11%;而当烧瓶上有个小开口时, 产率可提高至81%.作者推测这可能是因为反应对浓度变化比较敏感.当反应在高温下进行时, 溶剂CH3CN从烧瓶开口蒸发出去, 使得反应过程中浓度逐渐增大, 产率也随之提高.该反应操作简单, 三氟甲基试剂(TFA)在反应过程中会放出CO2.为在芳环上C—H键上引入三氟甲基化提供了新的合成思路.
|
|
(23) |
2015年, Zhang等[37]设计了一种新型的三卤甲基试剂ClCF2CO2TMS (84, TCDA), 84是由二氟氯乙酸钠(82)与氯化三甲基硅烷83在60 ℃下混合反应来制备(Eq. 24).利用此试剂, 作者在银参与下成功实现了碘代芳烃85的三氟甲基化反应(Eq. 25).当底物85上R=p-COOEt时, 产物3的产率为91%.此外, 吡啶、嘧啶、噻吩等碘代杂环芳烃也具有很好的底物适应性.如当以2-氯-5-碘吡啶作为底物时, 产率高达98%.与目前常用的三氟甲基试剂相比, TCDA对缺电子化合物、中性化合物以及一些富电子的碘代(杂环)芳烃等都具有很好的普适性.
|
|
(24) |
|
|
(25) |
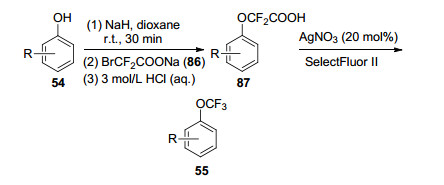
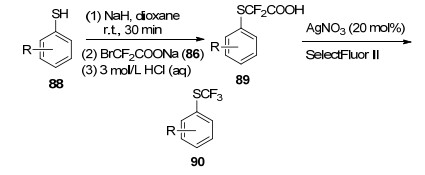
2016年, Hu课题组[38]也报道了苯酚类衍生物54的O-三氟甲基化反应, 成功合成出芳基三氟甲基醚55 (Scheme 8).作者采取分步法来进行该反应:首先选用廉价易得的溴代二氟乙酸钠(86)与苯酚类衍生物54反应, 得到中间产物87, 接着在AgNO3催化下, 加入氟试剂(SelectFluor Ⅱ)使87发生脱羧氧化, 得到目标产物芳基三氟甲基醚55.产率为26%~83%.作者还将该反应进行了扩展, 以硫酚类化合物88作为底物, 在相同的条件下进行反应, 以中等程度的产率(51%~84%)实现了目标产物三氟甲基苯硫醚90 (Scheme 9).
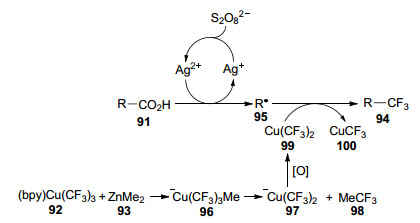
2017年, Li等[39]以AgNO3为催化剂, 报道了脂肪族羧酸91的脱羧三氟甲基化反应(Eq. 26), 得到产率为52%~88%的目标产物94.作者没有使用传统的三氟甲基试剂, 而是用(bpy)Cu(CF3)3 (92)与ZnMe2 (93)反应来制备三氟甲基源.
与绝大部分三氟甲基化反应的机理不同, 作者认为该反应经历了烷基自由基历程:首先脂肪族羧酸91在银催化下发生氧化脱羧, 得到烷基自由基95; 同时(bpy)Cu(CF3)3与ZnMe2相互作用生成-Cu(CF3)3Me (96), 其随后发生还原消除得到-Cu(CF3)2 (97)和MeCF3 (98), 前者继而被K2S2O8氧化成Cu(CF3)2 (99), 最后再与烷基自由基95反应得到目标产物94 (Scheme 10).
2008年, Yamakawa等[40]以FeSO4、H2O2和H2SO4组成催化体系, 高效实现了尿嘧啶(101)与三氟碘甲烷(102)的三氟甲基化反应(Eq. 27).产物103的产率高达97%.作者对一系列铁盐进行了研究, 发现只有以Fe(Ⅱ)化合物作为催化剂才能得到预期产物103, 其中FeSO4表现出最高的反应性.同时, 作者还发现H2O2在该反应中不可缺少.这一特性表明反应可能是以Feton氧化方式进行.该反应条件较温和, 通过使用这种催化体系, 各种尿嘧啶衍生物包括尿苷等都能够被选择性地进行5号位的三氟甲基化.
|
|
(27) |
2010年, Yamakawa等[41]在之前报道的基础上, 仍以Fe(Ⅱ)盐、H2O2和H2SO4组成催化体系, 实现了各种芳香化合物的三氟甲基化.他们同样以CF3I作为三氟甲基试剂, 以二甲基亚砜(DMSO)为溶剂, 根据底物类型的不同, 将催化体系分为四种(表 1).
 下载:
导出CSV
下载:
导出CSV
| Method | FeSO4 | Cp2Fe | H2O2 | H2SO4 |
| A | √ | √ | √ | |
| B | √ | √ | ||
| C | √ | √ | √ | |
| D | √ | √ | ||
| √ represents the presence of this compound in the system. | ||||
通过这些催化体系, 作者在温和的条件下合成了各种三氟甲基化的苯衍生物、五元杂环化合物及六元含氮化合物等, 产率为2%~96% (表 2).由于该反应为常规的一步反应, 反应条件较温和, 试剂也比较便宜, 因此具有潜在的工业化潜力.
下载:
导出CSV
| Entry | Substrate | Product | Method |
| 1 |  |
 |
D |
| 2 |  |
 |
B |
| 3 |  |
 |
A |
| 4 |  |
 |
C |
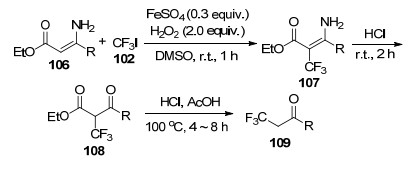
2016年, Yamakawa等[42]以Feton试剂为催化剂, 催化烯胺106与三氟碘甲烷(102)发生三氟甲基化反应, 继而在盐酸下水解得到目标产物3-氧代-2-(三氟甲基)羧酸酯108.在乙酸存在下, 羧酸酯108容易再发生脱羧反应转化为(2, 2, 2-三氟乙基)酮109 (Scheme 11).当R=Ph时, 底物的反应性最好, 所得产物108的产率高达94%, 在酸性条件下的脱羧产物109产率为88%;而当R=Me时, 产物108的产率只有64%.
2012年, Buchwald等[43]以FeCl2为催化剂, 实现乙烯三氟硼酸钾110与Togni试剂111的三氟甲基化反应(Eq. 28).研究表明:富电子化合物(如2-(杂)芳基乙烯三氟硼酸钾)是该反应的良好底物.当R为对氯苯基时, 所得产物112的产率为78% (E:Z>95:5);此外, 直链脂肪族底物虽也能很好地进行反应, 但产物的E/Z选择性较差.值得注意的是:当R为吸电子基作为底物时, 产率明显下降.如当R为环己基时, 相应产物112的产率只有34%.初步的机理研究表明:该反应是通过路易斯酸催化的碳正离子中间体进行的, 但作者也无法排除可能的自由基历程机理.
|
|
(28) |
2013年, Sodeoka等[44]报道了在Fe(OAc)2的催化下, 实现了二芳基烯丙醇113和Togni试剂的三氟甲基化反应(Eq. 29).研究表明:无论底物113上所连的芳基是否相同, 都能观察到芳基的选择性1, 2-迁移以及新的C(Ar)—C(sp3)键的形成.当Ar1=Ph, Ar2=C6F5时, 产物114的产率为79%;当Ar1=Ph, Ar2=2-BrC6H4时, 产率为81%.该反应条件较温和, 对官能团也具有很好的兼容性, 利用此方法可以从二芳基烯丙醇113得到一系列α-取代-β-三氟甲基化的羰基化合物114.
|
|
(29) |
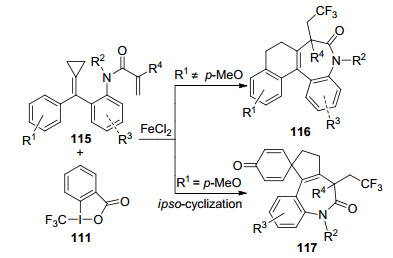
2016年, Shi等[45]以FeCl2为催化剂, 实现了N-取代丙烯酰胺115与Togni试剂的三氟甲基化反应(Scheme 12).目标产物为三氟甲基化的四环苯骈吖庚因衍生物116, 产率为34%~85%.这是首个合成七元环化合物的三氟甲基化反应.有趣的是, 当R1为对甲氧基时, 底物115先后经过ipso-环化和脱芳构化, 形成了螺环产物117, 产率为65%.
2015年, Gillaizeau等[46]首次报道了烯胺118在亚铁盐催化下的三氟甲基化反应(Eq. 30).作者以FeCl2为催化剂, 以Togni试剂为三氟甲基源, 在温和的反应条件下实现了底物的三氟甲基化, 产物119产率为19%~95%.该反应的底物适用范围比较广泛, 官能团耐受性也较好.研究表明:该转化在烯胺的3号位上表现出完全的区域选择性, 因此在后期氟化策略中具有重要的作用.
|
|
(30) |
2015年, Yi等[47]以Togni试剂为三氟甲基源, 在无过渡金属催化体系下实现了α, β-不饱和羧酸120的脱羧三氟甲基化反应(Eq. 31).该反应具有较高的立体选择性, 官能团耐受性也较好, 所得产物73的产率为68%~96%.作者以β-硝基苯乙烯72作为底物, 在相同的条件下进行反应, 虽然成功得到了三氟甲基化产物73, 但产率过低.因此, 作者尝试添加金属催化剂来解决该问题.经过仔细筛选后, 作者选用三乙酰丙酮铁121作为催化剂, 以良好的收率(61%~84%)得到相应的三氟甲基化产物73 (Eq. 32).
|
|
(31) |
|
|
(32) |
2016年, Deng等[48]同样以FeCl2为催化剂, 在温和的条件下实现了乙烯基环丙烷122的三氟甲基化及分子内环化反应(Eq. 33), 实现了目标产物三氟甲基的二氢萘衍生物123, 产率为17%~96%.实验结果表明:底物在40 ℃下只需10~30 min就能高效地实现转化; 在室温条件下, 减少催化剂的用量, 都会增延反应的时间, 降低产率.该反应对许多官能团具有良好的耐受性:当R1=H, R2=p-Toly, R3=Me时, 产物123的产率高达96%;然而, 当R1=R2=H, R3=Et时, 产率仅有17%.目标产物123可进一步催化氢化可以转化为相应的四氢化萘衍生物.
|
|
(33) |
2013年, Maiti等[49]尝试了以FeCl3为催化剂, 实现了α, β-不饱和羧酸120与三氟甲基亚磺酸钠(60)的脱羧三氟甲基化反应(Eq. 34).产物73的产率为54%~82%.研究表明:带有富电子基或缺电子基的肉桂酸都能够有效地进行反应, 并且相应产物73具有良好的立体选择性(E:Z>99:1).当苯环邻位连有羟基时, 所得产物73的产率为74%;由于具有未保护的Ar—OH基团, 其可进一步应用于C—O交叉偶联反应以及一系列其他的转化.值得注意的是:苯环邻位连有甲氧基时所得产物的产率要比对位连有甲氧基所得产物的产率低.
|
|
(34) |
2016年, Qing课题组[50]以CF3SO2Na (60)为三氟甲基源, 在FeCl3催化下实现了苯乙烯或脂肪族烯烃124的三氟甲基化反应, 成功合成出氯三氟甲基化合物125, 产率为51%~93% (Eq. 35).
|
|
(35) |
2014年, Plietker等[51]报道了以亲核的高铁酸盐Bu4N[Fe(CO)3(NO)] (127, TBAFe)为催化剂, 实现醛、酮化合物126与2的三氟甲基化反应(Eq. 36).产物128的产率为42%~99%.当底物126上R1=Ph, R2=H时, 产物128的产率高达97%.研究表明:低价态的高铁酸盐能够活化Me3SiCF3中的C—Si σ-键, 基于此, 作者用醋酸烯丙酯130来截断C—Si σ-键活化, 实现了缺电子烯烃129的串联三氟甲基化-烯丙基化反应, 生成目标产物131 (Eq. 37).
|
|
(36) |
|
|
(37) |
2015年, Goosen等[52]以FeCl2为催化剂, 报道了有机硫氰酸酯132与三氟乙酸钾(133)的三氟甲基化反应(Eq. 38).作者认为:三氟乙酸钾(133)首先在FeCl2强烈促进下发生脱羧反应, 形成了关键的亲核三氟甲基源, 随后再与硫氰酸酯132反应得到三氟甲基硫醚134, 反应的副产物KCN被FeCl2吸收形成无毒的六氰合铁酸钾.当R为(对苯氧基)苯基时, 相应产物134产率高达98%.作者将该反应进行了扩展, 以醛化合物135为底物, 在相同的反应条件下得到产物三氟甲基醇136, 产率为60% (Eq. 39).
|
|
(38) |
|
|
(39) |
早在2004年, Hartwig和Culkin[53]就合成了DPPBz- ligated芳基钯三氟甲基复合物, 这种复合物在高达130 ℃的温度下能够稳定存在数天, 并且不会引起还原消除反应, 而是生成三氟甲基取代产物138 (Eq. 40).
|
|
(40) |
之后, 在2006年, Grushin和Marshall[54]报道了一种Xantphos-based钯络合物体系, 该体系在过量Xantphos存在的温和条件下(50~80 ℃), 能够得到三氟甲基化产物(Eq. 41).在这里Xantphos对该络合体系的还原消除至关重要.如果将Xantphos替换成DPPE, 在碘苯存在下, 反应将生成联苯化合物142 (Eq. 42).
|
|
(41) |
|
|
(42) |
2010年, Sanford及其同事[55]报道了由PdⅣ复合物介导的Ar—CF3键形成反应.在温和条件下, 这些转化采用不同的含氮、膦辅助配体进行.由于PdⅡ(Ar)(CF3)络合物的还原消除会受到抑制, 因此反应中需加入N- fluoro-2, 4, 6-trimethylpyridinium triflate (简称: NFTPT).因为氧化形成的PdⅣ络合物可以通过还原消除来释放ArCF3, 产率在28%~89%之间(Scheme 13).

2011年, Buchwald课题组[56]就报道了钯参与的三氟甲磺酸环己烯酯的三氟甲基化反应.他们用Pd(dba)2或[(allyl)PdCl]2和单齿联芳基膦配体tBuXPhos (147)组成催化体系, 在温和的反应条件下, 在底物146上实现了三氟甲基化, 生成产物148 (Eq. 43).
2012年, Liu等[57]报道了钯与配体150络合形成的催化体系, 催化活化烯烃149的三氟甲基化-环化反应(Eq. 44).作者发现:在三氟甲基磺酸镱存在下, 反应能够更有效地进行, 得到目标产物三氟甲基取代的吲哚酮衍生物151, 产率为43%~89%.该反应对卤素官能团具有很好的兼容性.当R1为卤素基团时, 所得产物的产率较高(79%~89%).值得注意的是:当R1=R3=H, R2=Ts(对甲苯磺酰基)时, 反应未得到预期产物.初步的机理研究表明:反应涉及到C—PdⅣ(CF3)中间体的生成, 其经历了还原消除生成了C—CF3键.
|
|
(43) |
|
|
(44) |
同年, Liu等[58]仍以醋酸钯和配体150组成的催化体系, 实现了吲哚衍生物152的氧化三氟甲基化反应(Eq. 45).研究表明:无论是延长反应时间, 增加反应物的浓度或是升高反应温度, 都不能提高目标产物154的产率.而在这些条件下原料往往被消耗殆尽; 与此同时, 作者还分离出少量苯环上含有三氟甲基的副产物.作者认为反应中可能涉及到自由基过程, 并导致了副反应的发生.因此, 他往反应体系中加入了自由基抑制剂153 (2, 2, 6, 6-tetramethylpiperidine-1-oxyl, TEMPO), 结果产物的产率显著提高.当R1=CH3, R2=CH3, R3=H时, 产物154的产率达到83%;而由于快速分解, 3-甲基吲哚(R1=CH3, R2=H, R3=H)与氧化条件不相容, 不能得到预期的产物.
2011年, Samant等[59]在反胶束介质中, 以钯和配体156组成催化体系LPd, 实现了溴代芳烃155与三氟甲基试剂2的三氟甲基化反应(Eq. 46).实验结果表明:往体系中加入十二烷基硫酸钠(157, Sodium dodecyl sulfate, 157, SDS), 能够显著提高产物158的产率, 产率为68%~80%.作者认为, 这可能是因为试剂是相互分离的. TMSCF3/CsF存在于亲水核中, 而ArBr存在于栅栏层中; 在亲水核中试剂发生金属转移反应, 而SDS反胶束的各向异性栅栏层则是为LPd和ArBr提供了一个有效的氧化加成反应位点, 形成了ArLPdBr.在金属转移反应过程中阻止了TMSCF3的分解, 并且对ArLPdCF3还原消除生成ArCF3过程有着促进作用.该反应的底物适用性较广, 适用于具有醛基、羰基、羟基和氨基等官能团的溴代芳族化合物.
|
|
(45) |
|
|
(46) |
2014年, Georg等[60]在无过渡金属催化体系下, 实现了底物161的三氟甲基化.作者首先以159作为底物, 尝试了Liu所采用的反应体系(Eq. 44), 成功得到三氟甲基化产物160, 产率达到69% (Eq. 47).然而, 作者发现:当不加入Pd(OAc)2时, 反应也能够进行, 产率为46%.基于此发现, 作者优化了反应条件, 改用MeCN作为溶剂, 以KF代替CsF, 发展了无过渡金属催化体系来进行反应, 最终以59%的产率得到产物160, 并将该反应条件应用于底物161的三氟甲基化(Eq. 48), 产物162的产率为23%~81%.该反应条件温和, 操作简单, 采用了无过渡金属催化体系, 避免了过渡金属对环境造成的污染, 是一种绿色环保的一种合成方法.
2009年, Yu等[61]采用C—H活化的方法, 报道了乙酸钯(Ⅱ)催化2-取代苯基吡啶163与Umemoto试剂164的三氟甲基化反应(Eq. 49).在该反应中, 底物163苯环上所连的吡啶基是作为导向基, 反应主要得到邻位三氟甲基化产物167.产物区域选择性好, 产率为54%~88%.研究表明: TFA的使用对于该反应的成功至关重要.同时, 往体系中加入氧化剂Cu(OAc)2 (166)也能够提高反应的产率.当苯环连有强吸电子基时, 产物167的产率比较低; 而连有给电子基时, 产率有所提高.此外, 萘底物在该条件下也能被三氟甲基化.反应还可以扩展到使用连有其他导向基(如嘧啶基)的底物来进行反应.
|
|
(49) |
2013年, Shi等[62]以Pd(OAc)2为催化剂, 报道了首例取代的乙酰苯胺168与Umemoto试剂的三氟甲基化反应(Eq. 50).该反应在不需要配体的情况下, 高效实现了底物168的邻位三氟甲基化.当R1=p-COOEt, R2=CH3时, 产物169产率达到83%;然而, 当N原子上连有甲基时, 反应未得到相应的产物.此外, 作者还发现:当苯环间位连有甲氧基或三氟甲基时, 反应明显受到抑制, 也得不到预期产物169.该反应对许多官能团具有良好的耐受性, 广泛的底物范围使得该方法潜在地适用于合成多种生物活性分子.
|
|
(50) |
2012年, Yu等[63]在之前课题的基础上(Eq. 49), 仍以Pd(OAc)2为催化剂, 实现了N-芳基苯甲酰胺170的三氟甲基化反应(Eq. 51).与之前的反应体系略有不同, 该反应必须在N-甲基甲酰胺添加剂172存在下才能发生.这表明了该添加剂可能在反应中起到作为配体及碱的重要作用.该反应能兼容多种官能团, 尤其对给电子基团的耐受性比较好.当苯环间位连有CH3时, 相应产物173的产率高达94%;然而, 当苯环对位连有强吸电子基团CF3时, 产率降低至32%.
|
|
(51) |
2013年, Yu等[64]仍以钯为催化剂, 报道了N-未取代苄胺化合物174与Umemoto试剂的三氟甲基化反应(Eq. 52).邻位三氟甲基产物175产率能达到83%.同样的, 反应中加入Cu(OAc)2 (166)能够提高产物的产率.作者认为:乙酸铜可以清除反应过程中释放的二苯并噻吩, 这些游离的杂质可能会妨碍C—H活化过程.研究表明:适当降低TFA的用量也能提高产物的产率.这表明游离胺和胺盐之间的平衡比率是比较重要的.与之前的反应(Eq. 51)不同, 甲酰胺配体作为促进剂对该底物174无效.经过反复试验, 作者指出加入水和氧化银能够有效促进反应的进行.
|
|
(52) |
2005年, Chen等[65]开发了一种合成三氟甲基化卟啉的简便方法.他们在催化量Pd2(dba)3•CHCl3/AsPh3的存在下, 用FSO2CF2COOMe/CuI与各种溴化卟啉进行反应, 成功实现了底物的三氟甲基化(Eq. 53), 产率最高达到95%.这些三氟甲基化的卟啉可进一步应用于许多重要的领域, 比如催化、材料、仿生学和药物学等等.
|
|
(53) |
目前, 将三氟甲基引入到卤代芳烃上的方法有很多, 其中大多数方法的反应条件比较苛刻, 同时反应还会受到底物范围的限制. 2010年, Buchward等[66]首次实现了Pd0/PdⅡ催化氯代芳烃178的三氟甲基化反应(Eq. 54).作者使用大位阻膦配体进行反应, 有利于钯催化过程的还原消除步骤, 有效攻克了ArLPdCF3配合物还原消除生成ArCF3的难题.在该反应中, 作者以TESCF3 179为三氟甲基试剂, 所得三氟甲基化产物3的产率为70%~94%.值得注意的是:因为KF具有吸湿性, 所以所有的反应都是在充满N2的手套箱中进行, 防止反应过程中TESCF3发生水解.该方法的底物适用性也比较广泛, 可适用于酯、酰胺、醚、缩醛、腈和叔胺等.
|
|
(54) |
2016年, Beller等[67]报道了钯与配体183络合形成的催化体系, 催化(杂环)芳烃181与三氟溴甲烷(182)的三氟甲基化反应(Eq. 55).这是钯催化体系下首个使用CF3Br试剂来实现芳烃三氟甲基化的反应.作者以Cs2CO3作为碱, 添加了TEMPO作为温和的氧化还原介体, 在130 ℃下反应40~50 h, 实现了各种(杂环)芳烃的三氟甲基化, 产率为25%~81%.与之前报道的大多数反应相比, 该反应的条件虽然不是很温和, 但其优点在于不需要使用昂贵的三氟甲基试剂和强氧化剂, 同时反应具有良好的区域选择性和化学选择性.
|
|
(55) |
2015年, Nevado等[68]报道了以双(三苯基磷)二氯化钯188为催化剂, 催化末端炔烃185与硼酸化合物186及全氟烷基碘化物187的三组分反应(Eq. 56), 以高度区域化学控制和立体化学控制的方式得到了三取代全氟烯烃(189).其中, 当底物R1=Ph, R2=p-tBu-C6H4, Rf=CF3时, 得到了相应的三氟甲基化产物189, 产率为47%.
|
|
(56) |
2014年, Fu等[69]首次报道了合成三氟甲基化噁唑啉的反应.该反应涉及到烯烃与三氟甲基亚磺酸钠的分子间三氟甲基化以及分子内环化.作者首先以4-甲基- N-(2-苯基烯丙基)苯甲酰胺(190)作为底物, 以一系列过渡金属盐作为催化剂, 成功合成出目标产物191 (Eq. 57).其中, 当以NiCl2作为催化剂时, 产物191的产率为63%;然而, 当不加入任何金属盐时, 在相同条件下, 产率最高达到72%(表 3).显然, 过渡金属盐在这里抑制了反应的进行.在此基础上, 作者进行了改进, 他以无过渡金属催化体系, 实现了各种N-烯丙基胺化合物192与CF3SO2Na的三氟甲基化-环化反应, 得到各种含有三氟甲基的噁唑啉衍生物193 (Eq. 58), 产率为38%~80%.
下载:
导出CSV
| Entry | Cat. | Oxidant | T/℃ | Yield/% |
| 1 | CuI | PhI(OAc)2 | 90 | 53 |
| 2 | Pd(OAc)2 | PhI(OAc)2 | 90 | 40 |
| 3 | FeCl3 | PhI(OAc)2 | 90 | 54 |
| 4 | NiCl2 | PhI(OAc)2 | 90 | 63 |
| 5 | — | PhI(OAc)2 | 90 | 72 |
|
|
(57) |
|
|
(58) |
2017年, Zhang等[70]首次报道了以镍为催化剂, 在水中实现了芳香胺195与三氟甲基亚磺酸钠的三氟甲基化反应(Eq. 59).在该转化中, 作者使用吡啶酰胺作为导向基, 在NiSO4•6H2O催化下, 以良好的产率实现了底物194的C—H三氟甲基化.当R=p-Me时, 产物195的产率为71%.作者发现:当底物194的吡啶环被替换为呋喃环或噻吩环时, 反应并不能得到相应的产物.这说明了氮杂环和酰胺在该反应中具有重要作用, 其可能是作为金属原子的双齿结合位点.该反应的优点在于这种含水的催化剂体系能够至少重复使用八次, 虽然在循环过程中催化剂的活性会略有降低.目前, 该反应已成功应用于酸性红266的高效合成.
|
|
(59) |
2016年, Wang等[71]首次报道了以[NiCl2(dppp)] 197为催化剂, 催化咪唑并[1, 2-a]吡啶化合物196与三氟碘甲烷的三氟甲基化反应(Eq. 60).研究表明: R2在苯环上的位置不同, 对目标产物198的产率有所影响.当R1=H, R2=o-Me时, 198的产率只有51%;当R1=H, R2=m-Me时, 产率为73%;当R1=H, R2=p-Me时, 产率则达到80%.
|
|
(60) |
作者认为可能的反应机理是:原位生成的Ni(I)配合物199和CF3I先通过单电子氧化形成•CF3, 接着与底物196发生加成反应得到自由基中间体200, 随后200被Ni(Ⅱ)Ln 197氧化生成碳正离子中间体201.最后, 在碱triethylenediamine(简称: DABCO)作用下发生脱质子化得到目标产物(Scheme 14).

2004年, Ando等[72]首次报道了以铑为催化剂, 实现了α, β-不饱和酮202与三氟碘甲烷(102)的三氟甲基化反应(Eq. 61).目标产物为α-三氟甲基酮(203), 产率为31%~77%.实验结果表明:底物202的双键β位上连有两个取代基时可能会抑制反应的进行.如当以β, β-二甲基烯酮作为底物时, 反应并不能得到预期的产物.此外, 当R1=Ph时, 产物的产率有所降低.这可能是因为苯环的作用导致羰基上的电子得到分散.作者将该反应进行了扩展, 他以卤代全氟烷基化合物(RfX)代替CF3I来进行反应, 成功合成出相应的α-全氟烷基酮.
|
|
(61) |
2008年, Ando等[73]在之前课题的基础上(Eq. 61), 仍以RhCl(PPh3)3和Et2Zn组成的催化体系, 催化烯醇硅醚204与三氟碘甲烷的三氟甲基化反应(Eq. 62).产物为α-三氟甲基酮205.在之前的课题中, 作者得不到季碳原子连接三氟甲基的产物.该反应则克服了这个问题:当底物204上R1=Ph, R2=R3=CH3时, 反应能够得到季碳原子连接三氟甲基的产物205, 产率为23%.值得注意的是:当R1=Ph, R2=R3=H时, 反应并没有得到预期产物, 而是得到其二聚产物207 (Eq. 63), 产率为84%.同样, 该反应也可以使用卤代全氟烷基化合物来实现底物的α-全氟烷基化.
|
|
(62) |
|
|
(63) |
2016年, Szabó等[74]以[Rh2(OAc)4]为催化剂, 报道了重氮羰基化合物208的一个双官能团化新反应.作者以稳定易得的苯并碘氧杂戊环试剂111作为氟源, 在反应体系中通过添加醇类化合物209来引入第二种官能团, 从而使底物208实现氟化和烷氧化反应.其中, 作者用Togni试剂作为三氟甲基源来进行反应, 在温和的条件下实现了底物的三氟甲基化-烷氧化(Eq. 64).产物210的产率为30%~85%.该反应在室温下只需15 min就能实现转化.因此, 这种方法适用于快速的后期氟化和三氟甲基化反应.
|
|
(64) |
2015年, Shen等[75]以铑为催化剂, 设计了一种新型的三氟甲基亲电试剂213, 通过一步反应实现了β-酮酸酯214的三氟甲基化反应(Scheme 15).产率为48%~75%.与常用的三氟甲基试剂相比, 这种新型的试剂较易合成, 成本低, 且在水分和空气中比较稳定.
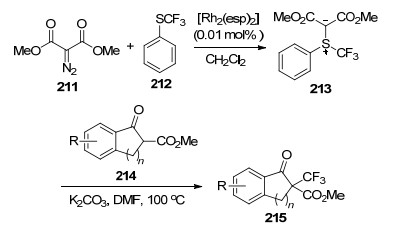
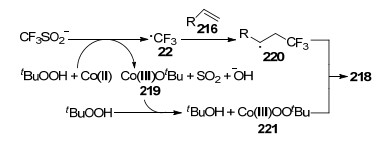
2017年, Zhang等[76]以钴为催化剂, 首次报道了烯烃216与三氟甲基试剂60和氢过氧化物217的三氟甲基化-过氧化反应(Eq. 65).目标产物218的产率为25%~79%.在该反应体系中, 氢过氧化物217主要作为自由基引发剂和偶合配体.值得注意的是, 该反应必须在氩气环境中进行.
|
|
(65) |
作者认为可能的反应机理是: Co(OAc)2和CF3SO2Na首先在TBHP存在下分别生成Co(Ⅲ)叔丁氧基配合物219和三氟甲基自由基(22).随后自由基进攻底物216的双键得到烷基自由基220.同时配合物219与TBHP反应得到Co(Ⅲ)中间体叔丁基过氧化合物221, 最后烷基自由基220和221之间发生交叉偶合得到目标产物218 (Scheme 16).
2017年, Hisaeda等[77]首次报道了以维生素B12衍生物作为钴催化剂, 实现芳烃1的电化学三氟甲基化及全氟烷基化反应(Eq. 66).作者以甲醇为溶剂, 通过控制电位在0.8 V (vs. Ag/AgCl), 电解制备出维生素B12衍生物Co(I)物质, 接着与氟烷基化试剂反应形成Co-Rf复合物.该复合物在可见光照射下会释放出Rf基团, 随后直接与未活化的(杂)芳烃1反应, 得到预期的氟烷基化产物222.由于维生素B12骨架固有的高稳定性, 因此该反应的TON相对较高(超过100).
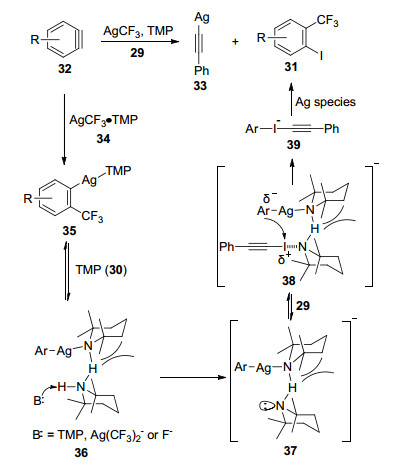
2011年, Weng等[78]同样以TMSCF3为三氟甲基试剂, 报道了AgF与CuI协同催化碘代芳烃85的三氟甲基化反应(Eq. 67), 并提出了可能的反应机理.反应体系中AgF首先与N, N-二甲基乙二胺(223, N, N-dimethyl- ethylenediamine, DMEDA)络合, 然后和三氟甲基试剂2发生转移金属化反应, 得到AgCF3双氮配体络合物224, 224随即与CuI进一步反应得到铜络合物226, 最后与碘代芳烃85发生反应, 从而实现碘代芳烃的三氟甲基化(Scheme 17).
|
|
(66) |
|
|
(67) |
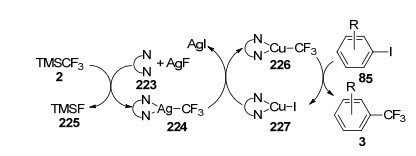
2012年, Fu等[79]同样以Cu盐为催化剂, 在AgF参与下实现了烷基硼酸的三氟甲基化反应.实验结果表明:伯烷基硼酸228在CuI/AgF作用下, 能够以良好的产率实现三氟甲基化, 产率最高可达到78% (Eq. 68).然而, 该体系并不适用于仲烷基硼酸231的转化.经过仔细筛选后, 作者得出:以[CuOTf]2•0.5C6H6为催化剂, AgOTs为氧化剂, 在50 ℃下反应才是仲烷基硼酸231实现三氟甲基化的最优条件, 产率最高只有54% (Eq. 69).这是首例烷基硼酸衍生物在Cu催化下实现C—C键的交叉偶联反应.通过使用该方法, 许多环状和非环状的烷烃都能成功被三氟甲基化.
|
|
(68) |
|
|
(69) |
Maiti等曾以铁盐为催化剂, 报道过肉桂酸的脱羧三氟甲基化反应(Eq. 41). 2014年, Li等[80]在此基础上, 以CuCl/Ag2CO3为催化剂, 在相对温和的条件下实现了α, β-不饱和羧酸120的脱羧三氟甲基化(Eq. 70).产物73表现出较高的E/Z立体选择性.作者发现:当不饱和羧酸的双键β位上连接的是甲基或苯基时, 反应可以更有效地进行.如当底物为120a时, 目标产物73的产率为58%, E/Z>99:1;当底物为120b时, 产率为62%, E:Z>98:2;当底物为120c时, 产率达到64%, E:Z>98:2.这些含有卤素基团的三氟甲基化产物73可进一步应用于其他的化学转化, 如Heck反应和Suzuki反应等.
|
|
(70) |
本文从过渡金属催化剂和三氟甲基化试剂的种类出发, 综述了近几年过渡金属催化三氟甲基化反应的研究进展.从以上的论述可以看出, 将三氟甲基引入到有机分子结构中已经引起化学家的广泛关注, 并且已经取得了很大的进展.目前常用的三氟甲基试剂有Ruppert试剂、Togni试剂、Umemoto试剂和Langlois试剂等.三氟甲基在反应中存在的中间体形式主要有三种:三氟甲基自由基和三氟甲基正负离子.所以随着反应中三氟甲基试剂的不同, 三氟甲基化反应的机理也有所不同.目前存在的主要问题有:对于钯催化的三氟甲基化反应, 体系中往往需要加入配体才能实现转化.由于钯与所用配体价格昂贵, 这将不利于大规模的工业化应用.所以, 寻找高效廉价的配体以及其他廉价的金属催化剂是一大挑战; 其次, 有些三氟甲基试剂也相对较贵.比如最常用的亲电三氟甲基试剂TMSCF3.虽然也有廉价易得的试剂比如CF3SO2Na, 但由于其在有机溶剂中溶解度较低, 会影响反应性, 所以应用范围也受到一些限制.所以寻找和发展新型的三氟甲基试剂仍是将来研究的方向; 另外, 尽管过渡金属催化三氟甲基化反应类型有很多, 但这些方法目前只适用于实验室的合成.因此寻找成本低、反应条件温和、催化剂用量少的三氟甲基化反应, 并将其应用于工业化生产, 是值得关注和发展的另一个方向; 最后, 三氟甲基化反应机理的进一步研究也值得深入探讨.
(a) Kirk, K. L. Org. Process Res. Dev. 2008, 12, 305.
(b) Kirsch, P. ; Bremer, M. Angew. Chem. , Int. Ed. 2000, 39, 4216.
Wong, D.-T.; Perry, K. W.; Bymaster, F. P. Nat. Rev. Drug. Discovery 2005, 4, 764. doi: 10.1038/nrd1821
Margot, P.; Huggenberger, F.; Amrein, J.; Weiss, B. BCPC Conf.-Pests Dis. 1998, 2, 375.
Reiffenrath, V.; Krause, J.; Plach, H. J.; Weber, G. Liq. Cryst. 1989, 5, 159. doi: 10.1080/02678298908026359
(a) Swarts, F. Bull. Acad. R. Belg. 1892, 24, 309.
(b) Boswell, G. A., Jr. ; Ripka, W. C. ; Scribner, R. M. ; Tullock, C. W. Org. React. 1974, 21, 1.
Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 470. doi: 10.1038/nature10108
Lundgren, R. J.; Stradiotto, M. Angew. Chem., Int. Ed. 2010, 49, 9322. doi: 10.1002/anie.201004051
Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475. doi: 10.1021/cr1004293
Roy, S.; Gregg, B. T.; Gribble, G. W.; Le, V. D. Tetrahedron Lett. 2011, 67, 2161. doi: 10.1016/j.tet.2011.01.002
王光祖, 赫侠平, 戴建军, 许华建, 有机化学, 2014, 34, 837. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract343728.shtmlWang, G.-Z.; He, X.-P.; Dai, J.-J.; Xu, H.-J. Chin. J. Org. Chem. 2014, 34, 837(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract343728.shtml
吕翠萍, 沈其龙, 刘丹, 有机化学, 2012, 32, 1380. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract341101.shtmlLü, C.-P.; Shen, Q.-L.; Liu, D. Chin. J. Org. Chem. 2012, 32, 1380(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract341101.shtml
戚自松, 董亚丽, 李亚明, 段春迎, 化学进展, 2012, 24, 2178.Qi, Z.-S.; Dong, Y.-L.; Li, Y.-M.; Duan, C.-Y. Prog. Chem. 2012, 24, 2178(in Chinese).
潘菲, 施章杰, 化学学报, 2012, 70, 1679.Pan, F.; Shi, Z.-J. Acta Chim. Sinica 2012, 70, 1679(in Chinese).
Ye, Y.-D.; Lee, S. H.; Sanford, M. S. Org. Lett. 2011, 13, 5464. doi: 10.1021/ol202174a
Han, J.-B.; Xu, B.; Hammond, G. B. Org. Lett. 2011, 13, 3450. doi: 10.1021/ol2011902
Hafner, A.; Br se, S. Angew. Chem., Int. Ed. 2012, 51, 3713. doi: 10.1002/anie.v51.15
Wang, X.; Xu, Y.; Zhou, Y.-J.; Zhang, Y.; Wang, J.-B. Synthesis 2014, 46, 2143. doi: 10.1055/s-00000084
Wu, X.-Y.; Chu, L.-L.; Qing, F.-L. Angew. Chem., Int. Ed. 2013, 52, 2198. doi: 10.1002/anie.201208971
Wang, K.-P.; Yun, S.-Y.; Mamidipalli, P.; Lee, D. Chem. Sci. 2013, 4, 3205. doi: 10.1039/c3sc50992c
Zeng, Y.-W.; Zhang, L.-J.; Zhao, Y.-C.; Ni, C.-F.; Zhao, J.-W.; Hu, J.-B. J. Am. Chem. Soc. 2013, 135, 2955. doi: 10.1021/ja312711c
Liu, Q.-L.; Wu, Y.-C.; Chen, P.-H.; Liu, G.-S. Org. Lett. 2013, 15, 6210. doi: 10.1021/ol403059z
Wang, X.-B.; Qiu, G.-Y.-S.; Zhang, L.; Wu, J. Tetrahedron Lett. 2014, 55, 962. doi: 10.1016/j.tetlet.2013.12.064
Yu, W.; Xu, X-H.; Qing, F-L. Adv. Synth. Catal. 2015, 357, 2039. doi: 10.1002/adsc.v357.9
SeO, S.; Taylor, J. B.; Greaney, M. F. Chem. Commun. 2013, 49, 6385. doi: 10.1039/c3cc41829d
Teng, F.; Cheng, J.; Bolm, C. Org. Lett. 2015, 17, 3166. doi: 10.1021/acs.orglett.5b01537
Liu, J.-B.; Xu, X.-H.; Qing, F.-L. Org. Lett. 2015, 17, 5048. doi: 10.1021/acs.orglett.5b02522
Liu, J.-B.; Chen, C.; Chu, L.; Chen, Z.-H.; Xu, X.-H.; Qing, F.-L. Angew. Chem., Int. Ed. 2015, 54, 11839. doi: 10.1002/anie.201506329
Wu, Y.-B.; Lu, G.-P.; Yuan, T.; Xu, Z.-B.; Wan, L.; Cai, C. Chem. Commun. 2016, 52, 13668. doi: 10.1039/C6CC08178A
Deb, A.; Manna, S.; Modak, A.; Patra, T.; Maity, S.; Maiti, D. Angew. Chem., Int. Ed. 2013, 52, 9747. doi: 10.1002/anie.201303576
Mai, W.-P.; Wang, J.-T.; Yang, L.-R.; Yuan, J.-W.; Xiao, Y.-M.; Mao, P.; Qu, L.-B. Org. Lett. 2014, 16, 204. doi: 10.1021/ol403196h
Liu, J.-D.; Zhuang, S.-B.; Gui, Q.-W.; Chen, X.; Yang, Z.-Y.; Tan, Z. Eur. J. Org. Chem. 2014, 3196.
Maji, A.; Hazra, A.; Maiti, D. Org. Lett. 2014, 16, 4524. doi: 10.1021/ol502071g
Monir, K.; Bagdi, A. K.; Ghosh, M.; Hajra, A. J. Org. Chem. 2015, 80, 1332. doi: 10.1021/jo502928e
Huang, P.; Li, Y.-M.; Fu, X.-M.; Zhang, R.; Jin, K.; Wang, W.-X.; Duan, C.-Y. Tetrahedron Lett. 2016, 57, 4705. doi: 10.1016/j.tetlet.2016.09.016
Liu, Y.-R.; Tu, H.-Y.; Zhang, X.-G. Synthesis 2015, 47, 3460. doi: 10.1055/s-00000084
Shi, G.-F.; Shao, C.-D.; Pan, S.-L.; Yu, J.-X.; Zhang, Y.-H. Org. Lett. 2015, 17, 38. doi: 10.1021/ol503189j
Zhang, X.-M.; Wang, J.; Wan, Z.-H. Org. Lett. 2015, 17, 2086. doi: 10.1021/acs.orglett.5b00619
Zhou, M.; Ni, C.-F.; He, Z.-B.; Hu, J.-B. Org. Lett. 2016, 18, 3754. doi: 10.1021/acs.orglett.6b01779
Tan, X.-Q.; Liu, Z.-L.; Shen, H.-G.; Zhang, P.; Zhang, Z.-Z.; Li, C.-Z. J. Am. Chem. Soc. 2017, 139, 12430. doi: 10.1021/jacs.7b07944
Uraguchi, D.; Yamamoto, K.; Ohtsuka, Y.; Tokuhisa, K.; Yamakawa, T. Appl. Catal., A 2008, 342, 137. doi: 10.1016/j.apcata.2008.03.009
Kino, T.; Nagase, Y.; Ohtsuka, Y.; Yamamoto, K.; Uraguchi, D.; Tokuhisa, K.; Yamakawa, T. J. Fluorine Chem. 2010, 131, 98. doi: 10.1016/j.jfluchem.2009.09.007
Ohtsuka, Y.; Uraguchi, D.; Yamamoto, K.; Tokuhisa, K.; Yamakawa, T. J. Fluorine Chem. 2016, 181, 1. doi: 10.1016/j.jfluchem.2015.10.013
Parsons, A. T.; Senecal, T. D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2012, 51, 2947. doi: 10.1002/anie.201108267
Egami, H.; Shimizu, R.; Usui, Y.; Sodeoka, M. Chem. Commun. 2013, 49, 7346. doi: 10.1039/c3cc43936d
Yu, L.-Z.; Xu, Q.; Tang, X.-Y.; Shi, M. ACS Catal. 2016, 6, 526. doi: 10.1021/acscatal.5b02400
Rey-Rodriguez, R.; Retailleau, P.; Bonnet, P.; Gillaizeau, I. Chem.-Eur. J. 2015, 21, 3572. doi: 10.1002/chem.v21.9
Ma, J.-J.; Yi, W.-B.; Lu, G.-P.; Cai, C. Adv. Synth. Catal. 2015, 357, 3447. doi: 10.1002/adsc.201500631
Li, Z.-R.; Bao, X.-X.; Sun, J.; Shen, J.; Wu, D.-Q.; Liu, Y.-K.; Deng, Q.-H.; Liu, F. Org. Chem. Front. 2016, 3, 934. doi: 10.1039/C6QO00166A
Patra, T.; Deb, A.; Manna, S.; Sharma, U.; Maiti, D. Eur. J. Org. Chem. 2013, 2013, 5247.
Yang, B.; Xu, X.-H.; Qing, F.-L. Chin. J. Chem. 2016, 34, 465. doi: 10.1002/cjoc.201500641
Klein, J. E. M. N.; Rommel, S.; Plietker, B. Organometallics 2014, 33, 5802. doi: 10.1021/om5005012
Exner, B.; Bayarmagnai, B.; Jia, F.; Goossen, L. J. Chem.-Eur. J. 2015, 21, 17220. doi: 10.1002/chem.201503915
Culkin, D. A.; Hartwig, J. F. Organometallics 2004, 23, 3398. doi: 10.1021/om049726k
Grushin, V. V.; Marshall, W. J. J. Am. Chem. Soc. 2006, 128, 4632. doi: 10.1021/ja0602389
Ball, N. D.; Kampf, J. W.; Sanford, M. S. J. Am. Chem. Soc. 2010, 132, 2878. doi: 10.1021/ja100955x
Cho, E. J.; Buchwald, S. L. Org. Lett. 2011, 13, 6552. doi: 10.1021/ol202885w
Mu, X.; Wu, T.; Wang, H.-Y.; Guo, Y.-L.; Liu, G.-S. J. Am. Chem. Soc. 2012, 134, 878. doi: 10.1021/ja210614y
Mu, X.; Chen, S.-J.; Zhen, X.-L.; Liu, G.-S. Chem.-Eur. J. 2011, 17, 6039. doi: 10.1002/chem.v17.22
Samant, B. S.; Kabalka, G. W. Chem. Commun. 2011, 47, 7236. doi: 10.1039/c1cc12098k
Yu, Y.-Y.; Ranade, A. R.; Georg, G. I. Adv. Synth. Catal. 2014, 356, 3510. doi: 10.1002/adsc.v356.17
Wang, X.-S.; Truesdale, L.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 3648. doi: 10.1021/ja909522s
Zhang, L.-S.; Chen, K.; Chen, G.-H.; Li, B.-J.; Luo, S.; Guo, Q.-Y.; Wei, J.-B.; Shi, Z.-J. Org. Lett. 2013, 15, 10. doi: 10.1021/ol302814x
Zhang, X.-G.; Dai, H.-X.; Wasa, M.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 11948. doi: 10.1021/ja305259n
Miura, M.; Feng, C.-G.; Ma, S.; Yu, J.-Q. Org. Lett. 2013, 15, 5258. doi: 10.1021/ol402471y
Liu, C.; Chen, Q.-Y. Eur. J. Org. Chem. 2005, 3680
Cho, E. J.; Senecal, T. D.; Kinzel, T.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679. doi: 10.1126/science.1190524
Natte, K.; Jagadeesh, R. V.; He, L.; Rabeah, J.; Chen, J.-B.; Taeschler, C.; Ellinger, S.; Zaragoza, F.; Neumann, H.; Brückner, A.; Beller, M. Angew. Chem., Int. Ed. 2016, 55, 2782. doi: 10.1002/anie.201511131
Li, Z.-D.; García-Domínguez, A.; Nevado, C. J. Am. Chem. Soc. 2015, 137, 11610. doi: 10.1021/jacs.5b07432
Yu, J.-P.; Yang, H.-J.; Fu, H. Adv. Synth. Catal. 2014, 356, 3669. doi: 10.1002/adsc.v356.17
Xu, J.; Qiao, L.; Shen, J.-B.; Chai, K.-J.; Shen, C.; Zhang, P.-F. Org. Lett. 2017, 19, 5661. doi: 10.1021/acs.orglett.7b02823
Wu, Y.; Zhang, H.-R.; Jin, R.-X.; Lan, Q.; Wang, X.-S. Adv. Synth. Catal. 2016, 358, 3528. doi: 10.1002/adsc.v358.22
Sato, K.; Omote, M.; Ando, A.; Kumadaki, I. Org. Lett. 2004, 6, 4359. doi: 10.1021/ol048134v
Sato, K.; Yuki, T.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Tetrahedron Lett. 2008, 49, 3558. doi: 10.1016/j.tetlet.2008.04.030
Yuan, W.-M.; Eriksson, L.; Szabó, K. J. Angew. Chem., Int. Ed. 2016, 55, 8410. doi: 10.1002/anie.201602137
Liu, Y.-F.; Shao, X.-X.; Zhang, P.-P.; Lu, L.; Shen, Q.-L. Org. Lett. 2015, 17, 2752. doi: 10.1021/acs.orglett.5b01170
Zhang, H.-Y.; Ge, C.; Zhao, J.-Q.; Zhang, Y.-C. Org. Lett. 2017, 19, 5260. doi: 10.1021/acs.orglett.7b02353
Hossain, M. J.; Ono, T.; Wakiya, K.; Hisaeda, Y. Chem. Commun. 2017, 53, 10878. doi: 10.1039/C7CC06221D
Weng, Z.-Q.; Lee, R.; Jia, W.-G.; Yuan, Y.-F.; Wang, W.-F.; Feng, X.; Huang, K.-W. Organometallics 2011, 30, 3229. doi: 10.1021/om200204y
Xu, J.; Xiao, B.; Xie, C.-Q, Luo, D.-F.; Liu, L.; Fu, Y. Angew. Chem., Int. Ed. 2012, 51, 12551. doi: 10.1002/anie.201206681
Yin, J.; Li, Y.-M.; Zhang, R.; Jin, K.; Duan, C.-Y. Synthesis 2014, 46, 607.

图式 1 氨基炔烃实现三氟甲基化及胺化的历程
Scheme 1 Process of trifluoromethylation and amination of aminoalkynes

图式 2 芳香胺15在Ag促进下实现三氟甲基化的过程
Scheme 2 Process of trifluoromethylation of aromatic amines 15 under the promotion of Ag

图式 3 Ag促进烯烃17三氟甲基化反应的可能机理
Scheme 3 Plausible mechanism of Ag-promoted trifluoromethylation of alkene 17

图式 4 Ag促进苯炔化合物32三氟甲基化反应的可能机理
Scheme 4 Plausible mechanism of Ag-promoted trifluoromethylation of benzyne compound 32

图式 5 1-(三氟甲基)-4-氟-1, 2-二氢异喹啉(45)的合成
Scheme 5 Synthesis of 1-(trifluoromethyl)-4-fluoro-1, 2-dihy- droisoquinoline (45)

图式 6 Ag促进苯酚化合物54三氟甲基化反应的可能机理
Scheme 6 Plausible mechanism of Ag-promoted trifluoromethylation of phenol compound 54

图式 7 Ag催化芳基异腈74三氟甲基化反应的可能机理
Scheme 7 Plausible mechanism of Ag-catalyzed trifluoromethylation of aryl isonitrile 74

图式 8 苯酚类化合物54在Ag促进下实现三氟甲基化的过程
Scheme 8 Process of trifluoromethylation of phenolic compound 54 promoted by Ag

图式 9 硫酚类化合物88在Ag促进下实现三氟甲基化的过程
Scheme 9 Process of trifluoromethylation of thiophenolic compound 88 promoted by Ag

图式 10 Ag催化羧酸91三氟甲基化反应的可能机理
Scheme 10 Plausible mechanism of Ag-catalyzed trifluoromethylation of carboxylic acid 91

图式 11 (2, 2, 2-三氟乙基)酮(109)的合成
Scheme 11 Synthesis of (2, 2, 2-trifluoroethyl)-one (109)

图式 12 N-取代丙烯酰胺115在Fe促进下的三氟甲基化反应
Scheme 12 Trifluoromethylation of N-substituted acrylamide 115 promoted by Fe

图式 13 PdIV催化下形成Ar—CF3键的反应
Scheme 13 Bond formation of Ar—CF3 in presence of PdIV complex catalyst

图式 14 Ni催化底物196三氟甲基化反应的可能机理
Scheme 14 Plausible mechanism of Ni-catalyzed trifluoromethylation of substrate 196

图式 15 β-酮酸酯214在Rh催化下的三氟甲基化反应
Scheme 15 Trifluoromethylation of β-ketoester 214 catalyzed by Rh catalyst

图式 16 Co催化烯烃216三氟甲基化反应的可能机理
Scheme 16 Plausible mechanism of Co-catalyzed trifluoromethylation of alkene 216

图式 17 Cu/Ag催化碘代芳烃85三氟甲基化反应的可能机理
Scheme 17 Plausible mechanism of Cu/Ag-catalyzed trifluoromethylation of aryl iodide 85
表 1 四种不同的催化体系
Table 1. Four different kinds of catalytic systems
| Method | FeSO4 | Cp2Fe | H2O2 | H2SO4 |
| A | √ | √ | √ | |
| B | √ | √ | ||
| C | √ | √ | √ | |
| D | √ | √ | ||
| √ represents the presence of this compound in the system. | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 不同底物的三氟甲基化产物及产率
Table 2. Trifluoromethylation products and yields of different substrates
| Entry | Substrate | Product | Method |
| 1 | |
|
D |
| 2 | |
|
B |
| 3 | |
|
A |
| 4 | |
|
C |
下载: 导出CSV
表 3 不同催化剂对产率的影响
Table 3. Effects of different catalysts on yield
| Entry | Cat. | Oxidant | T/℃ | Yield/% |
| 1 | CuI | PhI(OAc)2 | 90 | 53 |
| 2 | Pd(OAc)2 | PhI(OAc)2 | 90 | 40 |
| 3 | FeCl3 | PhI(OAc)2 | 90 | 54 |
| 4 | NiCl2 | PhI(OAc)2 | 90 | 63 |
| 5 | — | PhI(OAc)2 | 90 | 72 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



































































 下载:
下载: